
The Enrichment of Specific Hair Follicle-Associated Cell Populations in Plucked Hairs Offers an Opportunity to Study Gene Expression Underlying Hair Traits

 , , , and
, , , and 
Abstract
1. Introduction
2. Results
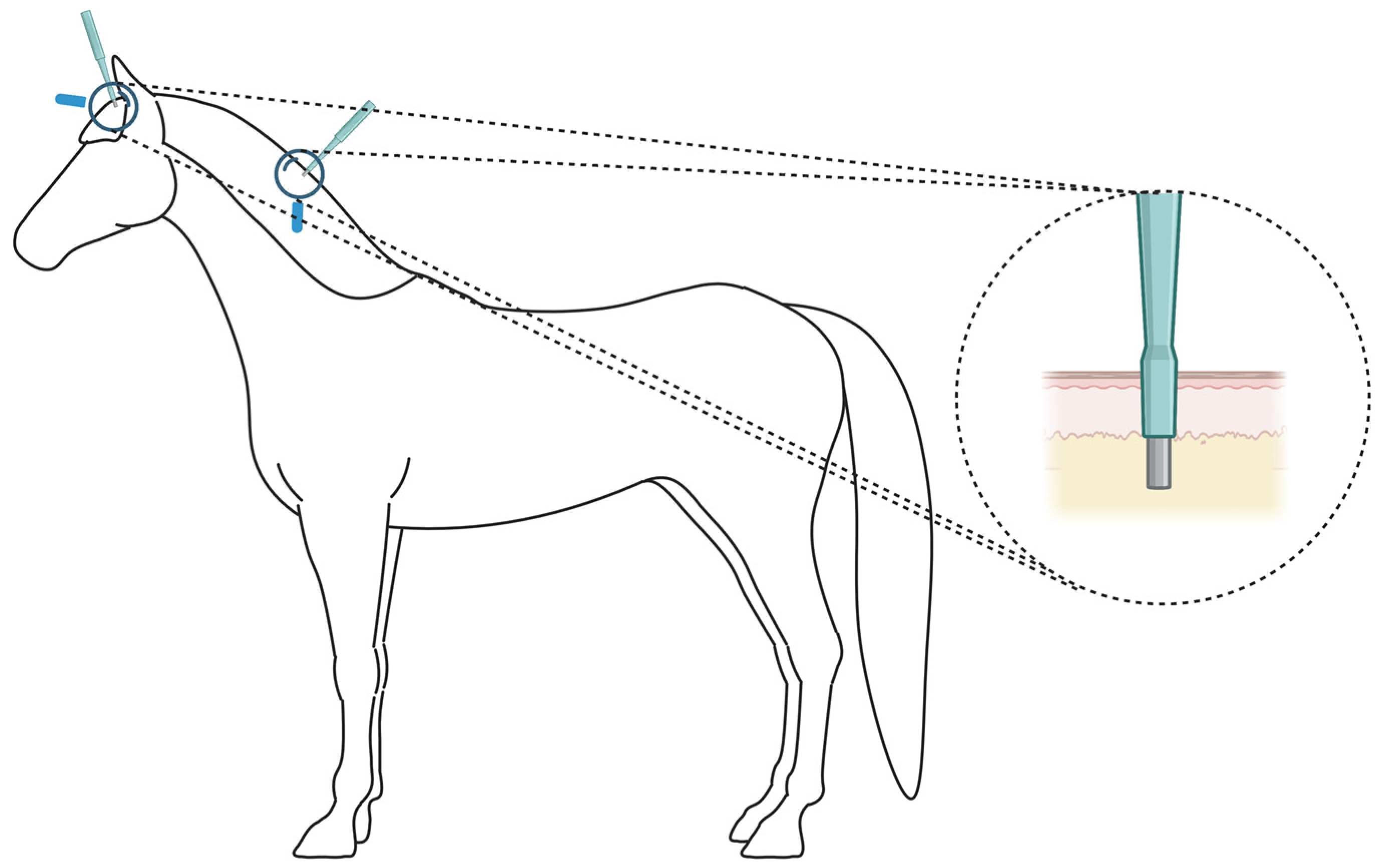
2.1. Non-Invasive Hair-Plucking and Invasive Skin-Biopsy Sample-Collection Methods Yield Comparable Quality and Quantity of RNA-Seq Results
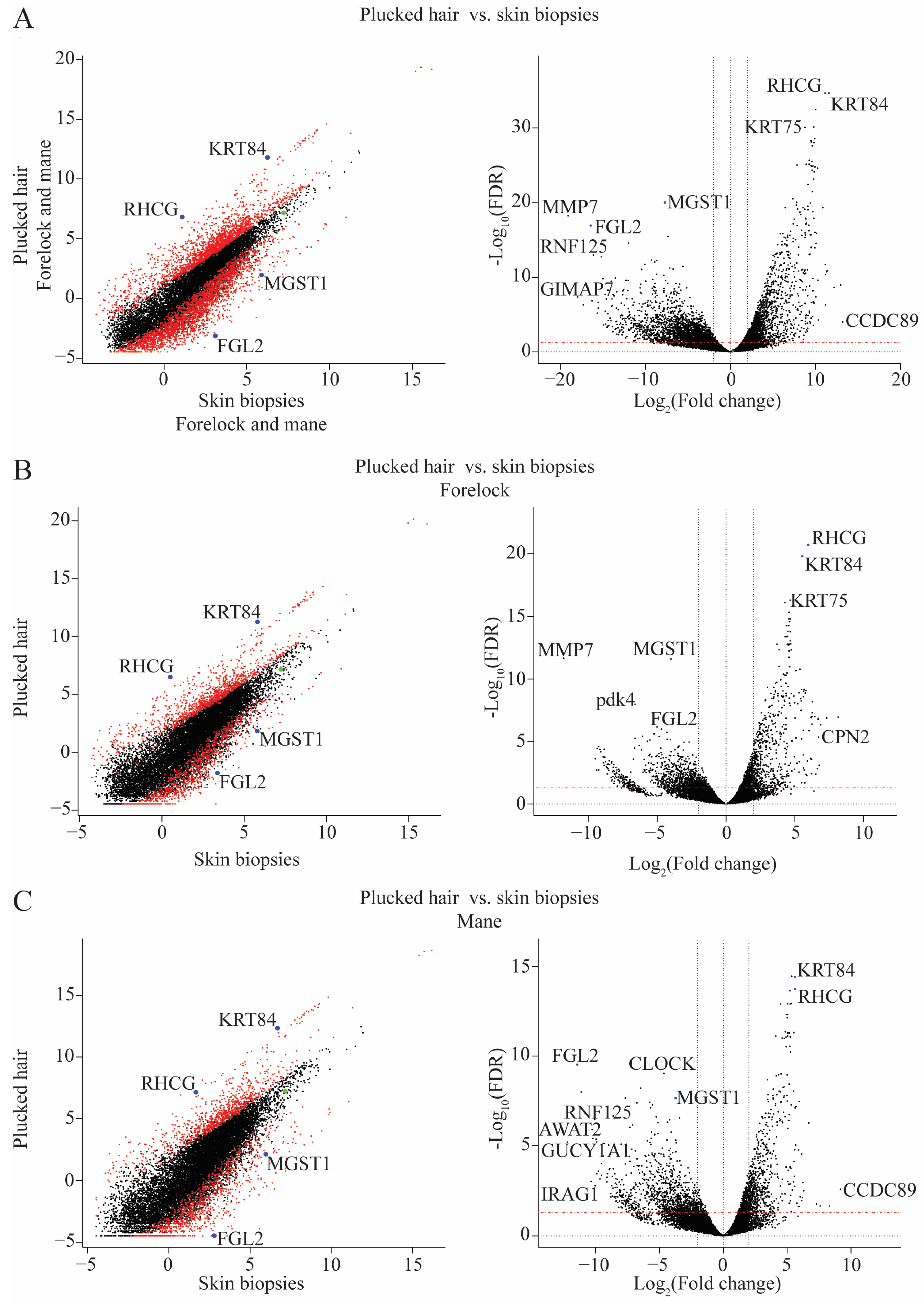
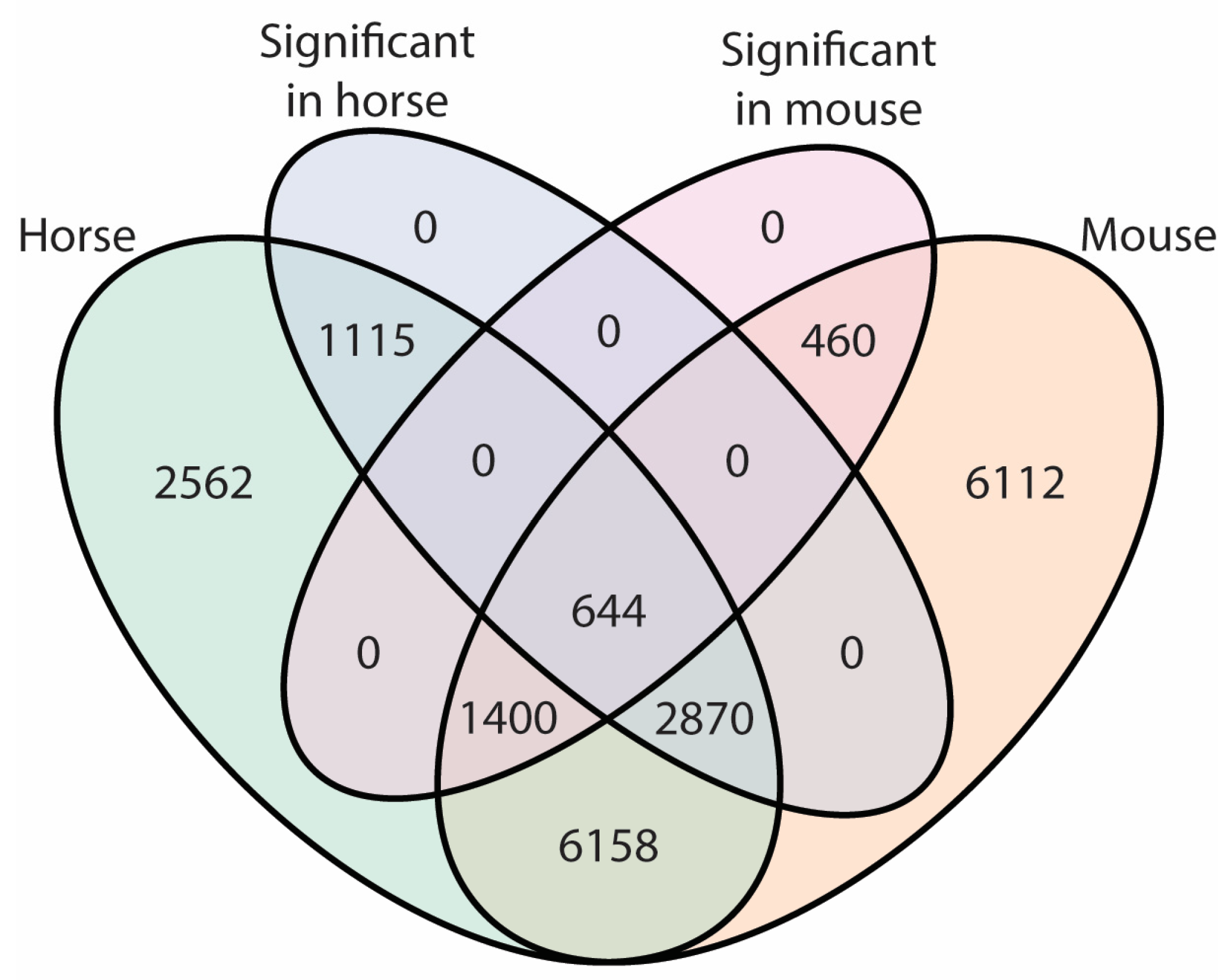
2.2. Differences in Transcriptome between Plucked Hairs and Skin Biopsies Affect Keratin-Related Genes and Cellular Process Enzymes
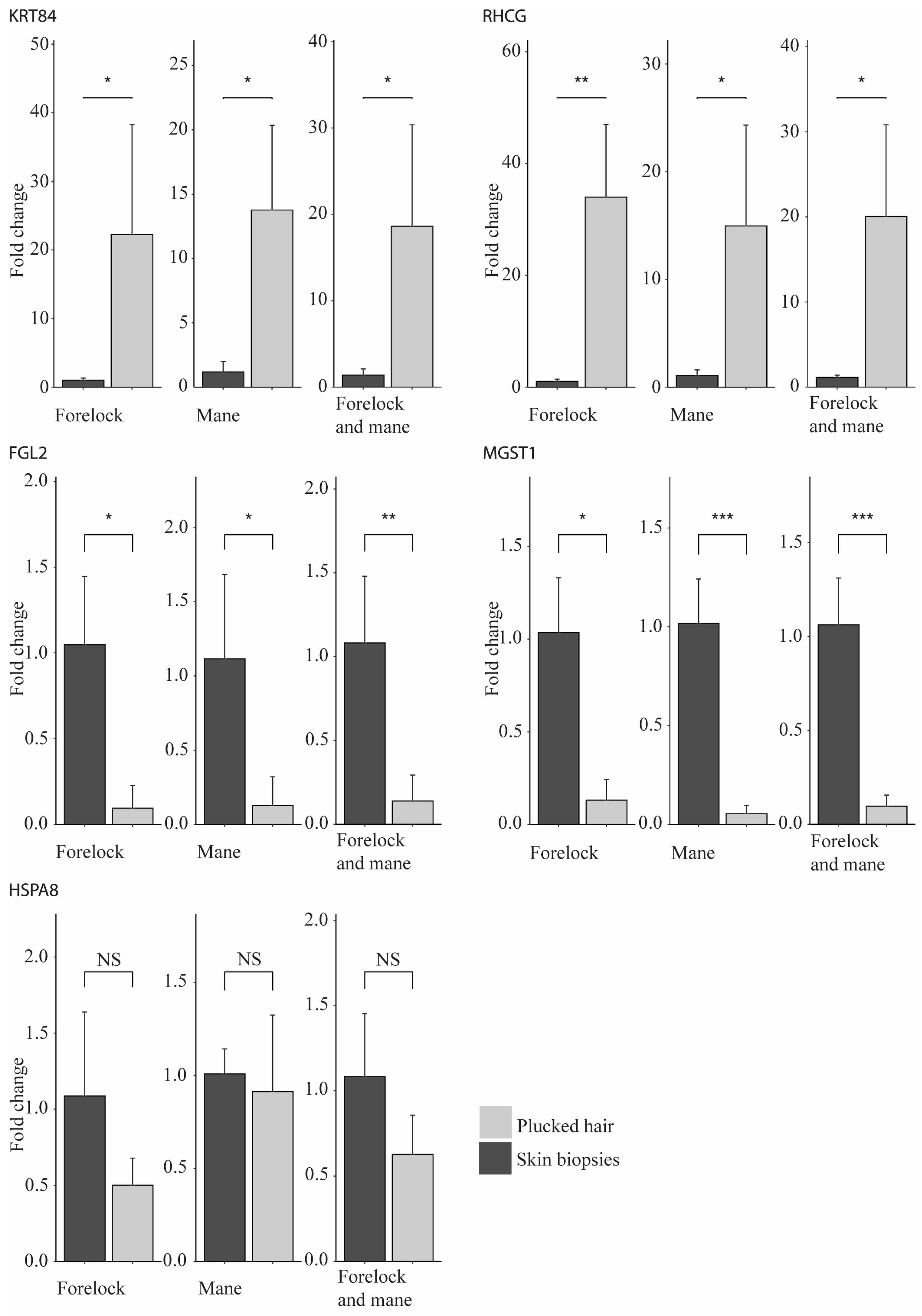
2.3. Validation of the RNA-Seq Results Using qPCR
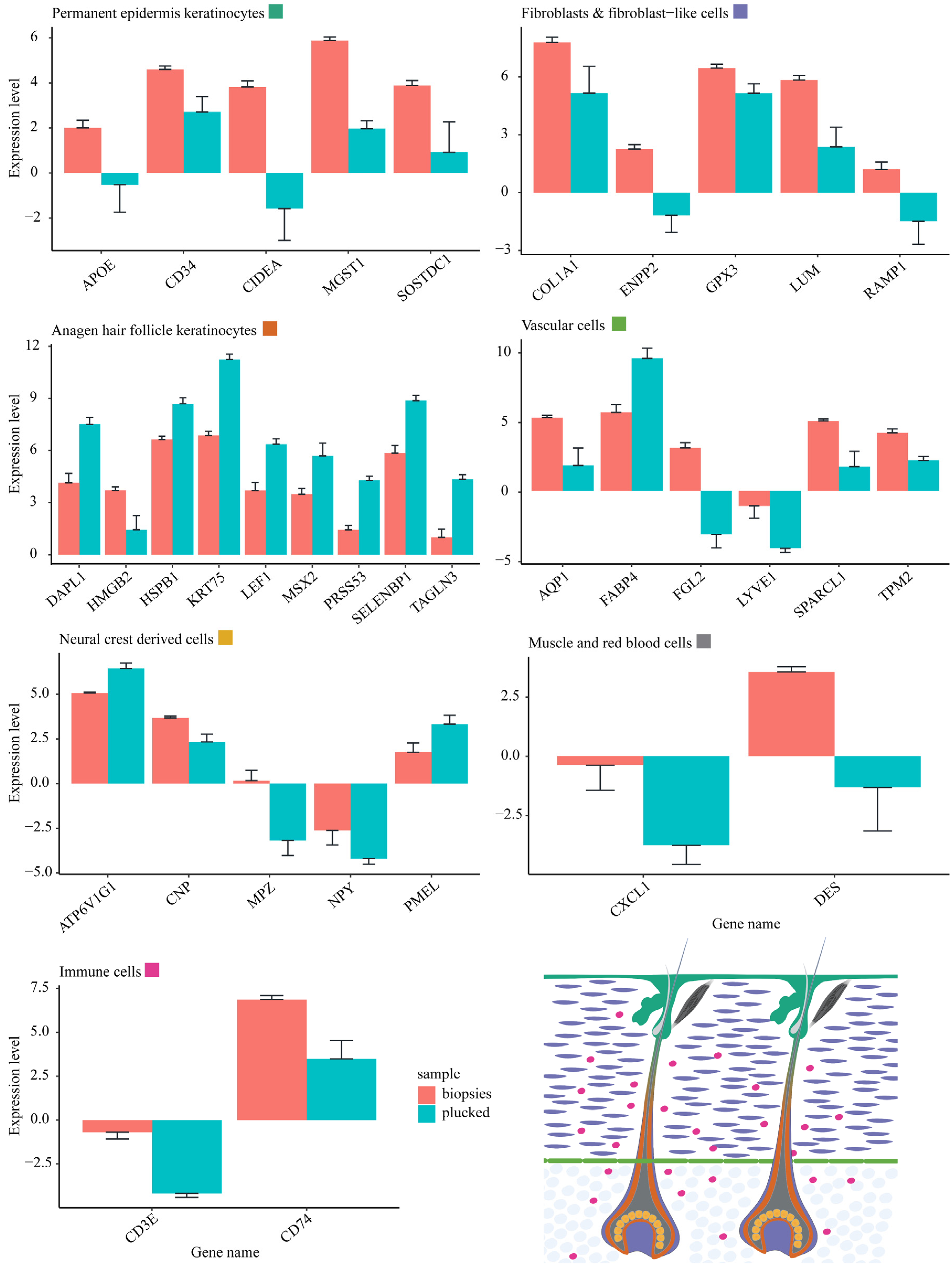
2.4. Differential mRNA Abundance Is Due to Different Amounts of HF-Associated and Interfollicular Cell Populations between Plucked Hair and Skin Biopsies
3. Discussion
4. Materials and Methods
4.1. Animals and Sample Collection
4.2. RNA Extraction
4.3. Library Prep
4.4. Sequencing
4.5. Analysis
4.6. qPCR Validation
4.7. Cell Type Idéntification
