
Posterior Polymorphous Corneal Dystrophy in a Patient with a Novel ZEB1 Gene Mutation

 , , ,
, , , 
Abstract
1. Introduction
2. Case Presentation
2.1. Clinical Case
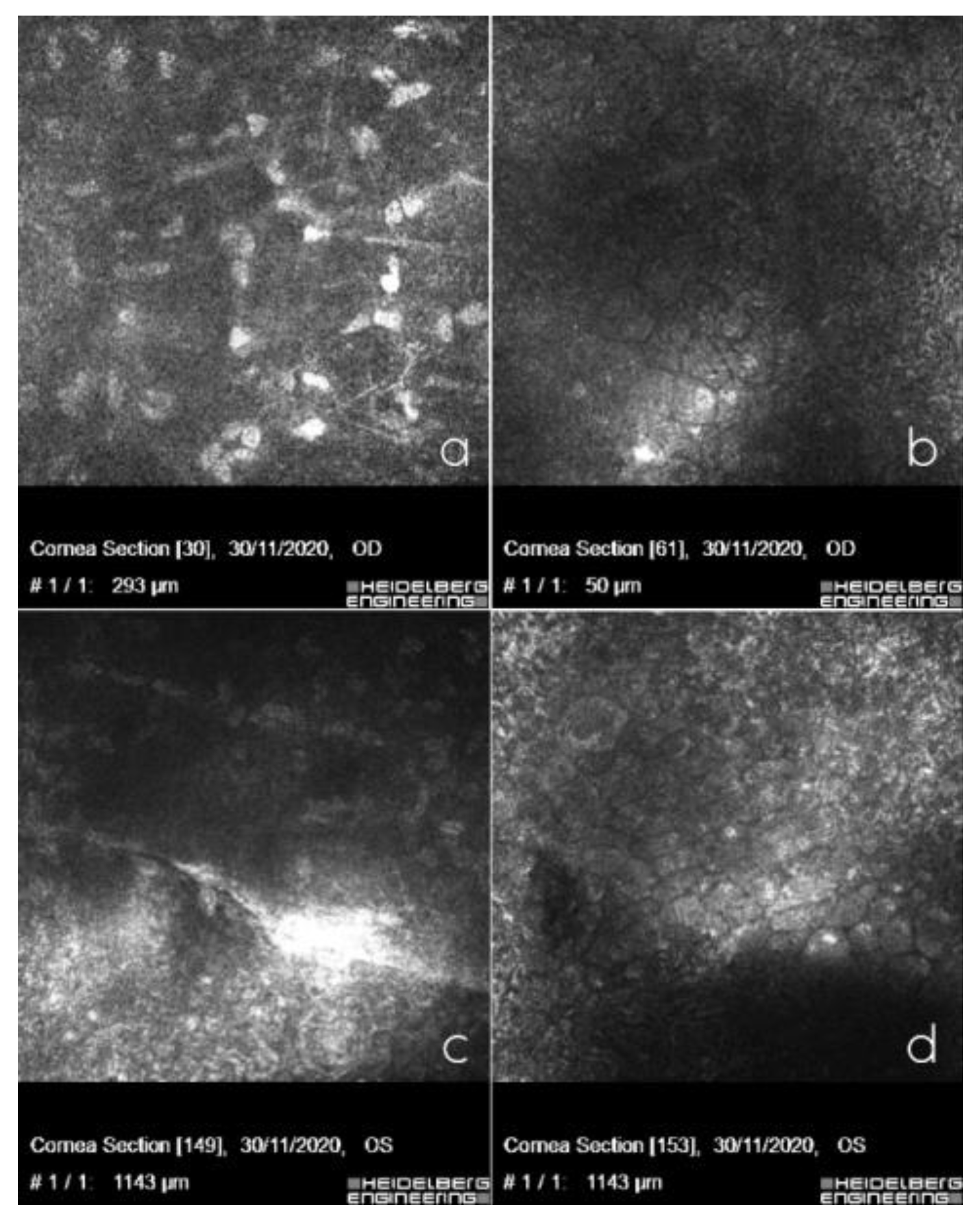
2.2. Clinical Findings
2.3. Molecular Genetics
2.4. Material and Methods
2.4.1. Ophthalmological Evaluation
2.4.2. Genetic Analysis
3. Discussion

4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PPCD | ICE Syndrome | ||
|---|---|---|---|
| Etiology | PPCD1: VSX1 PPCD2: COL8A2 PPCD3: ZEB1 | Unknown Probable viral etiology | |
| Inheritance | Autosomal dominant | Sporadic | |
| Presentation | More frequently bilateral | More frequently unilateral | |
| Onset | Early childhood | Adulthood | |
| Signs | Cornea | Endothelial lesions
| “Beaten metal” appearance Chandler: early marked edema APS: possible edema Cogan-Reese: possible edema |
| Anterior chamber | Peripheral iridocorneal adhesions in 25% | Peripheral anterior synechiae | |
| Iris | Atrophy | Chandler: iris atrophy PIA: Full thickness holes Cogan-Reese: nodules + atrophy | |
| Pupil | Corectopia | Chandler: corectopia PIA: polycoria Cogan-Reese: uncommon changes | |
| Cytokeratins | CK7 CK19 | 34BE12 Pkk1 KL1 CK19 | |
Genes Included in the OFTv2.1 Panel
| Code | Description |
|---|---|
| PVS1 | Null variant (nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single or multiexon deletion) in a gene where LOF is a known mechanism of disease. |
| PS1 | Same amino acid change as a previously established pathogenic variant regardless of nucleotide change. |
| PS2 | De novo (both maternity and paternity confirmed) in a patient with the disease and no family history. |
| PS3 | Well-established in vitro or in vivo functional studies supportive of a damaging effect on the gene or gene product. |
| BS1 | Allele frequency is greater than expected for disorder. |
| PM1 | Located in a mutational hot spot and/or critical and well-established functional domain (e.g., active site of an enzyme) without benign variation. |
