
Regulatory Mechanisms of Prg4 and Gdf5 Expression in Articular Cartilage and Functions in Osteoarthritis

 , ,
, ,
Abstract
:1. Introduction
2. Prg4 Is Essential for Maintaining the Health and Homeostasis of Cartilage
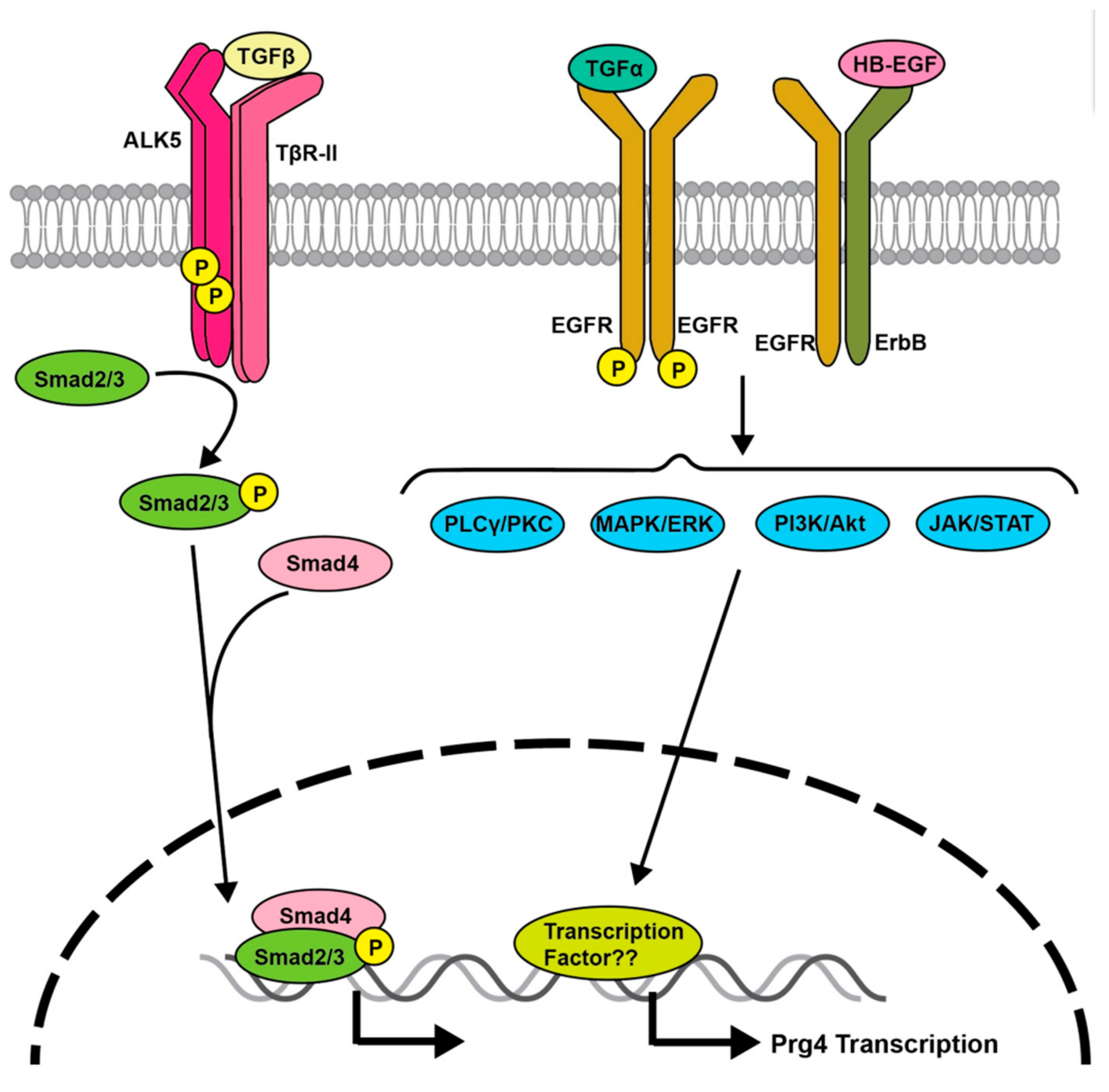
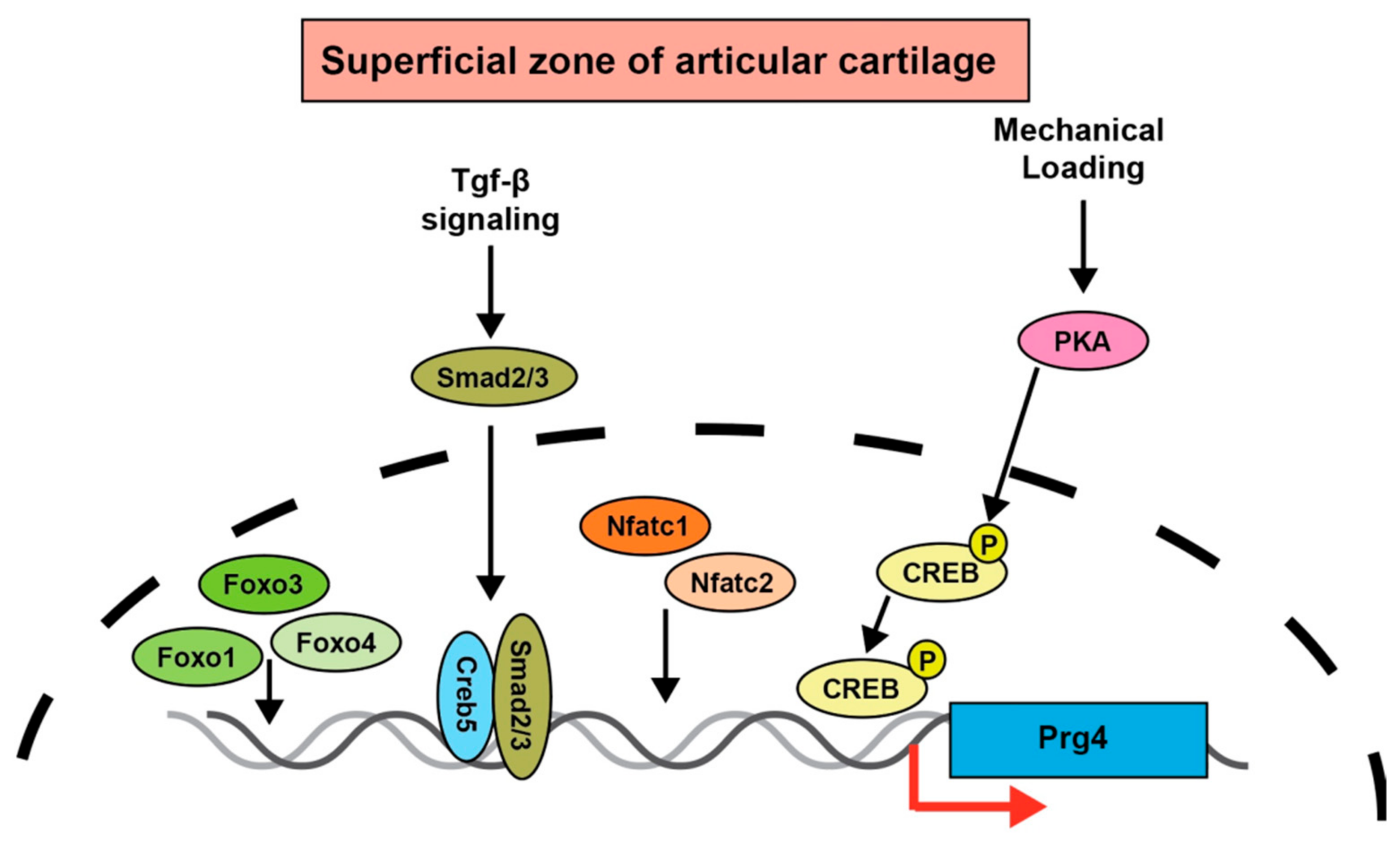
3. Transforming Growth Factor (TGF)-β Signaling Regulates Prg4 Expression
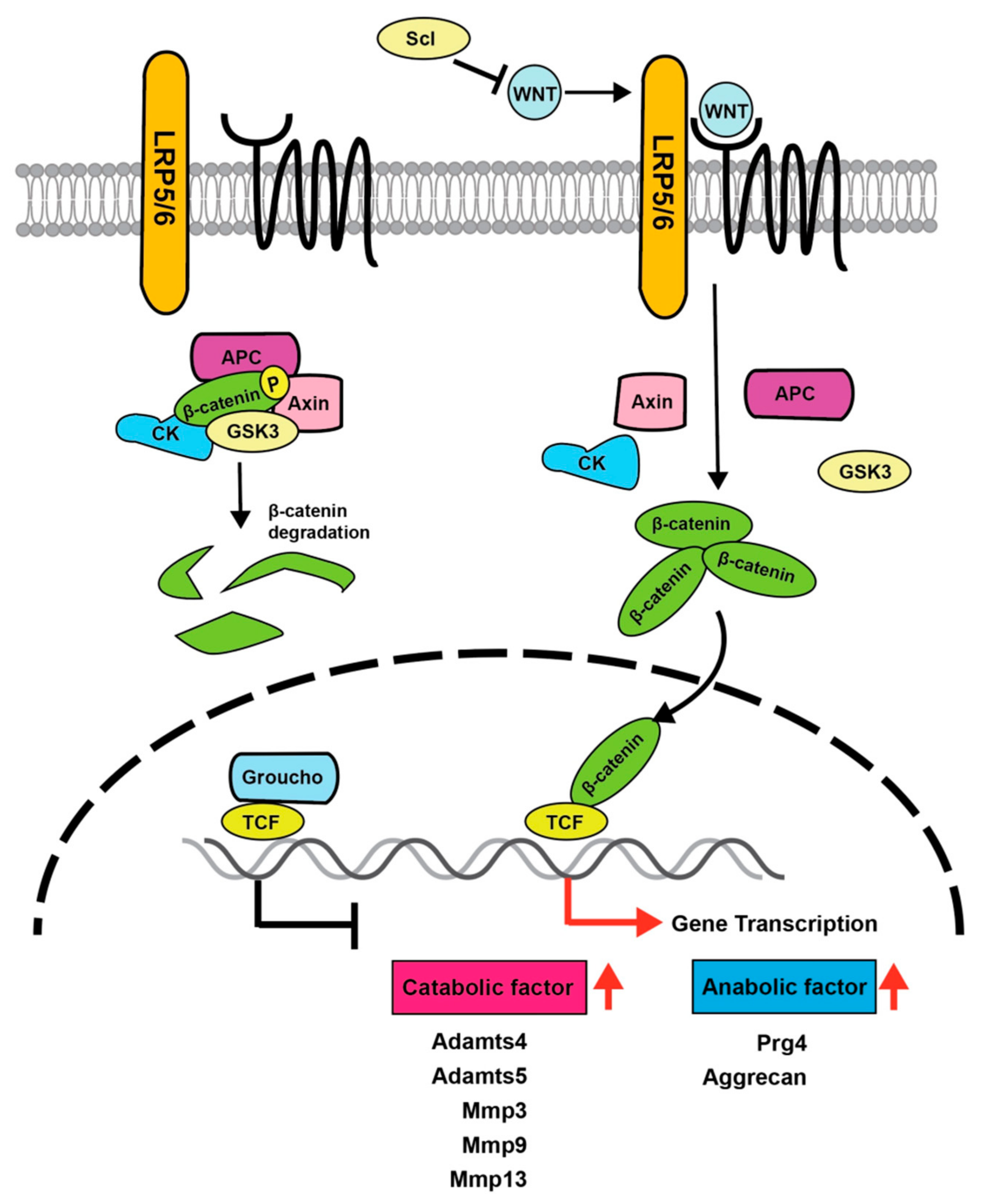
4. Importance of Wnt Signaling in Articular Cartilage
5. EGFR Signaling Regulates Prg4
6. Prg4 Regulation by Transcription Factors
7. GDF5 Signaling
8. Gdf5 Functions in Cartilage Development
9. Gdf5 Is Associated with the Development of OA
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- James, C.B.; Uhl, T.L. A review of articular cartilage pathology and the use of glucosamine sulfate. J. Athl. Train. 2001, 36, 413–419. [Google Scholar] [PubMed]
- Kolasinski, S.L.; Neogi, T.; Hochberg, M.C.; Oatis, C.; Guyatt, G.; Block, J.; Callahan, L.; Copenhaver, C.; Dodge, C.; Felson, D.; et al. 2019 American College of Rheumatology/Arthritis Foundation Guideline for the Management of Osteoarthritis of the Hand, Hip, and Knee. Arthritis Rheumatol. 2020, 72, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Boer, C.G.; Hatzikotoulas, K.; Southam, L.; Stefánsdóttir, L.; Zhang, Y.; Coutinho de Almeida, R.; Wu, T.T.; Zheng, J.; Hartley, A.; Teder-Laving, M.; et al. Deciphering osteoarthritis genetics across 826,690 individuals from 9 populations. Cell 2021, 184, 4784–4818.e4717. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Fukai, A.; Mabuchi, A.; Ikeda, T.; Yano, F.; Ohba, S.; Nishida, N.; Akune, T.; Yoshimura, N.; Nakagawa, T.; et al. Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat. Med. 2010, 16, 678–686. [Google Scholar] [CrossRef]
- Yano, F.; Ohba, S.; Murahashi, Y.; Tanaka, S.; Saito, T.; Chung, U.I. Runx1 contributes to articular cartilage maintenance by enhancement of cartilage matrix production and suppression of hypertrophic differentiation. Sci. Rep. 2019, 9, 7666. [Google Scholar] [CrossRef]
- Goldring, M.B.; Marcu, K.B. Epigenomic and microRNA-mediated regulation in cartilage development, homeostasis, and osteoarthritis. Trends Mol. Med. 2012, 18, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Jules, J.; Feng, X. In vitro investigation of the roles of the proinflammatory cytokines tumor necrosis factor-α and interleukin-1 in murine osteoclastogenesis. Methods Mol. Biol. 2014, 1155, 109–123. [Google Scholar] [CrossRef]
- Takahata, Y.; Nakamura, E.; Hata, K.; Wakabayashi, M.; Murakami, T.; Wakamori, K.; Yoshikawa, H.; Matsuda, A.; Fukui, N.; Nishimura, R. Sox4 is involved in osteoarthritic cartilage deterioration through induction of ADAMTS4 and ADAMTS5. FASEB J. 2019, 33, 619–630. [Google Scholar] [CrossRef]
- Wang, M.; Sampson, E.R.; Jin, H.; Li, J.; Ke, Q.H.; Im, H.J.; Chen, D. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res. Ther. 2013, 15, R5. [Google Scholar] [CrossRef] [Green Version]
- Little, C.B.; Barai, A.; Burkhardt, D.; Smith, S.M.; Fosang, A.J.; Werb, Z.; Shah, M.; Thompson, E.W. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheumatol. 2009, 60, 3723–3733. [Google Scholar] [CrossRef]