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rognoni, E.; Watt, F.M. Skin cell heterogeneity in development, wound healing, and cancer. Trends Cell Biol. 2018, 28, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, G.; Naboulsi, R.; Frey, R.; Solé, M. Genetics of Skin Disease in Horses. Vet. Clin. Equine Pract. 2020, 36, 323–339. [Google Scholar] [CrossRef]
- Zhang, J.; Wallace, S.J.; Shiu, M.Y.; Smith, I.; Rhind, S.G.; Langlois, V.S. Human hair follicle transcriptome profiling: A minimally invasive tool to assess molecular adaptations upon low-volume, high-intensity interval training. Physiol. Rep. 2017, 5, e13534. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E. Scratching the surface of skin development. Nature 2007, 445, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Joost, S.; Annusver, K.; Jacob, T.; Sun, X.; Dalessandri, T.; Sivan, U.; Sequeira, I.; Sandberg, R.; Kasper, M. The molecular anatomy of mouse skin during hair growth and rest. Cell Stem Cell 2020, 26, 441–457.e7. [Google Scholar] [CrossRef]
- Claussnitzer, M.; Cho, J.H.; Collins, R.; Cox, N.J.; Dermitzakis, E.T.; Hurles, M.E.; Kathiresan, S.; Kenny, E.E.; Lindgren, C.M.; MacArthur, D.G. A brief history of human disease genetics. Nature 2020, 577, 179–189. [Google Scholar] [CrossRef]
- Bailey, E.; Petersen, J.L.; Kalbfleisch, T.S. Genetics of Thoroughbred Racehorse Performance. Annu. Rev. Anim. Biosci. 2022, 10, 131–150. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.M.; Beeson, S.K.; Rubin, C.-J.; Andersson, L.; Caputo, P.; Lykkjen, S.; Moore, A.; Piercy, R.J.; Mickelson, J.R.; McCue, M.E. Identification and validation of genetic variants predictive of gait in standardbred horses. PLoS Genet. 2019, 15, e1008146. [Google Scholar] [CrossRef]
- Horváth, M.B.; Martínez-Cruz, B.; Negro, J.J.; Kalmár, L.; Godoy, J.A. An overlooked DNA source for non-invasive genetic analysis in birds. J. Avian Biol. 2005, 36, 84–88. [Google Scholar] [CrossRef]
- Lorange, J.B. WorldFengur—The studbook of origin for the Icelandic horse. Acta Vet. Scand. 2011, 53, S5. [Google Scholar] [CrossRef]
