
αN-Acetyl β-Endorphin Is an Endogenous Ligand of σ1Rs That Regulates Mu-Opioid Receptor Signaling by Exchanging G Proteins for σ2Rs in σ1R Oligomers

Abstract
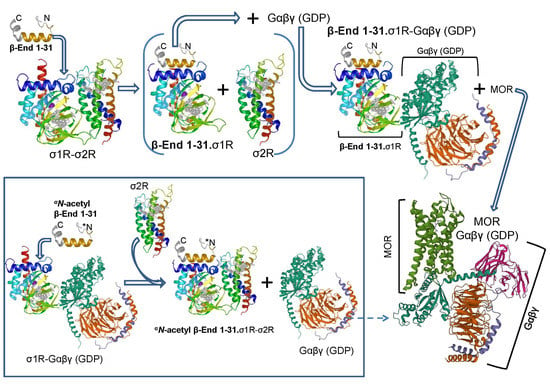
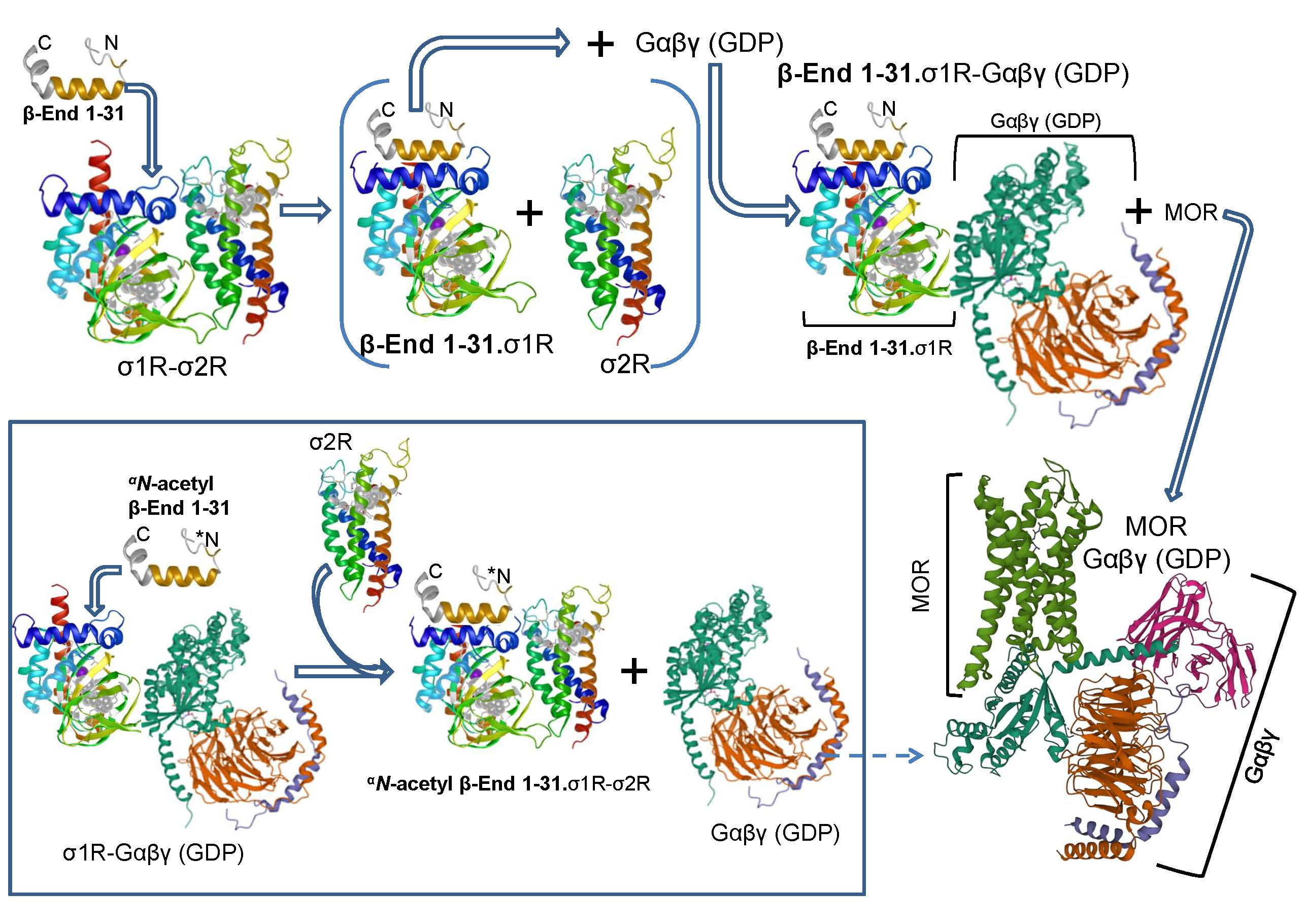{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. αN-Acetyl β-End 1–31 Diminishes the Neural Damage Caused by Permanent Unilateral Middle Cerebral Artery Occlusion (pMCAO)
2.2. Anticonvulsant Activity of αN-Acetyl β-End 1–31
2.3. Anti-Allodynia Effect of αN-Acetyl β-End 1–31
2.4. αN-Acetyl Derivatives and C-Terminal Sequences of β-End 1–31 Diminished Supraespinal Analgesia Produced by Morphine and β-End 1–31 in Mice
2.5. σ1R Binds to Gi/o/q-11/z Proteins: Effect of β-End 1–31 and αN-Acetyl β-End 1–31
2.6. In Vitro Activity of β-End 1–31 and Physiological Derivatives on σ1R Associations with σ2Rs, NMDAR NR1 C1 Subunits and BiP
3. Discussion
4. Materials and Methods
4.1. Animals and Drugs
4.2. Permanent Unilateral Middle Cerebral Artery Occlusion (pMCAO) and the Determination of Infarct Size
4.3. NMDA-Induced Seizures
4.4. Nerve Injury Pain Model
4.5. Evaluation of Analgesia
4.6. Immunoprecipitation and Western Blotting
4.7. Recombinant Protein Expression
4.8. In Vitro Interactions between Recombinant Proteins: Effect of Ligands on the σ1R-Proteins Interactions
4.9. Fishing Assays Using Agarose-σ1R as Bait and Solubilized Mouse Brain Synaptosomes: Effect of Opioid Peptides
4.10. Cell Culture
4.11. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic Lateral Sclerosis |
| β-End 1–31 | β-endorphin 1–31 |
| BD1047 | N′-[2-(3,4-dichlorophenyl)ethyl]-N,N,N′-trimethylethane-1,2-diamine;dihydrobromide |
| BD1063 | 1-[2-(3,4-dichlorophenyl)ethyl]-4-methylpiperazine |
| BiP | 78-kDa glucose-regulated binding protein (GRP78) |
| CCI | Unilateral sciatic nerve chronic contrition injury |
| CHO | Chinese hamster ovarian cells |
| CNS | Central nervous system |
| DADLE | ([D-Ala2, D-Leu5]-Enkephalin |
| D-Ala2 Deltorphin II | Tyr-D-Ala-Phe-Glu-Val-Val-Gly amide |
| DAMGO | ([D-Ala2, N-MePhe4, Gly-ol]-enkephalin |
| DOR | Delta-opioid receptor |
| DPDPE | ([D-Pen2,D-Pen5]enkephalin) |
| ε | Epsilon receptor |
| EC50 | Efficacious concentration to reduce 50% a given effect |
| ER | Endoplasmic reticulum |
| HINT1 | Histidine triad nucleotide binding protein 1 |
| GPCR | G-protein-coupled receptor |
| Icv | Intracerebroventricular |
| KOR | Kappa-opioid receptor |
| MOR | Mu-opioid receptor |
| αN-acetyl | Acetylated β-End N terminal Y1, α position |
| NMDA | N-methyl-d-aspartate acid |
| NMDAR | N-methyl-d-aspartate acid glutamate receptor |
| PAG | Periaqueductal grey matter |
| pMCAO | Permanent unilateral middle cerebral artery occlusion |
| POMC | Proopiomelanocortin |
| (+/−)PPCC | (1R,2S/1S,2R)-2-[4-hydroxy-4-phenylpiperidin-1-yl)methyl]-1-(4-methylphenyl) cyclopropanecarboxylate |
| PRE084 | 2-(4-morpholinethyl)-1-phenylcyclohexanecarboxylate hydrochloride |
| PTX | Pertussis toxin |
| RVD | Rat vas deferens |
| σ1R | Type 1 sigma receptor |
| σ2R | Type 2 sigma receptor |
| SBM | Solubilized brain membranes enriched in synaptosomes |
| SKF-10,047 | (2S,6S,11S)-3-Allyl-1,2,3,4,5,6-hexahydro-6,11-dimethyl-2,6-methano-3-benzazocine-8-ol |
| U-50,488H | Trans-3,4-Dichloro-N-methyl-N-[2-(1-pyrrolidinyl)cyclohexyl]-benzeneacetamide |