- Stanton, H.; Rogerson, F.M.; East, C.J.; Golub, S.B.; Lawlor, K.E.; Meeker, C.T.; Little, C.B.; Last, K.; Farmer, P.J.; Campbell, I.; et al. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature 2005, 434, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Botter, S.M.; Glasson, S.S.; Hopkins, B.; Clockaerts, S.; Weinans, H.; van Leeuwen, J.P.; van Osch, G.J. ADAMTS5-/- mice have less subchondral bone changes after induction of osteoarthritis through surgical instability: Implications for a link between cartilage and subchondral bone changes. Osteoarthr. Cartil. 2009, 17, 636–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glasson, S.S.; Askew, R.; Sheppard, B.; Carito, B.; Blanchet, T.; Ma, H.L.; Flannery, C.R.; Peluso, D.; Kanki, K.; Yang, Z.; et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 2005, 434, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Glasson, S.S.; Askew, R.; Sheppard, B.; Carito, B.A.; Blanchet, T.; Ma, H.L.; Flannery, C.R.; Kanki, K.; Wang, E.; Peluso, D.; et al. Characterization of and osteoarthritis susceptibility in ADAMTS-4-knockout mice. Arthritis Rheumatol. 2004, 50, 2547–2558. [Google Scholar] [CrossRef]
- Song, R.H.; Tortorella, M.D.; Malfait, A.M.; Alston, J.T.; Yang, Z.; Arner, E.C.; Griggs, D.W. Aggrecan degradation in human articular cartilage explants is mediated by both ADAMTS-4 and ADAMTS-5. Arthritis Rheumatol. 2007, 56, 575–585. [Google Scholar] [CrossRef]
- Ruan, M.Z.; Erez, A.; Guse, K.; Dawson, B.; Bertin, T.; Chen, Y.; Jiang, M.M.; Yustein, J.; Gannon, F.; Lee, B.H. Proteoglycan 4 expression protects against the development of osteoarthritis. Sci. Transl. Med. 2013, 5, 176ra134. [Google Scholar] [CrossRef] [Green Version]
- Mang, T.; Kleinschmidt-Dörr, K.; Ploeger, F.; Lindemann, S.; Gigout, A. The GDF-5 mutant M1673 exerts robust anabolic and anti-catabolic effects in chondrocytes. J. Cell Mol. Med. 2020, 24, 7141–7150. [Google Scholar] [CrossRef]
- Marcelino, J.; Carpten, J.D.; Suwairi, W.M.; Gutierrez, O.M.; Schwartz, S.; Robbins, C.; Sood, R.; Makalowska, I.; Baxevanis, A.; Johnstone, B.; et al. CACP, encoding a secreted proteoglycan, is mutated in camptodactyly-arthropathy-coxa vara-pericarditis syndrome. Nat. Genet. 1999, 23, 319–322. [Google Scholar] [CrossRef]
- Bahabri, S.A.; Suwairi, W.M.; Laxer, R.M.; Polinkovsky, A.; Dalaan, A.A.; Warman, M.L. The camptodactyly-arthropathy-coxa vara-pericarditis syndrome: Clinical features and genetic mapping to human chromosome 1. Arthritis Rheumatol. 1998, 41, 730–735. [Google Scholar] [CrossRef]
- Rhee, D.K.; Marcelino, J.; Baker, M.; Gong, Y.; Smits, P.; Lefebvre, V.; Jay, G.D.; Stewart, M.; Wang, H.; Warman, M.L.; et al. The secreted glycoprotein lubricin protects cartilage surfaces and inhibits synovial cell overgrowth. J. Clin. Invest. 2005, 115, 622–631. [Google Scholar] [CrossRef] [Green Version]
- Kozhemyakina, E.; Zhang, M.; Ionescu, A.; Ayturk, U.M.; Ono, N.; Kobayashi, A.; Kronenberg, H.; Warman, M.L.; Lassar, A.B. Identification of a Prg4-expressing articular cartilage progenitor cell population in mice. Arthritis Rheumatol. 2015, 67, 1261–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nugent-Derfus, G.E.; Takara, T.; O’neill, J.K.; Cahill, S.B.; Görtz, S.; Pong, T.; Inoue, H.; Aneloski, N.M.; Wang, W.W.; Vega, K.I.; et al. Continuous passive motion applied to whole joints stimulates chondrocyte biosynthesis of PRG4. Osteoarthr. Cartil. 2007, 15, 566–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jay, G.D.; Torres, J.R.; Rhee, D.K.; Helminen, H.J.; Hytinnen, M.M.; Cha, C.J.; Elsaid, K.; Kim, K.S.; Cui, Y.; Warman, M.L. Association between friction and wear in diarthrodial joints lacking lubricin. Arthritis Rheumatol. 2007, 56, 3662–3669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coles, J.M.; Zhang, L.; Blum, J.J.; Warman, M.L.; Jay, G.D.; Guilak, F.; Zauscher, S. Loss of cartilage structure, stiffness, and frictional properties in mice lacking PRG4. Arthritis Rheumatol. 2010, 62, 1666–1674. [Google Scholar] [CrossRef]
