
The Genomic Landscape of Corticotroph Tumors: From Silent Adenomas to ACTH-Secreting Carcinomas

 ,
,  , , ,
, , ,  and add
Show full author list
and add
Show full author list
Abstract
1. Introduction
2. Results
2.1. Clinical and Demographic Characteristics of the Patients
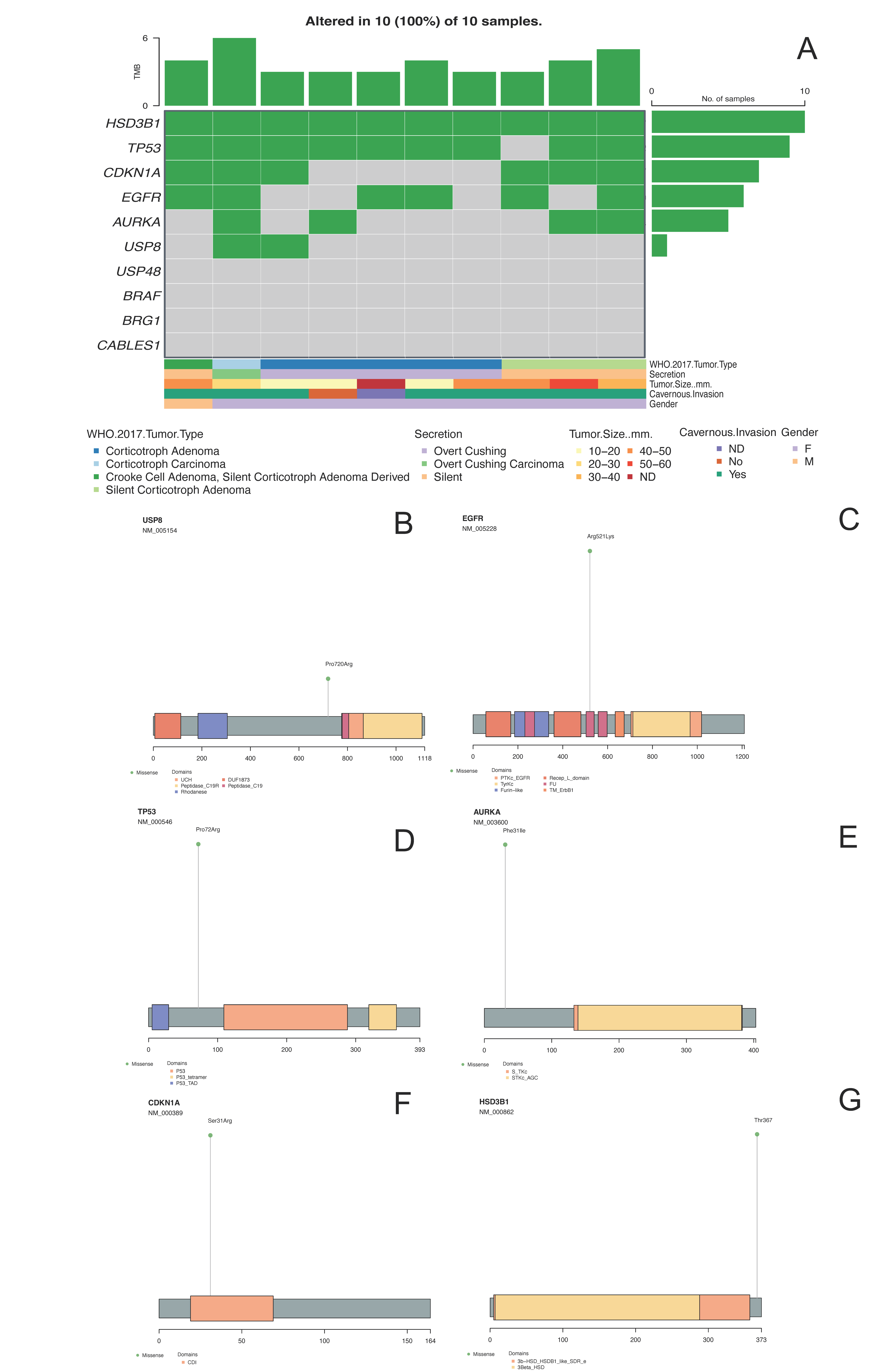
2.2. General Genomic Characteristics of Neoplasms of Corticotrophic Lineage
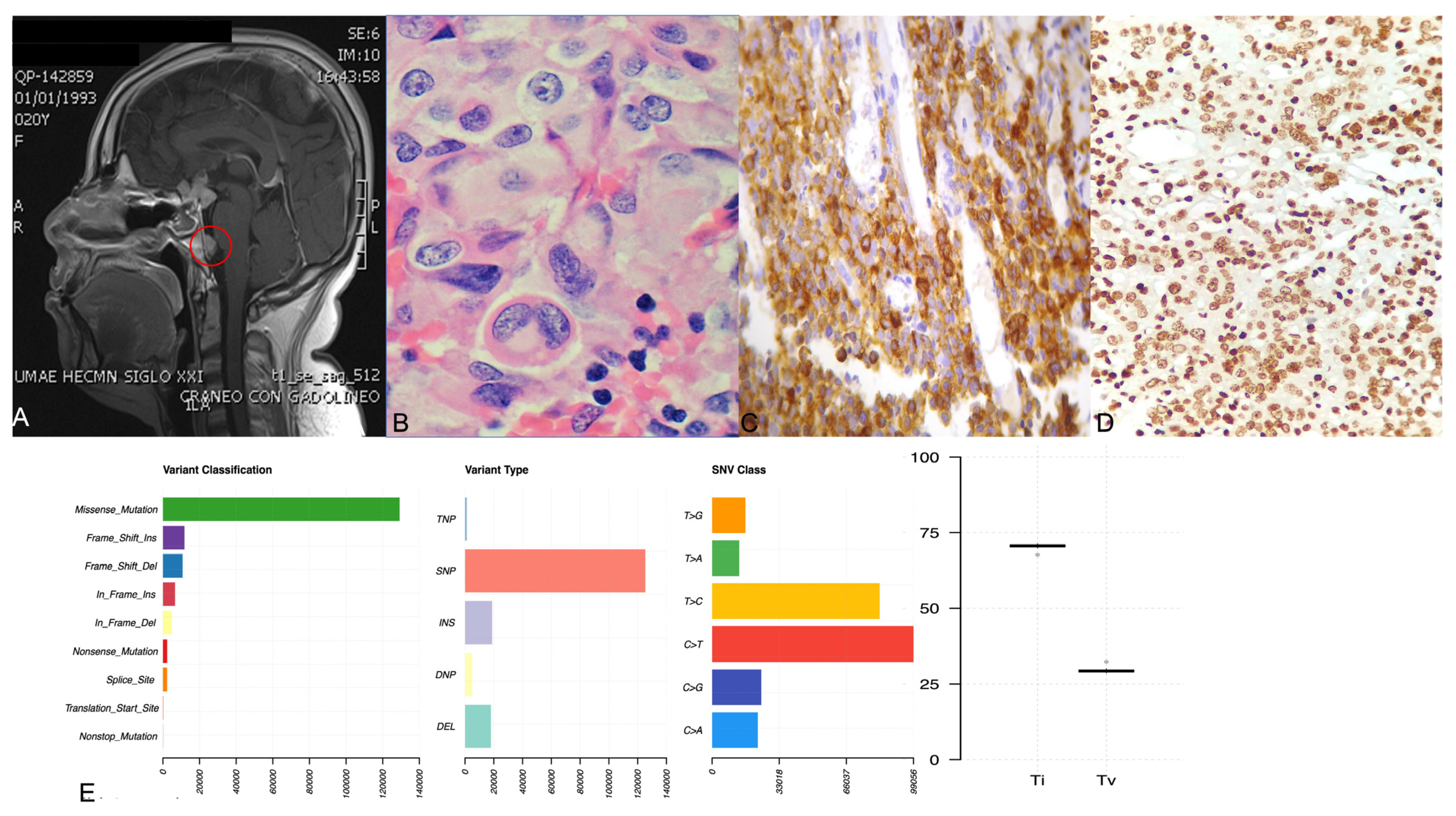
2.3. ACTH-Secreting Carcinoma (Tumor 1)
2.4. Crooke Cell Adenoma (Tumor 2)
2.5. Silent Corticotroph Adenomas (Tumors 3–5)
2.6. ACTH-Secreting Adenomas (Cushing Disease) (Tumors 6–9)
2.7. ACTH-Secreting Adenoma Causing Nelson Syndrome (Tumor 10)
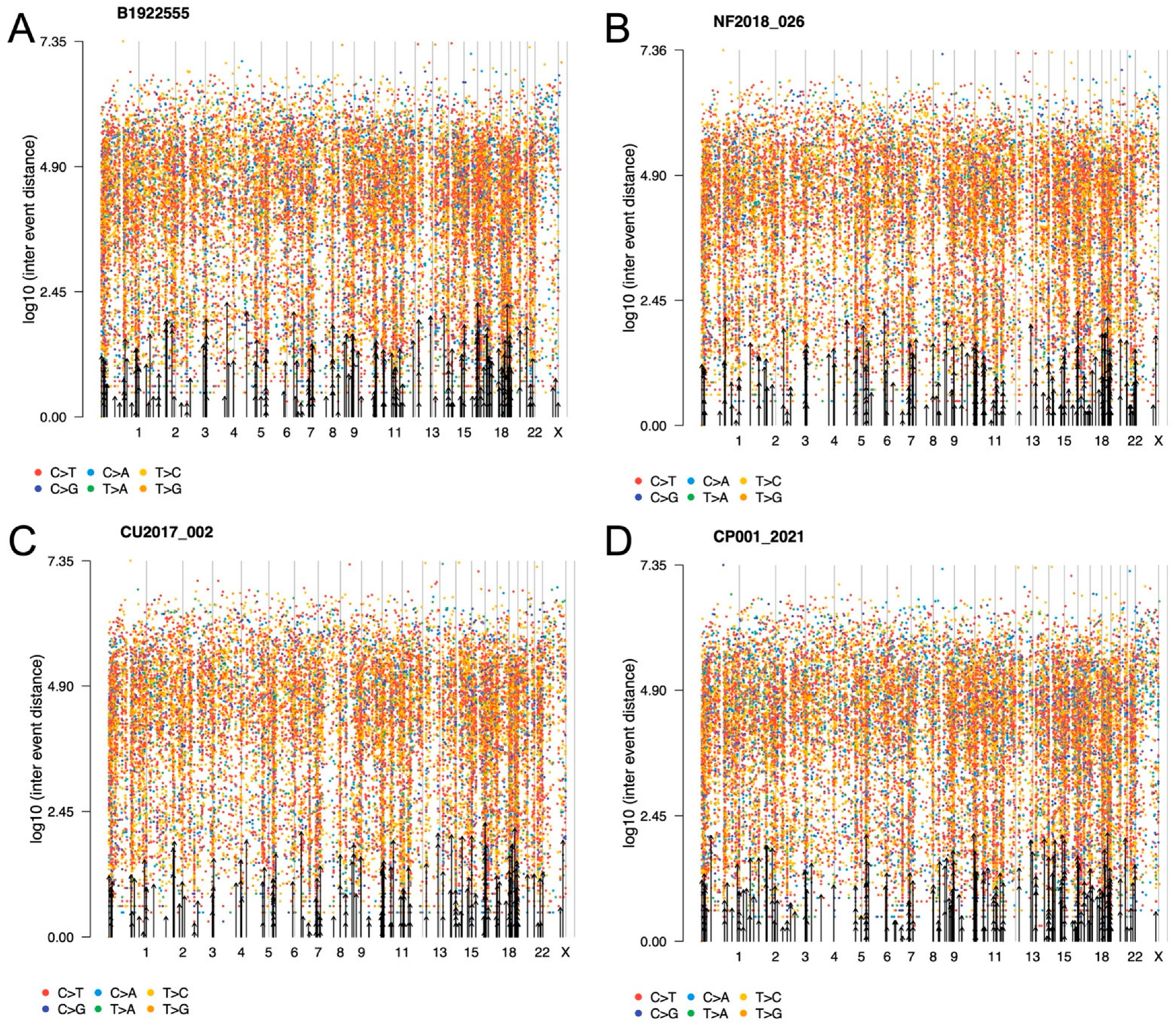
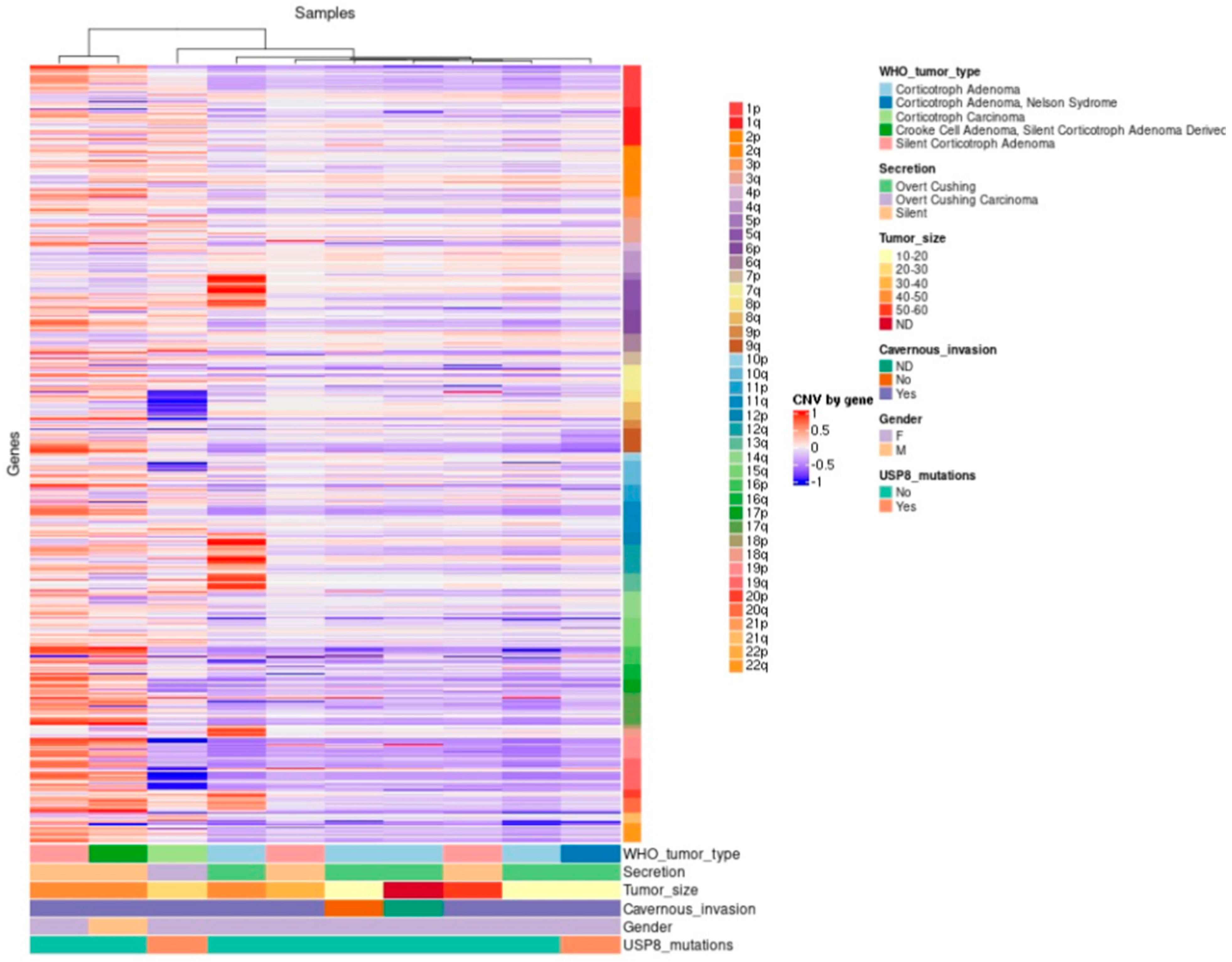
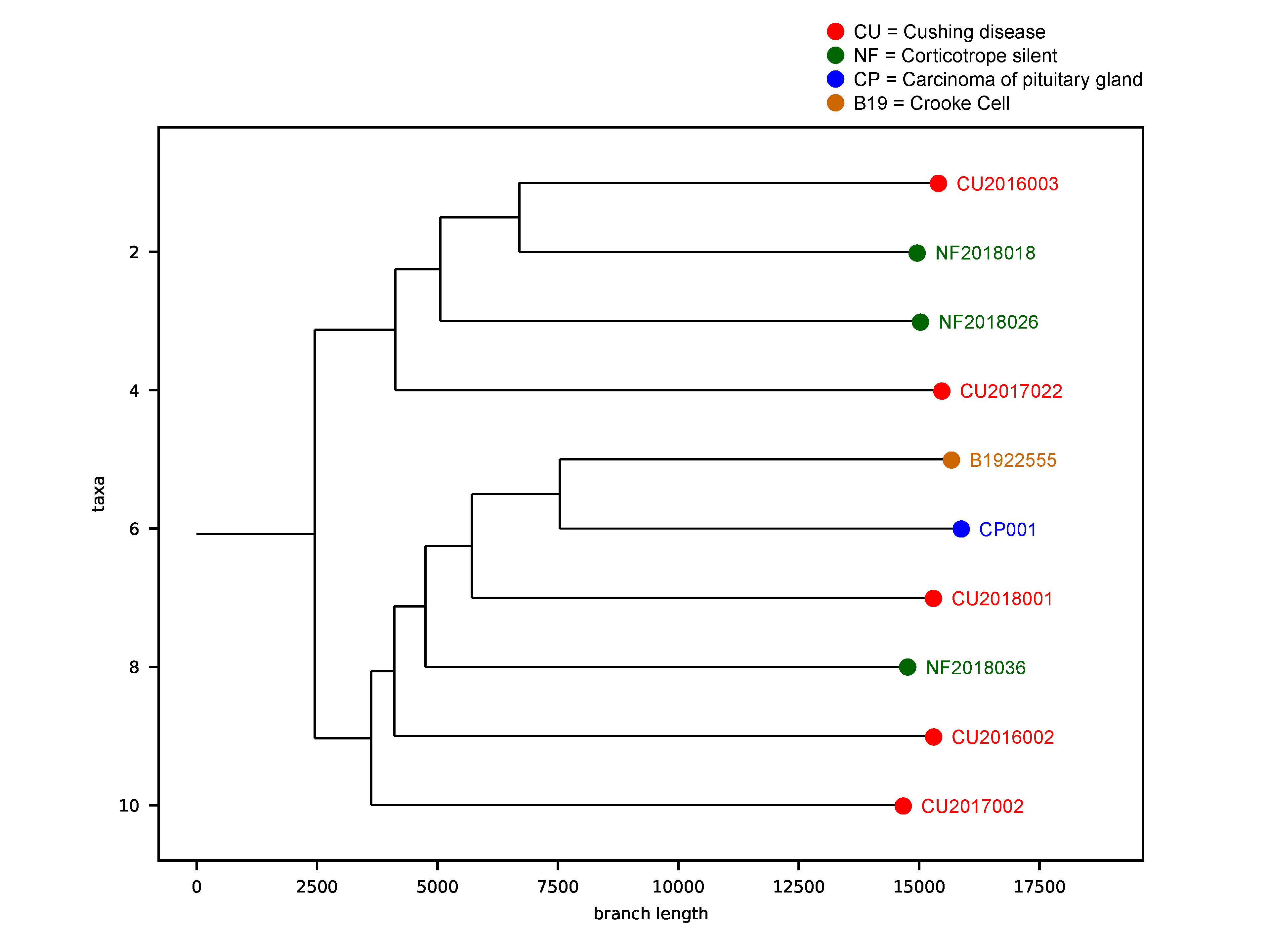
2.8. Tumor Phylogenic Analysis
2.9. Correlation between Gene Variants and Clinicopathological Features
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Patients and Tumor Tissue Samples
5.2. Construction and Sequencing of Whole Exome Libraries
5.3. Bioinformatics Analysis
5.4. Sanger Sequencing forConfirmation of Exome Findings
5.5. Hormone and Transcription Factor Immunohistochemistry