| WT | Wild-type |
References
- Li, C.H.; Chung, D. Isolation and structure of an untriakontapeptide with opiate activity from camel pituitary glands. Proc. Natl. Acad. Sci. USA 1976, 73, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Akil, H.; Shiomi, H.; Matthews, J. Induction of the intermediate pituitary by stress: Synthesis and release of a nonopioid form of beta-endorphin. Science 1985, 227, 424–426. [Google Scholar] [CrossRef] [PubMed]
- Patey, G.; Rossier, J. Discovery, anatomical mapping and biosynthesis of various families of endogenous opioid peptides. Ann. Endocrinol. 1986, 47, 71–87. [Google Scholar]
- Nicolas, P.; Li, C.H. Beta-endorphin-(1-27) is a naturally occurring antagonist to etorphine-induced analgesia. Proc. Natl. Acad. Sci. USA 1985, 82, 3178–3181. [Google Scholar] [CrossRef]
- Smyth, D.G.; Massey, D.E.; Zakarian, S.; Finnie, M.D. Endorphins are stored in biologically active and inactive forms: Isolation of α-N-acetyl peptides. Nature 1979, 279, 252–254. [Google Scholar] [CrossRef]
- Parish, D.C.; Smyth, D.G.; Normanton, J.R.; Wolstencroft, J.H. Glycyl glutamine, an inhibitory neuropeptide derived from beta-endorphin. Nature 1983, 306, 267–270. [Google Scholar] [CrossRef]
- Ng, T.B.; Chung, D.; Li, C.H. Isolation and properties of beta-endorphin-(1-27), N alpha-acetyl-beta-endorphin, corticotropin, gamma-lipotropin and neurophysin from equine pituitary glands. Int. J. Pept. Protein Res. 1981, 18, 443–450. [Google Scholar] [CrossRef]
- Smyth, D.G. β-Endorphin and related peptides in pituitary, brain, pancreas and antrum. Br. Med. Bull. 1983, 39, 25–30. [Google Scholar] [CrossRef]
- Zakarian, S.; Smyth, D. Distribution of active and inactive forms of endorphins in rat pituitary and brain. Proc. Natl. Acad. Sci. USA 1979, 76, 5972–5976. [Google Scholar] [CrossRef]
- Li, C.H.; Tseng, L.F.; Jibson, M.D.; Hammonds, R.G., Jr.; Yamashiro, D.; Zaoral, M. beta-Endorphin- (1-27): Acetylation of alpha-amino groups enhances immunoreactivity but diminishes analgesic and receptor-binding activities with no changes in circular dichroism spectra. Biochem. Biophys. Res. Commun. 1980, 97, 932–938. [Google Scholar] [CrossRef]
- Zaoral, M.; Yamashiro, D.; Hammonds, R.G., Jr.; Li, C.H. Beta-Endorphin: Synthesis and radioreceptor binding activity of Beta h-endorphin-(1-27) and its analogs. Int. J. Pept. Protein Res. 1981, 17, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Hammonds, R.G., Jr.; Nicolas, P.; Li, C.H. beta-endorphin-(1-27) is an antagonist of beta-endorphin analgesia. Proc. Natl. Acad. Sci. USA 1984, 81, 1389–1390. [Google Scholar] [CrossRef] [PubMed]
- Unal, C.B.; Owen, M.D.; Millington, W.R. Beta-endorphin-induced cardiorespiratory depression is inhibited by glycyl-L-glutamine, a dipeptide derived from beta-endorphin processing. J. Pharmacol. Exp. Ther. 1994, 271, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Garzón, J.; Sánchez-Blázquez, P. αN-acetyl derivatives of β-endorphin-(1-31) and -(1-27) regulate the supraspinal antinociceptive activity of different opioids in mice. Life Sci. 1991, 48, 1417–1427. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sánchez-Blázquez, P.; Garzón, J. Further characterization of αN-acetyl β-endorphin-(1-31) regulatory activity, I: Effect on opioid- and α2-mediated supraspinal antinociception in mice. Life Sci. 1992, 50, 2083–2097. [Google Scholar] [CrossRef]