- Waller, K.A.; Zhang, L.X.; Elsaid, K.A.; Fleming, B.C.; Warman, M.L.; Jay, G.D. Role of lubricin and boundary lubrication in the prevention of chondrocyte apoptosis. Proc. Natl. Acad. Sci. USA 2013, 110, 5852–5857. [Google Scholar] [CrossRef] [Green Version]
- Chavez, R.D.; Sohn, P.; Serra, R. Prg4 prevents osteoarthritis induced by dominant-negative interference of TGF-ß signaling in mice. PLoS ONE 2019, 14, e0210601. [Google Scholar] [CrossRef]
- Miyatake, K.; Iwasa, K.; McNary, S.M.; Peng, G.; Reddi, A.H. Modulation of Superficial Zone Protein/Lubricin/PRG4 by Kartogenin and Transforming Growth Factor-β1 in Surface Zone Chondrocytes in Bovine Articular Cartilage. Cartilage 2016, 7, 388–397. [Google Scholar] [CrossRef] [Green Version]
- Panahipour, L.; Omerbasic, A.; Nasirzade, J.; Gruber, R. TGF-β Activity of a Demineralized Bone Matrix. Int. J. Mol. Sci. 2021, 22, 664. [Google Scholar] [CrossRef]
- Jones, A.R.; Flannery, C.R. Bioregulation of lubricin expression by growth factors and cytokines. Eur. Cell Mater. 2007, 13, 40–45, discussion 45. [Google Scholar] [CrossRef]
- Chavez, R.D.; Coricor, G.; Perez, J.; Seo, H.S.; Serra, R. SOX9 protein is stabilized by TGF-β and regulates PAPSS2 mRNA expression in chondrocytes. Osteoarthr. Cartil. 2017, 25, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, T.A.; Gastelum, N.S.; Han, E.H.; Nugent-Derfus, G.E.; Schumacher, B.L.; Sah, R.L. Differential regulation of proteoglycan 4 metabolism in cartilage by IL-1alpha, IGF-I, and TGF-beta1. Osteoarthr. Cartil. 2008, 16, 90–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Summa, F.; Kargarpour, Z.; Nasirzade, J.; Stähli, A.; Mitulović, G.; Panić-Janković, T.; Koller, V.; Kaltenbach, C.; Müller, H.; Panahipour, L. TGFβ activity released from platelet-rich fibrin adsorbs to titanium surface and collagen membranes. Sci. Rep. 2020, 10, 10203. [Google Scholar] [CrossRef]
- Kazemi, D.; Fakhrjou, A.; Dizaji, V.M.; Alishahi, M.K. Effect of autologous platelet rich fibrin on the healing of experimental articular cartilage defects of the knee in an animal model. Biomed. Res. Int. 2014, 2014, 486436. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, K.N.; Peifer, M. Wnt/Beta-Catenin Signaling Regulation and a Role for Biomolecular Condensates. Dev. Cell 2019, 48, 429–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Gunawardena, A.T.; Iwamoto, M.; Enomoto-Iwamoto, M. Wnt signaling in cartilage development and diseases: Lessons from animal studies. Lab. Investig. 2016, 96, 186–196. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Tang, D.; Wu, Q.; Hao, S.; Chen, M.; Xie, C.; Rosier, R.N.; O’Keefe, R.J.; Zuscik, M.; Chen, D. Activation of beta-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult beta-catenin conditional activation mice. J. Bone Miner. Res. 2009, 24, 12–21. [Google Scholar] [CrossRef]
- Yuasa, T.; Otani, T.; Koike, T.; Iwamoto, M.; Enomoto-Iwamoto, M. Wnt/beta-catenin signaling stimulates matrix catabolic genes and activity in articular chondrocytes: Its possible role in joint degeneration. Lab. Investig. 2008, 88, 264–274. [Google Scholar] [CrossRef] [Green Version]
- Dell’accio, F.; De Bari, C.; Eltawil, N.M.; Vanhummelen, P.; Pitzalis, C. Identification of the molecular response of articular cartilage to injury, by microarray screening: Wnt-16 expression and signaling after injury and in osteoarthritis. Arthritis Rheumatol. 2008, 58, 1410–1421. [Google Scholar] [CrossRef]
- Lories, R.J.; Peeters, J.; Bakker, A.; Tylzanowski, P.; Derese, I.; Schrooten, J.; Thomas, J.T.; Luyten, F.P. Articular cartilage and biomechanical properties of the long bones in Frzb-knockout mice. Arthritis Rheumatol. 2007, 56, 4095–4103. [Google Scholar] [CrossRef] [PubMed]