5.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heaney, A. Clinical review: Pituitary carcinoma: Difficult diagnosis and treatment. J. Clin. Endocrinol. Metab. 2011, 96, 3649–3660. [Google Scholar] [CrossRef] [PubMed]
- Raverot, G.; Burman, P.; McCormack, A.; Heaney, A.; Petersenn, S.; Popovic, V.; Trouillas, J.; Dekkers, O.M.; The European Society of Endocrinology. European Society of Endocrinology Clinical Practice Guidelines for the management of aggressive pituitary tumours and carcinomas. Eur. J. Endocrinol. 2018, 178, G1–G24. [Google Scholar] [CrossRef]
- Todeschini, A.B.; Beer-Furlan, A.; Montaser, A.S.; Jamshidi, A.O.; Ghalib, L.G.; Chavez, J.A.; Lehman, N.L.; Prevedello, D.M. Pituitary carcinomas: Review of the current literature and report of atypical case. Br. J. Neurosurg. 2020, 34, 528–533. [Google Scholar] [CrossRef]
- Dudziak, K.; Honegger, J.; Bornemann, A.; Horger, M.; Müssig, K. Pituitary carcinoma with malignant growth from first presentation and fulminant clinical course-case report and review of the literature. J. Clin. Endocrinol. Metab. 2011, 96, 2665–2669. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Di Ieva, A.; Davidson, J.; Syro, L.; Rotondo, F.; Montoya, J.; Horvath, E.; Cusimano, M.D.; Kovacs, K. Crooke’s cell tumors of the pituitary. Neurosurgery 2015, 76, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Fountas, A.; Lavrentaki, A.; Subramanian, A.; Toulis, K.; Nirantharakumar k Karavitaki, N. Recurrence in silent corticotroph adenomas after primary treatment: A systematic review and meta-analysis. J. Clin. Endocrinol. Metab. 2019, 104, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Molitch, M. Diagnosis and Treatment of Pituitary Adenomas: A Review. JAMA 2017, 317, 516–524. [Google Scholar] [CrossRef]
- Melmed, S. Pituitary-Tumor Endocrinopathies. N. Engl. J. Med. 2020, 382, 937–950. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.B.S. The 2017 World Health Organization classification of tumors of the pituitary gland: A sumary. Acta Neuropathol. 2017, 134, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.-J.; Reitman, Z.; Ma, Z.-Y.; Chen, J.-H.; Zhang, Q.-L.; Shou, X.-F.; Huang, C.X.; Wang, Y.F.; Li, S.Q.; Mao, Y.; et al. The genome-wide mutational landscape of pituitary adenomas. Cell Res. 2016, 26, 1255–1259. [Google Scholar] [CrossRef]
- NCBI. Available online: https://www.ncbi.nlm.nih.gov/snp/rs1047303#frequency_tab (accessed on 23 February 2022).
- Shiota, M.; Narita, S.; Akamatsu, S.; Fujimoto, N.; Sumiyoshi, T.; Fujiwara, M.; Uchiumi, T.; Habuchi, T.; Ogawa, O.; Eto, M. Association of Missense Polymorphism in HSD3B1 With Outcomes Among Men With Prostate Cancer Treated With Androgen-Deprivation Therapy or Abiraterone. JAMA Netw. Open 2019, 2, e190115. [Google Scholar] [CrossRef]