| PM2 | Absent from controls (or at extremely low frequency if recessive) in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium. |
| PM3 | For recessive disorders, detected in trans with a pathogenic variant |
| PM4 | Protein length changes as a result of in-frame deletions/insertions in a non-repeat region or stop-loss variants. |
| PM5 | Novel missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before. |
| PP1 | Cosegregation with disease in multiple affected family members in a gene definitively known to cause the disease. |
| PP2 | Missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease. |
| PP3 | Multiple lines of computational evidence support a deleterious effect on the gene or gene product (conservation, evolutionary, splicing impact, etc.) |
| PP5 | Reputable source recently reports variant as pathogenic, but the evidence is not available to the laboratory to perform an independent evaluation. |
| BP4 | Multiple lines of computational evidence suggest no impact on gene or gene product (conservation, evolutionary, splicing impact, etc.) |
| BP6 | Reputable source recently reports variant as benign, but the evidence is not available to the laboratory to perform an independent evaluation. |
References
- Weiss, J.; Møller, H.; Lisch, W.; Kinoshita, S.; Aldave, A.J.; Belin, M.W.; Kivelä, T.; Busin, M.; Munier, F.L.; Seitz, B.; et al. La clasificación IC3D de las distrofias corneales. Córnea 2008, 27 (Suppl. 2), 43–83. Available online: https://corneasociety.org/sites/default/files/publications/ic3d_spanish_version.pdf (accessed on 9 March 2022).
- Heon, E.; Greenberg, A.; Kopp, K.K.; Rootman, D.; Vincent, A.L.; Billingsley, G.; Priston, M.; Dorval, K.M.; Chow, R.L.; McInnes, R.R.; et al. VSX1: A gene for posterior polymorphous dystrophy and keratoconus. Hum. Mol. Genet. 2002, 11, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.N.; Chen, J.; Cui, H.P. Characteristics of corneal dystrophies: A review from clinical, histological and genetic perspectives. Int. J. Ophthalmol. 2016, 9, 904–913. Available online: http://europepmc.org/article/MED/27366696 (accessed on 9 March 2022).