- Thysen, S.; Luyten, F.P.; Lories, R.J. Loss of Frzb and Sfrp1 differentially affects joint homeostasis in instability-induced osteoarthritis. Osteoarthr. Cartil. 2015, 23, 275–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Zhu, M.; Awad, H.; Li, T.F.; Sheu, T.J.; Boyce, B.F.; Chen, D.; O’Keefe, R.J. Inhibition of beta-catenin signaling causes defects in postnatal cartilage development. J. Cell Sci. 2008, 121, 1455–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, M.; Chen, M.; Zuscik, M.; Wu, Q.; Wang, Y.J.; Rosier, R.N.; O’Keefe, R.J.; Chen, D. Inhibition of beta-catenin signaling in articular chondrocytes results in articular cartilage destruction. Arthritis Rheumatol. 2008, 58, 2053–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuhara, R.; Ohta, Y.; Yuasa, T.; Kondo, N.; Hoang, T.; Addya, S.; Fortina, P.; Pacifici, M.; Iwamoto, M.; Enomoto-Iwamoto, M. Roles of β-catenin signaling in phenotypic expression and proliferation of articular cartilage superficial zone cells. Lab. Investig. 2011, 91, 1739–1752. [Google Scholar] [CrossRef] [Green Version]
- Xuan, F.; Yano, F.; Mori, D.; Chijimatsu, R.; Maenohara, Y.; Nakamoto, H.; Mori, Y.; Makii, Y.; Oichi, T.; Taketo, M.; et al. Wnt/β-catenin signaling contributes to articular cartilage homeostasis through lubricin induction in the superficial zone. Arthritis Res. Ther. 2019, 21, 247. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zuo, Z.; Kuang, Y. An Emerging Target in the Battle against Osteoarthritis: Macrophage Polarization. Int. J. Mol. Sci. 2020, 21, 8513. [Google Scholar] [CrossRef]
- Chan, B.Y.; Fuller, E.S.; Russell, A.K.; Smith, S.M.; Smith, M.M.; Jackson, M.T.; Cake, M.A.; Read, R.A.; Bateman, J.F.; Sambrook, P.N.; et al. Increased chondrocyte sclerostin may protect against cartilage degradation in osteoarthritis. Osteoarthr. Cartil. 2011, 19, 874–885. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.C.; Christiansen, B.A.; Murugesh, D.K.; Sebastian, A.; Hum, N.R.; Collette, N.M.; Hatsell, S.; Economides, A.N.; Blanchette, C.D.; Loots, G.G.; et al. SOST/Sclerostin Improves Posttraumatic Osteoarthritis and Inhibits MMP2/3 Expression After Injury. J. Bone Miner. Res. 2018, 33, 1105–1113. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Day, T.F.; Jiang, X.; Garrett-Beal, L.; Topol, L.; Yang, Y. Wnt/beta-catenin signaling is sufficient and necessary for synovial joint formation. Genes Dev. 2004, 18, 2404–2417. [Google Scholar] [CrossRef]
- Citri, A.; Yarden, Y. EGF-ERBB signalling: Towards the systems level. Nat. Rev. Mol. Cell Biol. 2006, 7, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Appleton, C.T.; Usmani, S.E.; Mort, J.S.; Beier, F. Rho/ROCK and MEK/ERK activation by transforming growth factor-alpha induces articular cartilage degradation. Lab. Investig. 2010, 90, 20–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Siclari, V.A.; Lan, S.; Zhu, J.; Koyama, E.; Dupuis, H.L.; Enomoto-Iwamoto, M.; Beier, F.; Qin, L. The critical role of the epidermal growth factor receptor in endochondral ossification. J. Bone Miner. Res. 2011, 26, 2622–2633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, H.; Ma, X.; Tong, W.; Doyran, B.; Sun, Z.; Wang, L.; Zhang, X.; Zhou, Y.; Badar, F.; Chandra, A.; et al. EGFR signaling is critical for maintaining the superficial layer of articular cartilage and preventing osteoarthritis initiation. Proc. Natl. Acad. Sci. USA 2016, 113, 14360–14365. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Luo, L.; Gui, T.; Yu, F.; Yan, L.; Yao, L.; Zhong, L.; Yu, W.; Han, B.; Patel, J.M.; et al. Targeting cartilage EGFR pathway for osteoarthritis treatment. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Qin, L.; Beier, F. EGFR Signaling: Friend or Foe for Cartilage? JBMR Plus 2019, 3, e10177. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, J.; Liu, F.; Li, Y.; Chandra, A.; Levin, L.S.; Beier, F.; Enomoto-Iwamoto, M.; Qin, L. Reduced EGFR signaling enhances cartilage destruction in a mouse osteoarthritis model. Bone Res. 2014, 2, 14015. [Google Scholar] [CrossRef] [Green Version]