- Yagnik, G.; Jahangiri, A.; Chen, R.; Wagner, J.; Aghi, M. Role of a p53 polymorphism in the development of nonfunctional pituitary adenomas. Mol. Cell Endocrinol. 2017, 446, 81–90. [Google Scholar] [CrossRef]
- Heidari, Z.; Harati-Sadegh, M.; Arian, A.; Maruei-Milan, R.; Salimi, S. The effect of TP53 and P21 gene polymorphisms on papillary thyroid carcinoma susceptibility and clinical/pathological features. IUBMB Life 2020, 72, 922–930. [Google Scholar] [CrossRef]
- Akhter, N.; Dar, S.; Haque, S.; Wahid, M.; Jawed, A.; Akhtar, M.S.; A Alharbi, R.; A A Sindi, A.; Alruwetei, A.; Choudhry, H.M.Z.; et al. Crosstalk of Cyclin-dependent kinase inhibitor 1A (CDKN1A) gene polymorphism with p53 and CCND1 polymorphism in breast cancer. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 4258–4273. [Google Scholar]
- Wang, C.; Nie, H.; Li, Y.; Liu, G.; Wang, X.; Xing, S.; Zhang, L.; Chen, X.; Chen, Y.; Li, Y. The study of the relation of DNA repair pathway genes SNPs and the sensitivity to radiotherapy and chemotherapy of NSCLC. Sci. Rep. 2016, 6, 26526. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi-Ponciano, K.; Portocarrero-Ortiz, L.A.; Guinto, G.; Moreno-Jimenez, S.; Gomez-Apo, E.; Chavez-Macias, L.; Peña-Martínez, E.; Silva-Román, G.; Vela-Patiño, S.; Ordoñez-García, J.; et al. The kinome, cyclins and cyclin-dependent kinases of pituitary adenomas, a look into the gene expression rofile among tumors different lineages. BMC Med. Genom. 2022, 15, 52. [Google Scholar] [CrossRef]
- Sobral-Leite, M.; Lips, E.; Vieira-Monteiro, H.; Giacomin, L.; Freitas-Alves, D.; Cornelissen, S.; Mulder, L.; Wesseling, J.; Schmidt, M.K.; Vianna-Jorge, R. Evaluation of the EGFR polymorphism R497K in two cohorts of neoadjuvantly treated breast cancer patients. PLoS ONE 2017, 12, e0189750. [Google Scholar] [CrossRef]
- Zhang, H.; Berezov, A.; Wang, Q.; Zhang, G.; Drebin, J.; Murali, R.; Greene, M. ErbB receptors: From oncogenes to targeted cancer therapies. J. Clin. Investig. 2007, 117, 2051–2058. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Feng, M.; Dai, C.; Bao, X.; Deng, K.; Yao, Y.; Wang, R. Expression of EGFR in Pituitary Corticotroph Adenomas and Its Relationship With Tumor Behavior. Front. Endocrinol. 2019, 10, 785. [Google Scholar] [CrossRef]
- NCBI. Available online: https://www.ncbi.nlm.nih.gov/snp/rs2227983#frequency_tab (accessed on 23 February 2022).