- Sánchez-Blázquez, P.; Garzón, J. N-acetyl-endorphin-(1-31) and substance P regulate the supraspinal antinociception mediated by mu opioid and alpha-2 adrenoceptors but not by delta opioid receptors in the mouse. J. Pharmacol. Exp. Ther. 1993, 265, 835–843. [Google Scholar]
- Garzón, J.; Sánchez-Blázquez, P., II. αN-acetyl human-endorphin-(1-31) alleviates the morphine withdrawal syndrome in rodents: A comparative study with clonidine. Life Sci. 1992, 50, 2099–2109. [Google Scholar] [CrossRef]
- Cavun, S.; Goktalay, G.; Millington, W.R. Glycyl-glutamine, an endogenous beta-endorphin-derived peptide, inhibits morphine-induced conditioned place preference, tolerance, dependence, and withdrawal. J. Pharmacol. Exp. Ther. 2005, 315, 949–958. [Google Scholar] [CrossRef]
- Schülz, R.; Faase, E.; Wüster, M.; Herz, A. Selective receptors for beta-endorphin on the rat vas deferens. Life Sci. 1979, 24, 843–849. [Google Scholar] [CrossRef]
- Schülz, R.; Wüster, M.; Herz, A. Pharmacological characterization of the epsilon-opiate receptor. J. Pharmacol. Exp. Ther. 1981, 216, 604–606. [Google Scholar]
- Suh, H.H.; Tseng, L.F.; Li, C.H. β-endorphin-(1-27) antagonizes β-endorphin- but not morphine-, D-Pen2-D-Pen5-enkephalin- and U50,488H-induced analgesia in mice. Neuropharmacology 1988, 27, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Suh, H.H.; Tseng, L.F. Lack of antinociceptive cross-tolerance between intracerebroventricularly administered beta-endorphin and morphine or DPDPE in mice. Life Sci. 1990, 46, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Tseng, L.F. Evidence for epsilon-opioid receptor-mediated beta-endorphin-induced analgesia. Trends Pharmacol. Sci. 2001, 22, 623–630. [Google Scholar] [CrossRef]
- Narita, M.; Tseng, L.F. Evidence for the existence of the β-endorphin-sensitive “ε-opioid receptor” in the brain: The mechanisms of ε-mediated antinociception. Jpn. J. Pharmacol. 1998, 76, 233–253. [Google Scholar] [CrossRef]
- Contet, C.; Matifas, A.; Kieffer, B.L. No evidence for G-protein-coupled epsilon receptor in the brain of triple opioid receptor knockout mouse. Eur. J. Pharmacol. 2004, 492, 131–136. [Google Scholar] [CrossRef]
- Livingston, K.E.; Traynor, J.R. Allostery at opioid receptors: Modulation with small molecule ligands. Br. J. Pharmacol. 2018, 175, 2846–2856. [Google Scholar] [CrossRef]
- Mizoguchi, H.; Narita, M.; Nagase, H.; Tseng, L.F. Activation of G-proteins in the mouse pons/medulla by beta-endorphin is mediated by the stimulation of mu- and putative epsilon-receptors. Life Sci. 2000, 67, 2733–2743. [Google Scholar] [CrossRef]
- Rodríguez-Muñoz, M.; Sánchez-Blázquez, P.; Herrero-Labrador, R.; Martínez-Murillo, R.; Merlos, M.; Vela, J.M.; Garzón, J. The sigma1 receptor engages the redox-regulated HINT1 protein to bring opioid analgesia under NMDA receptor negative control. Antioxid. Redox. Signal. 2015, 22, 799–818. [Google Scholar] [CrossRef]
- Itzhak, Y.; Kalir, A.; Sarne, Y. On the opioid nature of phencyclidine and its 3-hydroxy derivative. Eur. J. Pharmacol. 1981, 73, 229–233. [Google Scholar] [CrossRef]