- Shepard, J.B.; Jeong, J.W.; Maihle, N.J.; O’Brien, S.; Dealy, C.N. Transient anabolic effects accompany epidermal growth factor receptor signal activation in articular cartilage in vivo. Arthritis Res. Ther. 2013, 15, R60. [Google Scholar] [CrossRef] [Green Version]
- Pest, M.A.; Russell, B.A.; Zhang, Y.W.; Jeong, J.W.; Beier, F. Disturbed cartilage and joint homeostasis resulting from a loss of mitogen-inducible gene 6 in a mouse model of joint dysfunction. Arthritis Rheumatol. 2014, 66, 2816–2827. [Google Scholar] [CrossRef] [Green Version]
- Bellini, M.; Pest, M.A.; Miranda-Rodrigues, M.; Qin, L.; Jeong, J.W.; Beier, F. Overexpression of MIG-6 in the cartilage induces an osteoarthritis-like phenotype in mice. Arthritis Res. Ther. 2020, 22, 119. [Google Scholar] [CrossRef]
- Burgering, B.M.; Medema, R.H. Decisions on life and death: FOXO Forkhead transcription factors are in command when PKB/Akt is off duty. J. Leukoc. Biol. 2003, 73, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Van der Horst, A.; Burgering, B.M. Stressing the role of FoxO proteins in lifespan and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Akasaki, Y.; Hasegawa, A.; Saito, M.; Asahara, H.; Iwamoto, Y.; Lotz, M.K. Dysregulated FOXO transcription factors in articular cartilage in aging and osteoarthritis. Osteoarthr. Cartil. 2014, 22, 162–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.I.; Choi, S.; Matsuzaki, T.; Alvarez-Garcia, O.; Olmer, M.; Grogan, S.P.; D’Lima, D.D.; Lotz, M.K. FOXO1 and FOXO3 transcription factors have unique functions in meniscus development and homeostasis during aging and osteoarthritis. Proc. Natl. Acad. Sci. USA 2020, 117, 3135–3143. [Google Scholar] [CrossRef]
- Matsuzaki, T.; Alvarez-Garcia, O.; Mokuda, S.; Nagira, K.; Olmer, M.; Gamini, R.; Miyata, K.; Akasaki, Y.; Su, A.I.; Asahara, H.; et al. FoxO transcription factors modulate autophagy and proteoglycan 4 in cartilage homeostasis and osteoarthritis. Sci. Transl. Med. 2018, 10, eaan0746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenblatt, M.B.; Ritter, S.Y.; Wright, J.; Tsang, K.; Hu, D.; Glimcher, L.H.; Aliprantis, A.O. NFATc1 and NFATc2 repress spontaneous osteoarthritis. Proc. Natl. Acad. Sci. USA 2013, 110, 19914–19919. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Gardner, B.M.; Lu, Q.; Rodova, M.; Woodbury, B.G.; Yost, J.G.; Roby, K.F.; Pinson, D.M.; Tawfik, O.; Anderson, H.C. Transcription factor Nfat1 deficiency causes osteoarthritis through dysfunction of adult articular chondrocytes. J. Pathol. 2009, 219, 163–172. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, H.; Kozhemyakina, E.; Hung, H.H.; Grodzinsky, A.J.; Lassar, A.B. Mechanical motion promotes expression of Prg4 in articular cartilage via multiple CREB-dependent, fluid flow shear stress-induced signaling pathways. Genes Dev. 2014, 28, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.H.; Gao, Y.; Jadhav, U.; Hung, H.H.; Holton, K.M.; Grodzinsky, A.J.; Shivdasani, R.A.; Lassar, A.B. Creb5 establishes the competence for Prg4 expression in articular cartilage. Commun. Biol. 2021, 4, 332. [Google Scholar] [CrossRef]
- Miyazono, K.; Maeda, S.; Imamura, T. BMP receptor signaling: Transcriptional targets, regulation of signals, and signaling cross-talk. Cytokine Growth Factor Rev. 2005, 16, 251–263. [Google Scholar] [CrossRef]