- Wang, S.; Qi, J.; Zhu, M.; Wang, M.; Nie, J. AURKA rs2273535 T>A Polymorphism Associated With Cancer Risk: A Systematic Review With Meta-Analysis. Front. Oncol. 2020, 10, 1040. [Google Scholar] [CrossRef] [PubMed]
- NCBI. Available online: https://www.ncbi.nlm.nih.gov/snp/rs2273535#frequency_tab (accessed on 23 February 2022).
- Reincke, M.; Sbiera, S.; Hayakawa, A.; Theodoropoulou, M.; Osswald, A.; Beuschlein, F.; Meitinger, T.; Mizuno-Yamasaki, E.; Kawaguchi, K.; Saeki, Y.; et al. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat. Genet. 2015, 47, 31–38. [Google Scholar]
- Perez-Rivas, L.; Theodoropoulou, M.; Ferraù, F.; Nusser, C.; Kawaguchi, K.; Faucz, F.; Nusser, C.; Kawaguchi, K.; Stratakis, C.A.; Faucz, F.R.; et al. The Gene of the Ubiquitin-Specific Protease 8 Is Frequently Mutated in Adenomas Causing Cushing’s Disease. J. Clin. Endocrinol. Metab 2015, 100, E997–E1004. [Google Scholar] [CrossRef] [PubMed]
- Albani, A.; Pérez-Rivas, L.G.; Dimopoulou, C.; Zopp, S.; Colón-Bolea, P.; Roeber, S.; Honegger, J.; Flitsch, J.; Rachinger, W.; Buchfelder, M.; et al. The USP8 mutational status may predict long-term remission in patients with Cushing’s disease. Clin. Endocrinol. 2018, 89, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Wanichi, I.Q.; de Paula Mariani, B.M.; Frassetto, F.P.; Siqueira, S.A.C.; de Castro Musolino, N.R.; Cunha-Neto, M.B.C.; Ochman, G.; Cescato, V.A.S.; Machado, M.C.; Trarbach, E.B.; et al. Cushing’s disease due to somatic USP8 mutations: A systematic review and meta-analysis. Pituitary 2019, 22, 435–442. [Google Scholar] [CrossRef]
- Uzilov, A.; Taik, P.; Cheesman, K.; Javanmard, P.; Ying, K.; Roehnelt, A.; Wang, H.; Fink, M.Y.; Lau, C.Y.; Moe, A.S.; et al. USP8 and TP53 Drivers are Associated with CNV in a Corticotroph Adenoma Cohort Enriched for Aggressive Tumors. J. Clin. Endocrinol. Metab. 2021, 106, 826–842. [Google Scholar] [CrossRef]
- Hayashi, K.; Inoshita, N.; Kawaguchi, K.; Ibrahim, A.; Suzuki, H.; Fukuhara, N.; Okada, M.; Nishioka, H.; Takeuchi, Y.; Komada, M.; et al. The USP8 mutational status may predict drug susceptibility in corticotroph adenomas of Cushing’s disease. Eur. J. Endocrinol. 2016, 174, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Lv, N.; Liao, J.; Long, J.; Xue, R.; Ai, N.; Xu, D.; Fan, X. Copy number variation is highly correlated with differential gene expression: A pan-cancer study. BMC Med Genet. 2019, 20, 175. [Google Scholar] [CrossRef] [PubMed]
- Shlien, A.; Malkin, D. Copy number variations and cancer. Genome Med. 2009, 1, 62. [Google Scholar] [CrossRef]
- Pös, O.; Radvanszky, J.; Styk, J.; Pös, Z.; Buglyó, G.; Kajsik, M.; Budis, J.; Nagy, B.; Szemes, T. Copy Number Variation: Methods and Clinical Applications. Appl. Sci. 2021, 11, 819. [Google Scholar] [CrossRef]
- Cui, Y.; Li, C.; Jiang, Z.; Zhang, S.; Li, Q.; Liu, X.; Zhou, Y.; Li, R.; Wei, L.; Li, L.; et al. Single-cell transcriptome and genome analyses of pituitary neuroendocrine tumors. Neuro-Oncol. 2021, 23, 1859–1871. [Google Scholar] [CrossRef]
- Neou, M.; Villa, C.; Armignacco, R.; Jouinot, A.; Raffin-Sanson, M.-L.; Septier, A.; Letourneur, F.; Diry, S.; Diedisheim, M.; Izac, B.; et al. Pangenomic Classification of Pituitary Neuroendocrine Tumors. Cancer Cell 2020, 37, 123–134.e5. [Google Scholar] [CrossRef] [PubMed]
- Varis, A.; Wolf, M.; Monni, O.; Vakkari, M.-L.; Kokkola, A.; Moskaluk, C.; Frierson, H.; Powell, S.M.; Knuutila, S.; Kallioniemi, A.; et al. Targets of gene amplification and overexpression at 17q in gastric cancer. Cancer Res. 2002, 62, 2625–2629. [Google Scholar] [PubMed]
- Shlien, A.; Tabori, U.; Marshall, C.; Pienkowska, M.; Feuk, L.; Novokmet, A.; Nanda, S.; Druker, H.; Scherer, S.W.; Malkin, D. Excessive genomic DNA copy number variation in the Li-Fraumeni cancer predisposition syndrome. Proc. Natl. Acad. Sci. USA 2008, 105, 11264–11269. [Google Scholar] [CrossRef] [PubMed]