- Martin, W.R.; Eades, C.G.; Thompson, J.A.; Huppler, R.E.; Gilbert, P.E. The effects of morphine- and nalorphine- like drugs in the nondependent and morphine-dependent chronic spinal dog. J. Pharmacol. Exp. Ther. 1976, 197, 517–532. [Google Scholar]
- Mei, J.; Pasternak, G.W. Sigma1 receptor modulation of opioid analgesia in the mouse. J. Pharmacol. Exp. Ther. 2002, 300, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Torres, A.; de la Puente, B.; Rocasalbas, M.; Tourino, C.; Bura, S.A.; Fernandez-Pastor, B.; Romero, L.; Codony, X.; Zamanillo, D.; Buschmann, H.; et al. Sigma-1 receptor antagonism as opioid adjuvant strategy: Enhancement of opioid antinociception without increasing adverse effects. Eur. J. Pharmacol. 2013, 711, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Gillan, M.G.; Kosterlitz, H.W.; Magnan, J. Unexpected antagonism in the rat vas deferens by benzomorphans which are agonists in other pharmacological tests. Br. J. Pharmacol. 1981, 72, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.J.; Blanchard, S.G.; Cuatrecasas, P. Benzomorphan sites are ligand recognition sites of putative epsilon-receptors. Mol. Pharmacol. 1984, 26, 484–488. [Google Scholar] [PubMed]
- Nock, B.; Giordano, A.L.; Cicero, T.J.; O’Connor, L.H. Affinity of drugs and peptides for U-69,593-sensitive and -insensitive kappa opiate binding sites: The U-69,593-insensitive site appears to be the beta endorphin-specific epsilon receptor. J. Pharmacol. Exp. Ther. 1990, 254, 412–419. [Google Scholar] [PubMed]
- Nock, B.; Giordano, A.L.; Moore, B.W.; Cicero, T.J. Properties of the putative epsilon opioid receptor: Identification in rat, guinea pig, cow, pig and chicken brain. J. Pharmacol. Exp. Ther. 1993, 264, 349–359. [Google Scholar]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef]
- Rodríguez-Muñoz, M.; Onetti, Y.; Cortés-Montero, E.; Garzón, J.; Sánchez-Blázquez, P. Cannabidiol enhances morphine antinociception, diminishes NMDA-mediated seizures and reduces stroke damage via the sigma 1 receptor. Mol. Brain. 2018, 11, 51. [Google Scholar] [CrossRef]
- Sánchez-Blázquez, P.; Pozo-Rodrigalvarez, A.; Merlos, M.; Garzón, J. The Sigma-1 Receptor Antagonist, S1RA, Reduces Stroke Damage, Ameliorates Post-Stroke Neurological Deficits and Suppresses the Overexpression of MMP-9. Mol. Neurobiol. 2018, 55, 4940–4951. [Google Scholar] [CrossRef]
- Mathis, C.; Ungerer, A. Comparative analysis of seizures induced by intracerebroventricular administration of NMDA, kainate and quisqualate in mice. Exp. Brain Res. 1992, 88, 277–282. [Google Scholar] [CrossRef]
- Moreau, J.L.; Pieri, L.; Prud’hon, B. Convulsions induced by centrally administered NMDA in mice: Effects of NMDA antagonists, benzodiazepines, minor tranquilizers and anticonvulsants. Br. J. Pharmacol. 1989, 98, 1050–1054. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Muñoz, M.; Sánchez-Blázquez, P.; Garzón, J. Fenfluramine diminishes NMDA receptor-mediated seizures via its mixed activity at serotonin 5HT2A and type 1 sigma receptors. Oncotarget 2018, 9, 23373–23389. [Google Scholar] [CrossRef] [PubMed]
- Bennett, G.J.; Xie, Y.K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988, 33, 87–107. [Google Scholar] [CrossRef] [PubMed]