- Lam, H.; Wiggs, J.; Jurkunas, U. Unusual presentation of presumed posterior polymorphous dystrophy associated with iris heterochromia, band keratopathy, and keratoconus. Cornea 2010, 29, 1180. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2945457/ (accessed on 10 March 2022). [CrossRef] [PubMed]
- Liskova, P.; Evans, C.J.; Davidson, A.E.; Zaliova, M.; Dudakova, L.; Trkova, M.; Stranecky, V.; Carnt, N.; Plagnol, V.; Vincent, A.L.; et al. Heterozygous deletions at the ZEB1 locus verify haploinsufficiency as the mechanism of disease for posterior polymorphous corneal dystrophy type 3. Eur. J. Hum. Genet. 2016, 24, 985–991. Available online: https://www.nature.com/articles/ejhg2015232 (accessed on 10 March 2022). [CrossRef] [PubMed]
- Jirsova, K.; Merjava, S.; Martincova, R.; Gwilliam, R.; Ebenezer, N.D.; Liskova, P.; Filipec, M. Immunohistochemical characterization of cytokeratins in the abnormal corneal endothelium of posterior polymorphous corneal dystrophy patients. Exp. Eye Res. 2007, 84, 680–686. Available online: https://www.sciencedirect.com/science/article/abs/pii/S0014483506004775 (accessed on 14 March 2022). [CrossRef]
- Silva, L.; Najafi, A.; Suwan, Y.; Teekhasaenee, C.; Ritch, R. The iridocorneal endothelial syndrome. Surv. Ophthalmol. 2018, 63, 665–676. Available online: https://pubmed.ncbi.nlm.nih.gov/29331589/ (accessed on 14 March 2022). [CrossRef] [PubMed]
- Chung DW, D.; Frausto, R.F.; Ann, L.B.; Jang, M.S.; Aldave, A.J. Functional impact of ZEB1 mutations associated with posterior polymorphous and Fuchs’ endothelial corneal dystrophies. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6159–6166. Available online: https://pubmed.ncbi.nlm.nih.gov/25190660/ (accessed on 14 March 2022). [CrossRef] [PubMed]
- Frausto, R.F.; Chung, D.D.; Boere, P.M.; Swamy, V.S.; Duong, H.N.; Kao, L.; Azimov, R.; Zhang, W.; Carrigan, L.; Wong, D.; et al. ZEB1 insufficiency causes corneal endothelial cell state transition and altered cellular 341 processing. PLoS ONE 2019, 14, e0218279. Available online: https://pubmed.ncbi.nlm.nih.gov/31194824/ (accessed on 14 March 2022). [CrossRef] [PubMed]
- Davidson, A.E.; Hafford-Tear, N.J.; Dudakova, L.; Sadan, A.N.; Pontikos, N.; Hardcastle, A.J.; Tuft, S.J.; Liskova, P. CUGC for posterior polymorphous corneal dystrophy (PPCD). Eur. J. Hum. Genet. EJHG 2020, 28, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.J.; Liskova, P.; Dudakova, L.; Hrabcikova, P.; Horinek, A.; Jirsova, K.; Filipec, M.; Hardcastle, A.J.; Davidson, A.E.; Tuft, S.J. Identification of six novel mutations in ZEB1 and description of the associated phenotypes in patients with posterior polymorphous corneal dystrophy 3. Ann. Hum. Genet. 2015, 79, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lechner, J.; Dash, D.P.; Muszynska, D.; Hosseini, M.; Segev, F.; George, S.; Frazer, D.G.; Moore, J.E.; Kaye, S.B.; Young, T.; et al. Mutational spectrum of the ZEB1 gene in corneal dystrophies supports a genotype–phenotype correlation. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3215–3223. Available online: https://iovs.arvojournals.org/article.aspx?articleid=2189147 (accessed on 14 March 2022). [CrossRef] [PubMed]