- Kalbfleisch, T.S.; Rice, E.S.; DePriest, M.S.; Walenz, B.P.; Hestand, M.S.; Vermeesch, J.R.; Fiddes, I.T.; Vershinina, A.O.; Saremi, N.F.; Petersen, J.L. Improved reference genome for the domestic horse increases assembly contiguity and composition. Commun. Biol. 2018, 1, 197. [Google Scholar] [CrossRef]
- Alonso, L.; Fuchs, E. The hair cycle. J. Cell Sci. 2006, 119, 391–393. [Google Scholar] [CrossRef]
- Schmidt-Ullrich, R.; Paus, R. Molecular principles of hair follicle induction and morphogenesis. Bioessays 2005, 27, 247–261. [Google Scholar] [CrossRef]
- Muller-Rover, S.; Handjiski, B.; van der Veen, C.; Eichmuller, S.; Foitzik, K.; McKay, I.A.; Stenn, K.S.; Paus, R. A comprehensive guide for the accurate classification of murine hair follicles in distinct hair cycle stages. J. Investig. Derm. 2001, 117, 3–15. [Google Scholar] [CrossRef]
- Van Scott, E.J.; Reinertson, R.P.; Steinmuller, R. The growing hair roots of the human scalp and morphologic changes therein following amethopterin therapy. J. Investig. Derm. 1957, 29, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, E. Removal of intact hair papilla and connective tissue sheath by plucking anagen hairs. J. Investig. Derm. 1967, 48, 595–597. [Google Scholar] [CrossRef]