- Garzón, J.; Herrero-Labrador, R.; Rodríguez-Muñoz, M.; Shah, R.; Vicente-Sánchez, A.; Wagner, C.R.; Sánchez-Blázquez, P. HINT1 protein: A new therapeutic target to enhance opioid antinociception and block mechanical allodynia. Neuropharmacology 2015, 89, 412–423. [Google Scholar] [CrossRef]
- Akil, H.; Young, E.; Watson, S.J.; Coy, D.H. Opiate binding properties of naturally occurring N- and C-terminus modified β-endorphins. Peptides 1981, 2, 289–292. [Google Scholar] [CrossRef]
- Deakin, J.F.; Dostrovsky, J.O.; Smyth, D.G. Influence of N-terminal acetylation and C-terminal proteolysis on the analgesic activity of -endorphin. Biochem. J. 1980, 189, 501–506. [Google Scholar] [CrossRef]
- Gromek, K.A.; Suchy, F.P.; Meddaugh, H.R.; Wrobel, R.L.; LaPointe, L.M.; Chu, U.B.; Primm, J.G.; Ruoho, A.E.; Senes, A.; Fox, B.G. The oligomeric states of the purified sigma-1 receptor are stabilized by ligands. J. Biol. Chem. 2014, 289, 20333–20344. [Google Scholar] [CrossRef]
- Mishra, A.K.; Mavlyutov, T.; Singh, D.R.; Biener, G.; Yang, J.; Oliver, J.A.; Ruoho, A.; Raicu, V. The sigma-1 receptors are present in monomeric and oligomeric forms in living cells in the presence and absence of ligands. Biochem. J. 2015, 466, 263–271. [Google Scholar] [CrossRef]
- Garzón, J.; de Antonio, I.; Sánchez-Blázquez, P. In vivo modulation of G proteins and opioid receptor function by antisense oligodeoxynucleotides. Methods Enzymol. 2000, 314, 3–20. [Google Scholar] [CrossRef]
- Rossi, G.C.; Standifer, K.M.; Pasternak, G.W. Differential blockade of morphine and morphine-6 beta-glucuronide analgesia by antisense oligodeoxynucleotides directed against MOR-1 and G-protein alpha subunits in rats. Neurosci. Lett. 1995, 198, 99–102. [Google Scholar] [CrossRef]
- Raffa, R.B.; Martinez, R.P.; Connelly, C.D. G-protein antisense oligodeoxyribonucleotides and mu-opioid supraspinal antinociception. Eur. J. Pharmacol. 1994, 258, R5–R7. [Google Scholar] [CrossRef] [PubMed]
- Downes, G.B.; Gautam, N. The G protein subunit gene families. Genomics 1999, 62, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Fong, H.K.; Yoshimoto, K.K.; Eversole-Cire, P.; Simon, M.I. Identification of a GTP-binding protein alpha subunit that lacks an apparent ADP-ribosylation site for pertussis toxin. Proc. Natl. Acad. Sci. USA 1988, 85, 3066–3070. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Blázquez, P.; Cortés-Montero, E.; Rodríguez-Muñoz, M.; Merlos, M.; Garzón-Niño, J. The Sigma 2 receptor promotes and the Sigma 1 receptor inhibits mu-opioid receptor-mediated antinociception. Mol. Brain 2020, 13, 150. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, S.; Imagawa, Y.; Ogawa, S.; Araki, H.; Ajima, A.; Tanaka, M.; Muramatsu, M.; Nakazato, A.; Yamaguchi, K.; Yoshida, M.; et al. NE-100, a novel sigma receptor ligand: In vivo. tests Life Sci. 1993, 53, L285–L290. [Google Scholar] [CrossRef]
- Rodríguez-Muñoz, M.; Cortés-Montero, E.; Pozo-Rodrigalvarez, A.; Sánchez-Blázquez, P.; Garzón-Niño, J. The ON:OFF switch, sigma1R-HINT1 protein, controls GPCR-NMDA receptor cross-regulation: Implications in neurological disorders. Oncotarget 2015, 6, 35458–35477. [Google Scholar] [CrossRef]
- Al-Saif, A.; Al-Mohanna, F.; Bohlega, S. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 2011, 70, 913–919. [Google Scholar] [CrossRef]