- McCormack, A.; Dekkers, O.; Petersenn, S.; Popovic, V.; Trouillas, J.; Raverot, G.; Burman, P. Treatment of aggressive pituitary tumours and carcinomas: Results of a European Society of Endocrinology (ESE) survey 2016. Eur. J. Endocrinol. 2018, 178, 265–276. [Google Scholar] [CrossRef]
- Trouillas, J.; Jaffrain-Rea, M.; Vasiljevic, A.; Raverot, G.; Roncaroli, F.; Villa, C. How to Classify the Pituitary Neuroendocrine Tumors (PitNET)s in 2020. Cancers 2020, 12, 514. [Google Scholar] [CrossRef]
- Hu, H.; Khodadadi-Jamayran, A.; Dolgalev, I.; Cho, H.; Badri, S.; Chiriboga, L.A.; Zeck, B.; Gregorio, M.L.D.R.; Dowling, C.M.; Labbe, K.; et al. Targeting the Atf7ip-Setdb1 Complex Augments Antitumor Immunity by Boosting Tumor Immunogenicity. Cancer Immunol. Res. 2021, 9, 1298–1315. [Google Scholar] [CrossRef]
- Park, J.; Huang, S.; Tougeron, D.; Sinicrope, F. MSH3 mismatch repair protein regulates sensitivity to cytotoxic drugs and a histone deacetylase inhibitor in human colon carcinoma cells. PLoS ONE 2013, 8, e65369. [Google Scholar] [CrossRef]
- Raverot, G.; Ilie, M.; Lasolle, H.; Amodru, V.; Trouillas, J.; Castinetti, F.; Brue, T. Aggressive pituitary tumours and pituitary carcinomas. Nat. Rev. Endocrinol. 2021, 17, 671–684. [Google Scholar] [CrossRef]
- Syro, L.; Rotondo, F.; Camargo, M.; Ortiz, L.; Serna, C.; Kovacs, K. Temozolomide and Pituitary Tumors: Current Understanding, Unresolved Issues, and Future Directions. Front. Endocrinol. 2018, 9, 318. [Google Scholar] [CrossRef]
- Mamidi, T.K.K.; Wu, J.; Hicks, C. Integrating germline and somatic variation information using genomic data for the discovery of biomarkers in prostate cancer. BMC Cancer 2019, 19, 229. [Google Scholar] [CrossRef]
- Caja, F.; Vodickova, L.; Kral, J.; Vymetalkova, V.; Naccarati, A.; Vodicka, P. Mismatch repair gene variant in sporadic solid cancers. Int. J. Mol. Sci. 2020, 21, 5561. [Google Scholar] [CrossRef]
- Song, G.G.; Kim, J.H.; Lee, H. Genome-wide pathway analysis in major depresive disorder. J. Mol. Neurosci. 2013, 51, 428–436. [Google Scholar] [CrossRef] [PubMed]
- GATK. Available online: https://gatk.broadinstitute.org/ (accessed on 6 October 2021).
- GDC. Available online: https://docs.gdc.cancer.gov/ (accessed on 6 October 2021).
- Chang, M.; Yang, C.; Bao, X.; Wang, R. Genetic and Epigenetic Causes of Pituitary Adenomas. Front. Endocrinol. 2021, 11, 596554. [Google Scholar] [CrossRef] [PubMed]
- Ballmann, C.; Thiel, A.; Korah, H.E.; Reis, A.C.; Saeger, W.; Stepanow, S.; Köhrer, K.; Reifenberger, G.; Knobbe-Thomsen, C.B.; Knappe, U.J.; et al. USP8 Mutations in Pituitary Cushing Adenomas-Targeted Analysis by Next-Generation Sequencing. J. Endocr. Soc. 2018, 2, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, P.; Naidoo, R.; Ramkaran, P.; Chuturgoon, A. Effect of maternal HIV infection, BMI and NOx air pollution exposure on birth outcomes in South African pregnant women genotyped for the p53 Pro72Arg (rs1042522). Int. J. Immunogenet. 2020, 47, 414–429. [Google Scholar] [CrossRef] [PubMed]
- Leite, M.; Giacomin, L.; Piranda, D.; Festa-Vasconcellos, J.; Indio-do-Brasil, V.; Koifman, S.; de Moura-Neto, R.S.; de Carvalho, M.A.; Vianna-Jorge, R. Epidermal growth factor receptor gene polymorphisms are associated with prognostic features of breast cancer. BMC Cancer 2014, 14, 190. [Google Scholar] [CrossRef]
- Baumann, A.; Buchberger, A.; Piontek, G.; Schüttler, D.; Rudelius, M.; Reiter, R.; Gebel, L.; Piendl, G.; Brockhoff, G.; Pickhard, A. The Aurora-Kinase A Phe31-Ile polymorphism as possible predictor of response to treatment in head and neck squamous cell carcinoma. Oncotarget 2018, 9, 12769–12780. [Google Scholar] [CrossRef]