- Nishitoh, H.; Ichijo, H.; Kimura, M.; Matsumoto, T.; Makishima, F.; Yamaguchi, A.; Yamashita, H.; Enomoto, S.; Miyazono, K. Identification of type I and type II serine/threonine kinase receptors for growth/differentiation factor-5. J. Biol. Chem. 1996, 271, 21345–21352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, S.; Kanno, S.; Gai, Z.; Suemoto, H.; Kawakatsu, M.; Tanishima, H.; Morimoto, Y.; Nishioka, K.; Hatamura, I.; Yoshida, M.; et al. Trps1 plays a pivotal role downstream of Gdf5 signaling in promoting chondrogenesis and apoptosis of ATDC5 cells. Genes Cells 2008, 13, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Yang, T.; Meng, W.; Liu, H.; Zhang, T.; Shi, R. Overexpression of GDF5 through an adenovirus vector stimulates osteogenesis of human mesenchymal stem cells in vitro and in vivo. Cells Tissues Organs 2012, 196, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Settle, S.H.; Rountree, R.B.; Sinha, A.; Thacker, A.; Higgins, K.; Kingsley, D.M. Multiple joint and skeletal patterning defects caused by single and double mutations in the mouse Gdf6 and Gdf5 genes. Dev. Biol. 2003, 254, 116–130. [Google Scholar] [CrossRef] [Green Version]
- Storm, E.E.; Kingsley, D.M. Joint patterning defects caused by single and double mutations in members of the bone morphogenetic protein (BMP) family. Development 1996, 122, 3969–3979. [Google Scholar] [CrossRef]
- Storm, E.E.; Kingsley, D.M. GDF5 coordinates bone and joint formation during digit development. Dev. Biol. 1999, 209, 11–27. [Google Scholar] [CrossRef] [Green Version]
- Coleman, C.M.; Vaughan, E.E.; Browe, D.C.; Mooney, E.; Howard, L.; Barry, F. Growth differentiation factor-5 enhances in vitro mesenchymal stromal cell chondrogenesis and hypertrophy. Stem. Cells Dev. 2013, 22, 1968–1976. [Google Scholar] [CrossRef] [Green Version]
- Tsumaki, N.; Tanaka, K.; Arikawa-Hirasawa, E.; Nakase, T.; Kimura, T.; Thomas, J.T.; Ochi, T.; Luyten, F.P.; Yamada, Y. Role of CDMP-1 in skeletal morphogenesis: Promotion of mesenchymal cell recruitment and chondrocyte differentiation. J. Cell Biol. 1999, 144, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Faiyaz-Ul-Haque, M.; Ahmad, W.; Wahab, A.; Haque, S.; Azim, A.C.; Zaidi, S.H.; Teebi, A.S.; Ahmad, M.; Cohn, D.H.; Siddique, T.; et al. Frameshift mutation in the cartilage-derived morphogenetic protein 1 (CDMP1) gene and severe acromesomelic chondrodysplasia resembling Grebe-type chondrodysplasia. Am. J. Med. Genet. 2002, 111, 31–37. [Google Scholar] [CrossRef]
- Hatzikotoulas, K.; Roposch, A.; Shah, K.M.; Clark, M.J.; Bratherton, S.; Limbani, V.; Steinberg, J.; Zengini, E.; Warsame, K.; Ratnayake, M.; et al. Genome-wide association study of developmental dysplasia of the hip identifies an association with. Commun. Biol. 2018, 1, 56. [Google Scholar] [CrossRef]
- Baghdadi, T.; Nejadhosseinian, M.; Shirkoohi, R.; Mostafavi Tabatabaee, R.; Tamehri, S.S.; Saffari, M.; Mortazavi, S.M.J. DNA hypermethylation of GDF5 in developmental dysplasia of the hip (DDH). Mol. Genet. Genomic. Med. 2019, 7, e887. [Google Scholar] [CrossRef] [Green Version]
- Hatazawa, Y.; Ono, Y.; Hirose, Y.; Kanai, S.; Fujii, N.L.; Machida, S.; Nishino, I.; Shimizu, T.; Okano, M.; Kamei, Y.; et al. Reduced Dnmt3a increases Gdf5 expression with suppressed satellite cell differentiation and impaired skeletal muscle regeneration. FASEB J. 2018, 32, 1452–1467. [Google Scholar] [CrossRef] [Green Version]
- Reynard, L.N.; Bui, C.; Canty-Laird, E.G.; Young, D.A.; Loughlin, J. Expression of the osteoarthritis-associated gene GDF5 is modulated epigenetically by DNA methylation. Hum. Mol. Genet. 2011, 20, 3450–3460. [Google Scholar] [CrossRef] [Green Version]