- Rodríguez-Muñoz, M.; Cortés-Montero, E.; Garzón-Niño, J.; Sánchez-Blázquez, P. The ALS-related sigma1R E102Q Mutant Eludes Ligand Control and Exhibits Anomalous Response to Calcium. Int. J. Mol. Sci. 2020, 21, 7339. [Google Scholar] [CrossRef]
- Kim, F.J.; Kovalyshyn, I.; Burgman, M.; Neilan, C.; Chien, C.C.; Pasternak, G.W. Sigma 1 receptor modulation of G-protein-coupled receptor signaling: Potentiation of opioid transduction independent from receptor binding. Mol. Pharmacol. 2010, 77, 695–703. [Google Scholar] [CrossRef]
- Palmer, C.P.; Mahen, R.; Schnell, E.; Djamgoz, M.B.; Aydar, E. Sigma-1 receptors bind cholesterol and remodel lipid rafts in breast cancer cell lines. Cancer Res. 2007, 67, 11166–11175. [Google Scholar] [CrossRef]
- Penke, B.; Fulop, L.; Szucs, M.; Frecska, E. The Role of Sigma-1 Receptor, an Intracellular Chaperone in Neurodegenerative Diseases. Curr. Neuropharmacol. 2018, 16, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Blanc, J.P.; Kaiser, E.T. Biological and physical properties of a beta-endorphin analog containing only D-amino acids in the amphiphilic helical segment 13-31. J. Biol. Chem. 1984, 259, 9549–9556. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Roldan, J.L.; Ossa, F.; Schnell, J.R. Characterization of the human sigma-1 receptor chaperone domain structure and binding immunoglobulin protein (BiP) interactions. J Biol. Chem. 2013, 288, 21448–21457. [Google Scholar] [CrossRef] [PubMed]
- Blanc, J.P.; Taylor, J.W.; Miller, R.J.; Kaiser, E.T. Examination of the requirement for an amphiphilic helical structure in beta-endorphin through the design, synthesis, and study of model peptides. J. Biol. Chem. 1983, 258, 8277–8284. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Blázquez, P.; Rodríguez-Muñoz, M.; Herrero-Labrador, R.; Burgueño, J.; Zamanillo, D.; Garzón, J. The calcium-sensitive Sigma-1 receptor prevents cannabinoids from provoking glutamate NMDA receptor hypofunction: Implications in antinociception and psychotic diseases. Int. J. Neuropsychopharmacol. 2014, 17, 1943–1955. [Google Scholar] [CrossRef]
- Chu, U.B.; Ruoho, A.E. Biochemical Pharmacology of the Sigma-1 Receptor. Mol. Pharmacol. 2016, 89, 142–153. [Google Scholar] [CrossRef]
- Li, S.; Okamoto, T.; Chun, M.; Sargiacomo, M.; Casanova, J.E.; Hansen, S.H.; Nishimoto, I.; Lisanti, M.P. Evidence for a regulated interaction between heterotrimeric G proteins and caveolin. J. Biol. Chem. 1995, 270, 15693–15701. [Google Scholar] [CrossRef]
- Higashijima, T.; Burnier, J.; Ross, E.M. Regulation of Gi and Go by mastoparan, related amphiphilic peptides, and hydrophobic amines. Mechanism and structural determinants of activity. J. Biol. Chem. 1990, 265, 14176–14186. [Google Scholar] [CrossRef]
- Sánchez-Blázquez, P.; Garzón, J. αN-acetyl-β-endorphin-(1-31) disrupts the diminishing effect of mastoparan on opioid- and clonidine-evoked supraspinal antinociception in mice. J. Pharmacol. Exp. Ther. 1995, 273, 787–792. [Google Scholar]
- Chapman, D.B.; Hu, J.; Way, E.L. Methionine-enkephalin antagonism and endorphin potentiation of narcotic-induced analgesia. Eur. J. Pharmacol. 1980, 65, 369–377. [Google Scholar] [CrossRef]