- Paus, R.; Cotsarelis, G. The biology of hair follicles. N. Engl. J. Med. 1999, 341, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.W. Skin of the neck, mane and tail of the curly horse. Equine Vet. Educ. 2004, 16, 201–206. [Google Scholar] [CrossRef]
- Rice, R.H.; Bradshaw, K.M.; Durbin-Johnson, B.P.; Rocke, D.M.; Eigenheer, R.A.; Phinney, B.S.; Sundberg, J.P. Differentiating inbred mouse strains from each other and those with single gene mutations using hair proteomics. PLoS ONE 2012, 7, e51956. [Google Scholar] [CrossRef]
- Jones, K.F.; Carlson, T.L.; Eckenrode, B.A.; Donfack, J. Assessing protein sequencing in human single hair shafts of decreasing lengths. Forensic Sci. Int. Genet. 2020, 44, 102145. [Google Scholar] [CrossRef]
- Galván, I.; Garrido-Fernández, J.; Ríos, J.; Pérez-Gálvez, A.; Rodríguez-Herrera, B.; Negro, J.J. Tropical bat as mammalian model for skin carotenoid metabolism. Proc. Natl. Acad. Sci. USA 2016, 113, 10932–10937. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of DEGs 1 | ||
|---|---|---|
| Comparison | Lower Abundance | Higher Abundance |
| All Plucked hair versus all skin biopsies | 1838 | 1964 |
| Forelock plucked hair versus forelock biopsies | 1094 | 1168 |
| Mane plucked hair versus mane biopsies | 1091 | 944 |
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| KRT84 | GGCTTTGGCTACAGAGTTGG | TGTCGATGAAGGAGGCAAAT |
| RHCG | TGATGATCTTCGTGGGCTTC | TGTAGCCTTCCCTGTAGCAGT |
| MGST1 | CCCACCTGAATGACCTTGAA | CAGCCTGTAAGCCATTGACA |
| FGL2 | ACTATTCAGCTCCCGAAGCA | TCTGTGCCAGGTAACAGCAG |
| HSPA8 | ATCTTGGCACCACCTACTCG | GGGTTCATTGCAACTTGGTT |
| ACTB | CGAGCACGATGAAGATCAAG | GTGGACAATGAGGCCAGAAT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naboulsi, R.; Cieślak, J.; Headon, D.; Jouni, A.; Negro, J.J.; Andersson, G.; Lindgren, G. The Enrichment of Specific Hair Follicle-Associated Cell Populations in Plucked Hairs Offers an Opportunity to Study Gene Expression Underlying Hair Traits. Int. J. Mol. Sci. 2023, 24, 561. https://doi.org/10.3390/ijms24010561