- Kleinschmidt, K.; Ploeger, F.; Nickel, J.; Glockenmeier, J.; Kunz, P.; Richter, W. Enhanced reconstruction of long bone architecture by a growth factor mutant combining positive features of GDF-5 and BMP-2. Biomaterials 2013, 34, 5926–5936. [Google Scholar] [CrossRef]
- Parrish, W.R.; Byers, B.A.; Su, D.; Geesin, J.; Herzberg, U.; Wadsworth, S.; Bendele, A.; Story, B. Intra-articular therapy with recombinant human GDF5 arrests disease progression and stimulates cartilage repair in the rat medial meniscus transection (MMT) model of osteoarthritis. Osteoarthr. Cartil. 2017, 25, 554–560. [Google Scholar] [CrossRef] [Green Version]
- Lan, R.; Ge, D.; Liu, Y.Z.; You, Z. Dcx expression defines a subpopulation of Gdf5 positive cells with chondrogenic potentials in E12.5 mouse embryonic limbs. Biochem. Biophys. Rep. 2022, 29, 101200. [Google Scholar] [CrossRef]
- Roelofs, A.J.; Zupan, J.; Riemen, A.H.K.; Kania, K.; Ansboro, S.; White, N.; Clark, S.M.; De Bari, C. Joint morphogenetic cells in the adult mammalian synovium. Nat. Commun. 2017, 8, 15040. [Google Scholar] [CrossRef] [Green Version]
- Shwartz, Y.; Viukov, S.; Krief, S.; Zelzer, E. Joint Development Involves a Continuous Influx of Gdf5-Positive Cells. Cell. Rep. 2016, 15, 2577–2587. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.T.; Lin, K.; Nandedkar, M.; Camargo, M.; Cervenka, J.; Luyten, F.P. A human chondrodysplasia due to a mutation in a TGF-beta superfamily member. Nat. Genet. 1996, 12, 315–317. [Google Scholar] [CrossRef] [Green Version]
- Faiyaz-Ul-Haque, M.; Ahmad, W.; Zaidi, S.H.; Haque, S.; Teebi, A.S.; Ahmad, M.; Cohn, D.H.; Tsui, L.C. Mutation in the cartilage-derived morphogenetic protein-1 (CDMP1) gene in a kindred affected with fibular hypoplasia and complex brachydactyly (DuPan syndrome). Clin. Genet. 2002, 61, 454–458. [Google Scholar] [CrossRef]
- Al-Yahyaee, S.A.; Al-Kindi, M.N.; Habbal, O.; Kumar, D.S. Clinical and molecular analysis of Grebe acromesomelic dysplasia in an Omani family. Am. J. Med. Genet. A 2003, 121A, 9–14. [Google Scholar] [CrossRef]
- Savarirayan, R.; White, S.M.; Goodman, F.R.; Graham, J.M.; Delatycki, M.B.; Lachman, R.S.; Rimoin, D.L.; Everman, D.B.; Warman, M.L. Broad phenotypic spectrum caused by an identical heterozygous CDMP-1 mutation in three unrelated families. Am. J. Med. Genet. A 2003, 117A, 136–142. [Google Scholar] [CrossRef]
- Everman, D.B.; Bartels, C.F.; Yang, Y.; Yanamandra, N.; Goodman, F.R.; Mendoza-Londono, J.R.; Savarirayan, R.; White, S.M.; Graham, J.M.; Gale, R.P.; et al. The mutational spectrum of brachydactyly type C. Am. J. Med. Genet. 2002, 112, 291–296. [Google Scholar] [CrossRef]
- Seemann, P.; Schwappacher, R.; Kjaer, K.W.; Krakow, D.; Lehmann, K.; Dawson, K.; Stricker, S.; Pohl, J.; Plöger, F.; Staub, E.; et al. Activating and deactivating mutations in the receptor interaction site of GDF5 cause symphalangism or brachydactyly type A2. J. Clin. Investig. 2005, 115, 2373–2381. [Google Scholar] [CrossRef] [Green Version]
- Abd Elazeem, M.I.; Abdelaleem, E.A.; Mohamed, R.A. Genetic influence of growth and differentiation factor 5 gene polymorphism (+104T/C) on the development of knee osteoarthritis and its association with disease severity. Eur. J. Rheumatol. 2017, 4, 98–103. [Google Scholar] [CrossRef]