- Garzón, J.; Rodríguez-Muñoz, M.; López-Fando, A.; Sánchez-Blázquez, P. Activation of mu-opioid receptors transfers control of Galpha subunits to the regulator of G-protein signaling RGS9-2: Role in receptor desensitization. J. Biol. Chem. 2005, 280, 8951–8960. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Blázquez, P.; García-España, A.; Garzón, J. In vivo injection of antisense oligodeoxynucleotides to G alpha subunits and supraspinal analgesia evoked by mu and delta opioid agonists. J. Pharmacol. Exp. Ther. 1995, 275, 1590–1596. [Google Scholar] [PubMed]
- Sánchez-Blázquez, P.; Gómez-Serranillos, P.; Garzón, J. Agonists determine the pattern of G-protein activation in mu-opioid receptor-mediated supraspinal analgesia. Brain Res. Bull. 2001, 54, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Garzon, J.; Rodriguez-Munoz, M.; Lopez-Fando, A.; Sanchez-Blazquez, P. The RGSZ2 protein exists in a complex with mu-opioid receptors and regulates the desensitizing capacity of Gz proteins. Neuropsychopharmacology 2005, 30, 1632–1648. [Google Scholar] [CrossRef] [PubMed]
- Hinton, D.R.; Blanks, J.C.; Fong, H.K.; Casey, P.J.; Hildebrandt, E.; Simons, M.I. Novel localization of a G protein, Gz-alpha, in neurons of brain and retina. J. Neurosci. 1990, 10, 2763–2770. [Google Scholar] [CrossRef] [PubMed]
- Delander, G.E.; Portoghese, P.S.; Takemori, A.E. Role of spinal mu opioid receptors in the development of morphine tolerance and dependence. J. Pharmacol. Exp. Ther. 1984, 231, 91–96. [Google Scholar]
- Leck, K.J.; Bartlett, S.E.; Smith, M.T.; Megirian, D.; Holgate, J.; Powell, K.L.; Matthaei, K.I.; Hendry, I.A. Deletion of guanine nucleotide binding protein alpha z subunit in mice induces a gene dose dependent tolerance to morphine. Neuropharmacology 2004, 46, 836–846. [Google Scholar] [CrossRef]
- Garzón, J.; Martínez-Peña, Y.; Sánchez-Blázquez, P. Dissimilar efficacy of opioids to produce mu-mediated analgesia: Role of Gx/z and Gi2 transducer proteins. Life Sci. 1994, 55, L205–L212. [Google Scholar] [CrossRef]
- Sánchez-Blázquez, P.; Garzon, J. Antibodies directed against alpha subunits of Gi, Gx/z, Go and Gs transducer proteins reduced the morphine withdrawal syndrome in mice. Life Sci. 1994, 55, L445–L450. [Google Scholar] [CrossRef]
- Vaught, J.L.; Takemori, A.E. Differential effects of leucine and methionine enkephalin on morphine-induced analgesia, acute tolerance and dependence. J. Pharmacol. Exp. Ther. 1979, 208, 86–90. [Google Scholar]
- Porreca, F.; Takemori, A.E.; Sultana, M.; Portoghese, P.S.; Bowen, W.D.; Mosberg, H.I. Modulation of mu-mediated antinociception in the mouse involves opioid delta-2 receptors. J. Pharmacol. Exp. Ther. 1992, 263, 147–152. [Google Scholar] [PubMed]
- Huidobro-Toro, J.P.; Caturay, E.M.; Ling, N.; Lee, N.M.; Loh, H.H.; Way, E.L. Studies on the structural prerequisites for the activation of the beta-endorphin receptor on the rat vas deferens. J. Pharmacol. Exp. Ther. 1982, 222, 262–269. [Google Scholar] [PubMed]
- Shook, J.E.; Kazmierski, W.; Wire, W.S.; Lemcke, P.K.; Hruby, V.J.; Burks, T.F. Opioid receptor selectivity of beta-endorphin in vitro and in vivo: Mu, delta and epsilon receptors. J. Pharmacol. Exp. Ther. 1988, 246, 1018–1025. [Google Scholar] [PubMed]
- Wüster, M.; Schülz, R.; Herz, A. The direction of opiodid agonists towards mu-, delta- and epsilon-receptors in the vas deferens of the mouse and the rat. Life Sci. 1980, 27, 163–170. [Google Scholar] [CrossRef]