Naboulsi R, Cieślak J, Headon D, Jouni A, Negro JJ, Andersson G, Lindgren G. The Enrichment of Specific Hair Follicle-Associated Cell Populations in Plucked Hairs Offers an Opportunity to Study Gene Expression Underlying Hair Traits. International Journal of Molecular Sciences. 2023; 24(1):561. https://doi.org/10.3390/ijms24010561
Chicago/Turabian StyleNaboulsi, Rakan, Jakub Cieślak, Denis Headon, Ahmad Jouni, Juan J. Negro, Göran Andersson, and Gabriella Lindgren. 2023. "The Enrichment of Specific Hair Follicle-Associated Cell Populations in Plucked Hairs Offers an Opportunity to Study Gene Expression Underlying Hair Traits" International Journal of Molecular Sciences 24, no. 1: 561. https://doi.org/10.3390/ijms24010561
APA StyleNaboulsi, R., Cieślak, J., Headon, D., Jouni, A., Negro, J. J., Andersson, G., & Lindgren, G. (2023). The Enrichment of Specific Hair Follicle-Associated Cell Populations in Plucked Hairs Offers an Opportunity to Study Gene Expression Underlying Hair Traits. International Journal of Molecular Sciences, 24(1), 561. https://doi.org/10.3390/ijms24010561