- Masuya, H.; Nishida, K.; Furuichi, T.; Toki, H.; Nishimura, G.; Kawabata, H.; Yokoyama, H.; Yoshida, A.; Tominaga, S.; Nagano, J.; et al. A novel dominant-negative mutation in Gdf5 generated by ENU mutagenesis impairs joint formation and causes osteoarthritis in mice. Hum. Mol. Genet. 2007, 16, 2366–2375. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Ma, S.; Yuan, L.; Wu, S.; Liu, S.; Wei, X.; Chen, L.; Ma, C.; Zhao, H. Knockout of miR-21-5p alleviates cartilage matrix degradation by targeting Gdf5 in temporomandibular joint osteoarthritis. Bone Joint Res. 2020, 9, 689–700. [Google Scholar] [CrossRef]
- Chen, H.; Capellini, T.D.; Schoor, M.; Mortlock, D.P.; Reddi, A.H.; Kingsley, D.M. Heads, Shoulders, Elbows, Knees, and Toes: Modular Gdf5 Enhancers Control Different Joints in the Vertebrate Skeleton. PLoS Genet. 2016, 12, e1006454. [Google Scholar] [CrossRef] [Green Version]
- Ohta, Y.; Okabe, T.; Larmour, C.; Di Rocco, A.; Maijenburg, M.W.; Phillips, A.; Speck, N.A.; Wakitani, S.; Nakamura, T.; Yamada, Y.; et al. Articular cartilage endurance and resistance to osteoarthritic changes require transcription factor Erg. Arthritis Rheumatol. 2015, 67, 2679–2690. [Google Scholar] [CrossRef] [Green Version]
- Hada, S.; Ishijima, M.; Kaneko, H.; Kinoshita, M.; Liu, L.; Sadatsuki, R.; Futami, I.; Yusup, A.; Takamura, T.; Arita, H.; et al. Association of medial meniscal extrusion with medial tibial osteophyte distance detected by T2 mapping MRI in patients with early-stage knee osteoarthritis. Arthritis Res. Ther. 2017, 19, 201. [Google Scholar] [CrossRef] [Green Version]
- Hirata, M.; Kugimiya, F.; Fukai, A.; Saito, T.; Yano, F.; Ikeda, T.; Mabuchi, A.; Sapkota, B.R.; Akune, T.; Nishida, N.; et al. C/EBPβ and RUNX2 cooperate to degrade cartilage with MMP-13 as the target and HIF-2α as the inducer in chondrocytes. Hum. Mol. Genet. 2012, 21, 1111–1123. [Google Scholar] [CrossRef]
- Wang, M.; Shen, J.; Jin, H.; Im, H.J.; Sandy, J.; Chen, D. Recent progress in understanding molecular mechanisms of cartilage degeneration during osteoarthritis. Ann. N. Y. Acad. Sci. 2011, 1240, 61–69. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Prg4 | |
|---|---|
| Upregulation factor | Wnt signal |
| EGFR signal | |
| Tgf-β signal | |
| Foxo1/3/4 transcription factor | |
| Nfatc1, Nfatc2 transcription factor | |
| Creb 5 | |
| Mechanical loading |
| Gdf5 | |
|---|---|
| Upregulation factor | DMM model Pathological condition of OA |
| Down regulation factor | DNA methylation microRNA21-5p Yap |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takahata, Y.; Hagino, H.; Kimura, A.; Urushizaki, M.; Yamamoto, S.; Wakamori, K.; Murakami, T.; Hata, K.; Nishimura, R. Regulatory Mechanisms of Prg4 and Gdf5 Expression in Articular Cartilage and Functions in Osteoarthritis. Int. J. Mol. Sci. 2022, 23, 4672. https://doi.org/10.3390/ijms23094672
Takahata Y, Hagino H, Kimura A, Urushizaki M, Yamamoto S, Wakamori K, Murakami T, Hata K, Nishimura R. Regulatory Mechanisms of Prg4 and Gdf5 Expression in Articular Cartilage and Functions in Osteoarthritis. International Journal of Molecular Sciences. 2022; 23(9):4672. https://doi.org/10.3390/ijms23094672
Chicago/Turabian StyleTakahata, Yoshifumi, Hiromasa Hagino, Ayaka Kimura, Mitsuki Urushizaki, Shiori Yamamoto, Kanta Wakamori, Tomohiko Murakami, Kenji Hata, and Riko Nishimura. 2022. "Regulatory Mechanisms of Prg4 and Gdf5 Expression in Articular Cartilage and Functions in Osteoarthritis" International Journal of Molecular Sciences 23, no. 9: 4672. https://doi.org/10.3390/ijms23094672
APA StyleTakahata, Y., Hagino, H., Kimura, A., Urushizaki, M., Yamamoto, S., Wakamori, K., Murakami, T., Hata, K., & Nishimura, R. (2022). Regulatory Mechanisms of Prg4 and Gdf5 Expression in Articular Cartilage and Functions in Osteoarthritis. International Journal of Molecular Sciences, 23(9), 4672. https://doi.org/10.3390/ijms23094672