- Vincent, A.L.; Niederer, R.L.; Richards, A.; Karolyi, B.; Patel, D.V.; McGhee, C.N. Phenotypic characterisation and ZEB1 mutational analysis in posterior polymorphous corneal dystrophy in a New Zealand population. Mol. Vis. 2009, 15, 2544–2553. [Google Scholar] [PubMed]
- Raber, I.M.; Fintelmann, R.; Chhabra, S.; Ribeiro, M.P.F.; Eagle, R.C., Jr.; Orlin, S.E. Posterior polymorphous dystrophy associated with nonkeratoconic steep corneal curvatures. Cornea 2011, 30, 1120–1124. Available online: https://journals.lww.com/corneajrnl/Abstract/2011/10000/Posterior_Polymorphous_Dystrophy_Associated_With.9.aspx (accessed on 14 March 2022). [CrossRef] [PubMed]
- Aldave, A.J.; Ann, L.B.; Frausto, R.F.; Nguyen, C.K.; Yu, F.; Raber, I.M. Classification of posterior polymorphous corneal dystrophy as a corneal ectatic disorder following confirmation of associated significant corneal steepening. JAMA Ophthalmol. 2013, 131, 1583–1590. Available online: https://jamanetwork.com/journals/jamaophthalmology/article-abstract/1748767 (accessed on 15 March 2022). [CrossRef] [PubMed][Green Version]
- Dudakova, L.; Evans, C.J.; Pontikos, N.; Hafford-Tear, N.J.; Malinka, F.; Skalicka, P.; Horinek, A.; Munier, F.L.; Voide, N.; Studeny, P.; et al. The utility of massively parallel sequencing for posterior polymorphous corneal dystrophy type 3 molecular diagnosis. Exp. Eye Res. 2019, 182, 160–166. Available online: https://www.sciencedirect.com/science/article/pii/S0014483518307899 (accessed on 15 March 2022). [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Lovd.nl. Available online: https://databases.lovd.nl/shared/variants/ZEB1 (accessed on 21 November 2022).
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- VCV000488897.2—ClinVar-NCBI. (s/f). Nih.gov. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/488897/?new_evidence=true (accessed on 2 December 2022).
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef] [PubMed]
- Pantherdb.org. Available online: http://www.pantherdb.org/tools/csnpScore.do (accessed on 2 December 2022).



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Gutiérrez, E.; Fernández-Pérez, P.; Boto-De-Los-Bueis, A.; García-Fernández, L.; Rodríguez-Solana, P.; Solís, M.; Vallespín, E. Posterior Polymorphous Corneal Dystrophy in a Patient with a Novel ZEB1 Gene Mutation. Int. J. Mol. Sci. 2023, 24, 209. https://doi.org/10.3390/ijms24010209
Fernández-Gutiérrez E, Fernández-Pérez P, Boto-De-Los-Bueis A, García-Fernández L, Rodríguez-Solana P, Solís M, Vallespín E. Posterior Polymorphous Corneal Dystrophy in a Patient with a Novel ZEB1 Gene Mutation. International Journal of Molecular Sciences. 2023; 24(1):209. https://doi.org/10.3390/ijms24010209
Chicago/Turabian StyleFernández-Gutiérrez, Eva, Pedro Fernández-Pérez, Ana Boto-De-Los-Bueis, Laura García-Fernández, Patricia Rodríguez-Solana, Mario Solís, and Elena Vallespín. 2023. "Posterior Polymorphous Corneal Dystrophy in a Patient with a Novel ZEB1 Gene Mutation" International Journal of Molecular Sciences 24, no. 1: 209. https://doi.org/10.3390/ijms24010209
APA StyleFernández-Gutiérrez, E., Fernández-Pérez, P., Boto-De-Los-Bueis, A., García-Fernández, L., Rodríguez-Solana, P., Solís, M., & Vallespín, E. (2023). Posterior Polymorphous Corneal Dystrophy in a Patient with a Novel ZEB1 Gene Mutation. International Journal of Molecular Sciences, 24(1), 209. https://doi.org/10.3390/ijms24010209