- Tseng, L.F.; Wang, Q. Forebrain sites differentially sensitive to beta-endorphin and morphine for analgesia and release of Met-enkephalin in the pentobarbital-anesthesized rat. J. Pharmacol. Exp. Ther. 1992, 261, 1028–1036. [Google Scholar]
- Sheehan, M.J.; Hayes, A.G.; Tyers, M.B. Lack of evidence for epsilon-opioid receptors in the rat vas deferens. Eur. J. Pharmacol. 1988, 154, 237–245. [Google Scholar] [CrossRef] [PubMed]
- HALEY, T.J.; MCCORMICK, W.G. Pharmacological effects produced by intracerebral injection of drugs in the conscious mouse. Br. J. Pharmacol. Chemother. 1957, 12, 12–15. [Google Scholar] [CrossRef]
- Garzon, J.; Juarros, J.L.; Castro, M.A.; Sanchez-Blazquez, P. Antibodies to the cloned mu-opioid receptor detect various molecular weight forms in areas of mouse brain. Mol. Pharmacol. 1995, 47, 738–744. [Google Scholar] [PubMed]
- Rodríguez-Muñoz, M.; Sánchez-Blázquez, P.; Vicente-Sánchez, A.; Berrocoso, E.; Garzón, J. The mu-opioid receptor and the NMDA receptor associate in PAG neurons: Implications in pain control. Neuropsychopharmacology 2012, 37, 338–349. [Google Scholar] [CrossRef]
- Rodríguez-Muñoz, M.; Cortés-Montero, E.; Onetti, Y.; Sánchez-Blázquez, P.; Garzón-Niño, J. The sigma1 Receptor and the HINT1 Protein Control alpha2delta1 Binding to Glutamate NMDA Receptors: Implications in Neuropathic Pain. Biomolecules 2021, 11, 1681. [Google Scholar] [CrossRef]
- Garzon, J.; Rodriguez-Munoz, M.; Sanchez-Blazquez, P. Morphine alters the selective association between mu-opioid receptors and specific RGS proteins in mouse periaqueductal gray matter. Neuropharmacology 2005, 48, 853–868. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Blázquez, P.; García-España, A.; Garzón, J. Antisense oligodeoxynucleotides to opioid mu and delta receptors reduced morphine dependence in mice: Role of delta-2 opioid receptors. J. Pharmacol. Exp. Ther. 1997, 280, 1423–1431. [Google Scholar] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garzón-Niño, J.; Cortés-Montero, E.; Rodríguez-Muñoz, M.; Sánchez-Blázquez, P. αN-Acetyl β-Endorphin Is an Endogenous Ligand of σ1Rs That Regulates Mu-Opioid Receptor Signaling by Exchanging G Proteins for σ2Rs in σ1R Oligomers. Int. J. Mol. Sci. 2023, 24, 582. https://doi.org/10.3390/ijms24010582
Garzón-Niño J, Cortés-Montero E, Rodríguez-Muñoz M, Sánchez-Blázquez P. αN-Acetyl β-Endorphin Is an Endogenous Ligand of σ1Rs That Regulates Mu-Opioid Receptor Signaling by Exchanging G Proteins for σ2Rs in σ1R Oligomers. International Journal of Molecular Sciences. 2023; 24(1):582. https://doi.org/10.3390/ijms24010582
Chicago/Turabian StyleGarzón-Niño, Javier, Elsa Cortés-Montero, María Rodríguez-Muñoz, and Pilar Sánchez-Blázquez. 2023. "αN-Acetyl β-Endorphin Is an Endogenous Ligand of σ1Rs That Regulates Mu-Opioid Receptor Signaling by Exchanging G Proteins for σ2Rs in σ1R Oligomers" International Journal of Molecular Sciences 24, no. 1: 582. https://doi.org/10.3390/ijms24010582
APA StyleGarzón-Niño, J., Cortés-Montero, E., Rodríguez-Muñoz, M., & Sánchez-Blázquez, P. (2023). αN-Acetyl β-Endorphin Is an Endogenous Ligand of σ1Rs That Regulates Mu-Opioid Receptor Signaling by Exchanging G Proteins for σ2Rs in σ1R Oligomers. International Journal of Molecular Sciences, 24(1), 582. https://doi.org/10.3390/ijms24010582