- Vargas-Torres, S.L.; Portari, E.A.; Silva, A.L.; Klumb, E.M.; da Rocha Guillobel, H.C.; de Camargo, M.J.; Santos-Rebouças, C.B.; Russomano, F.B.; Macedo, J.M.B. Roles of CDKN1A gene polymorphisms (rs1801270 and rs1059234) in the development of cervical neoplasia. Tumour Biol. 2016, 37, 10469–10478. [Google Scholar] [CrossRef]
- Taniguchi-Ponciano, K.; Andonegui-Elguera, S.; Peña-Martínez, E.; Silva-Román, G.; Vela-Patiño, S.; Gomez-Apo, E.; Chavez-Macias, L.; Vargas-Ortega, G.; Espinosa-de-Los-Monteros, L.; Gonzalez-Virla, B.; et al. Transcriptome and methylome analysis reveals three cellular origins of pituitary tumors. Sci. Rep. 2020, 10, 19373. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Number | Age/Sex | Clinical Diagnosis | Pathological Diagnosis | Max. Tumor Diameter (mm) | Cav. Sinus Invasion | Vision Abnormal | Pituitary Surgery | SNV |
|---|---|---|---|---|---|---|---|---|
| 1 | 28/F | Cushing disease | ACTH-Carcinoma | 28 | Yes | Yes | 4 TSS, 1 TCS | USP8, TP53, AURKA, EGFR, HSD3B1, CDKN1A |
| 2 | 52/M | Non-Functioning | Crooke Cell Adenoma | 44 | Yes | Yes | 1 TSS | TP53, EGFR, HSD3B1, CDKN1A |
| 3 | 61/F | Non-Functioning | Silent ACTH-Adenoma | 51 | Yes | Yes | 2 TSS | CDKN1A, HSD3B1, AURKA, TP53 |
| 4 | 45/F | Non-Functioning | Silent ACTH-Adenoma | 45 | Yes | Yes | 2 TSS, 1 TCS | CDKN1A, EGFR, HSD3B1 |
| 5 | 52/F | Non-Functioning | Silent ACTH-Adenoma | 31 | Yes | Yes | 1 TSS | CDKN1A, AURKA, TP53, EGFR, HSD3B1 |
| 6 | 54/F | Cushing Disease | ACTH-Adenoma | 44 | Yes | Yes | 1 TCS | HSD3B1, TP53, |
| 7 | 40/F | Cushing Disease | ACTH-Adenoma | 18 | Yes | No | 1 TSS | EGFR, TP53, HSDB3B1 |
| 8 | 18/F | Cushing Disease | ACTH-Adenoma | 18 | No | No | 2 TSS | HSD3B1, TP53, AURKA, |
| 9 | 17/F | Cushing Disease | ACTH-Adenoma | 23 | Yes | Yes | 1 TCS | HSD3B1, TP53, EGFR |
| 10 | 21/F | Nelson Syndrome | ACTH-Adenoma | 17 | Yes | Yes | 2 TSS | USP8, TP53, HSD3B1, CDKN1A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andonegui-Elguera, S.; Silva-Román, G.; Peña-Martínez, E.; Taniguchi-Ponciano, K.; Vela-Patiño, S.; Remba-Shapiro, I.; Gómez-Apo, E.; Espinosa-de-los-Monteros, A.-L.; Portocarrero-Ortiz, L.A.; Guinto, G.; et al. The Genomic Landscape of Corticotroph Tumors: From Silent Adenomas to ACTH-Secreting Carcinomas. Int. J. Mol. Sci. 2022, 23, 4861. https://doi.org/10.3390/ijms23094861
Andonegui-Elguera S, Silva-Román G, Peña-Martínez E, Taniguchi-Ponciano K, Vela-Patiño S, Remba-Shapiro I, Gómez-Apo E, Espinosa-de-los-Monteros A-L, Portocarrero-Ortiz LA, Guinto G, et al. The Genomic Landscape of Corticotroph Tumors: From Silent Adenomas to ACTH-Secreting Carcinomas. International Journal of Molecular Sciences. 2022; 23(9):4861. https://doi.org/10.3390/ijms23094861
Chicago/Turabian StyleAndonegui-Elguera, Sergio, Gloria Silva-Román, Eduardo Peña-Martínez, Keiko Taniguchi-Ponciano, Sandra Vela-Patiño, Ilan Remba-Shapiro, Erick Gómez-Apo, Ana-Laura Espinosa-de-los-Monteros, Lesly A. Portocarrero-Ortiz, Gerardo Guinto, and et al. 2022. "The Genomic Landscape of Corticotroph Tumors: From Silent Adenomas to ACTH-Secreting Carcinomas" International Journal of Molecular Sciences 23, no. 9: 4861. https://doi.org/10.3390/ijms23094861
APA StyleAndonegui-Elguera, S., Silva-Román, G., Peña-Martínez, E., Taniguchi-Ponciano, K., Vela-Patiño, S., Remba-Shapiro, I., Gómez-Apo, E., Espinosa-de-los-Monteros, A.-L., Portocarrero-Ortiz, L. A., Guinto, G., Moreno-Jimenez, S., Chavez-Macias, L., Saucedo, R., Basurto-Acevedo, L., Lopez-Felix, B., Gonzalez-Torres, C., Gaytan-Cervantes, J., Ayala-Sumuano, J. T., Burak-Leipuner, A., ... Mercado, M. (2022). The Genomic Landscape of Corticotroph Tumors: From Silent Adenomas to ACTH-Secreting Carcinomas. International Journal of Molecular Sciences, 23(9), 4861. https://doi.org/10.3390/ijms23094861