
Mitochondrial DNA Consensus Calling and Quality Filtering for Constructing Ancient Human Mitogenomes: Comparison of Two Widely Applied Methods


Abstract
:1. Introduction
2. Results
2.1. Derived Mitogenome Consensus Sequences
2.1.1. Schmutzi Pipeline
2.1.2. ANGSD Pipeline
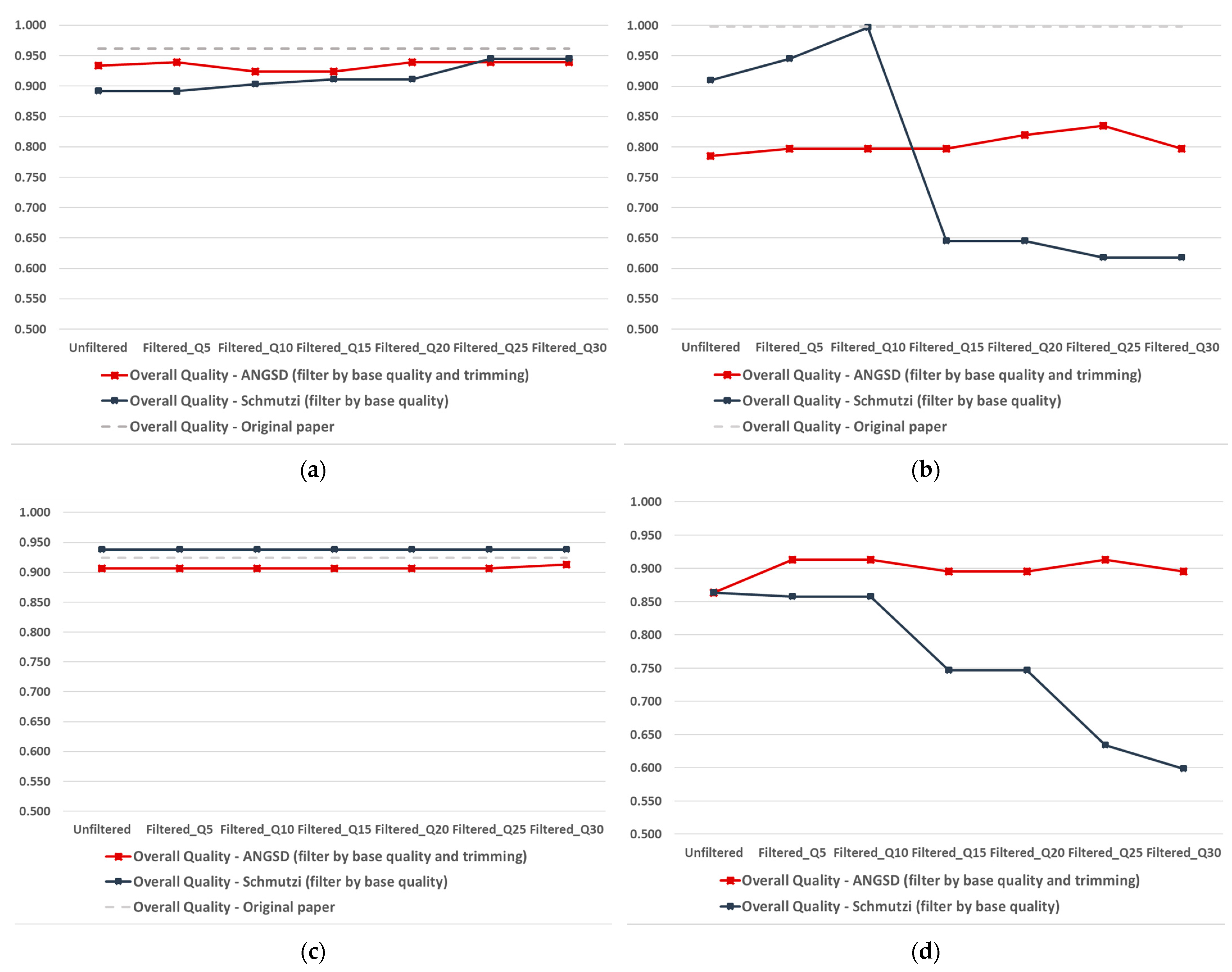
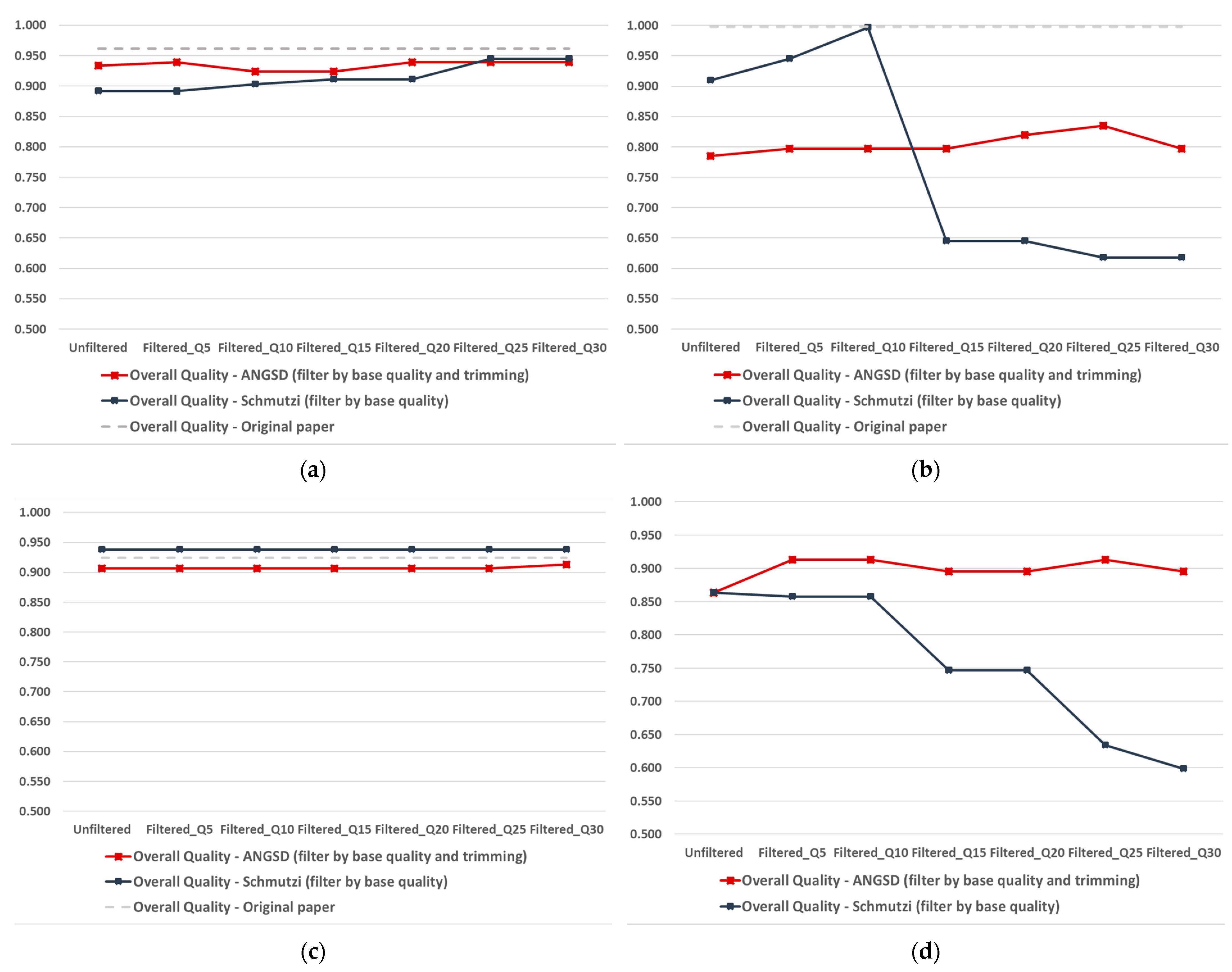
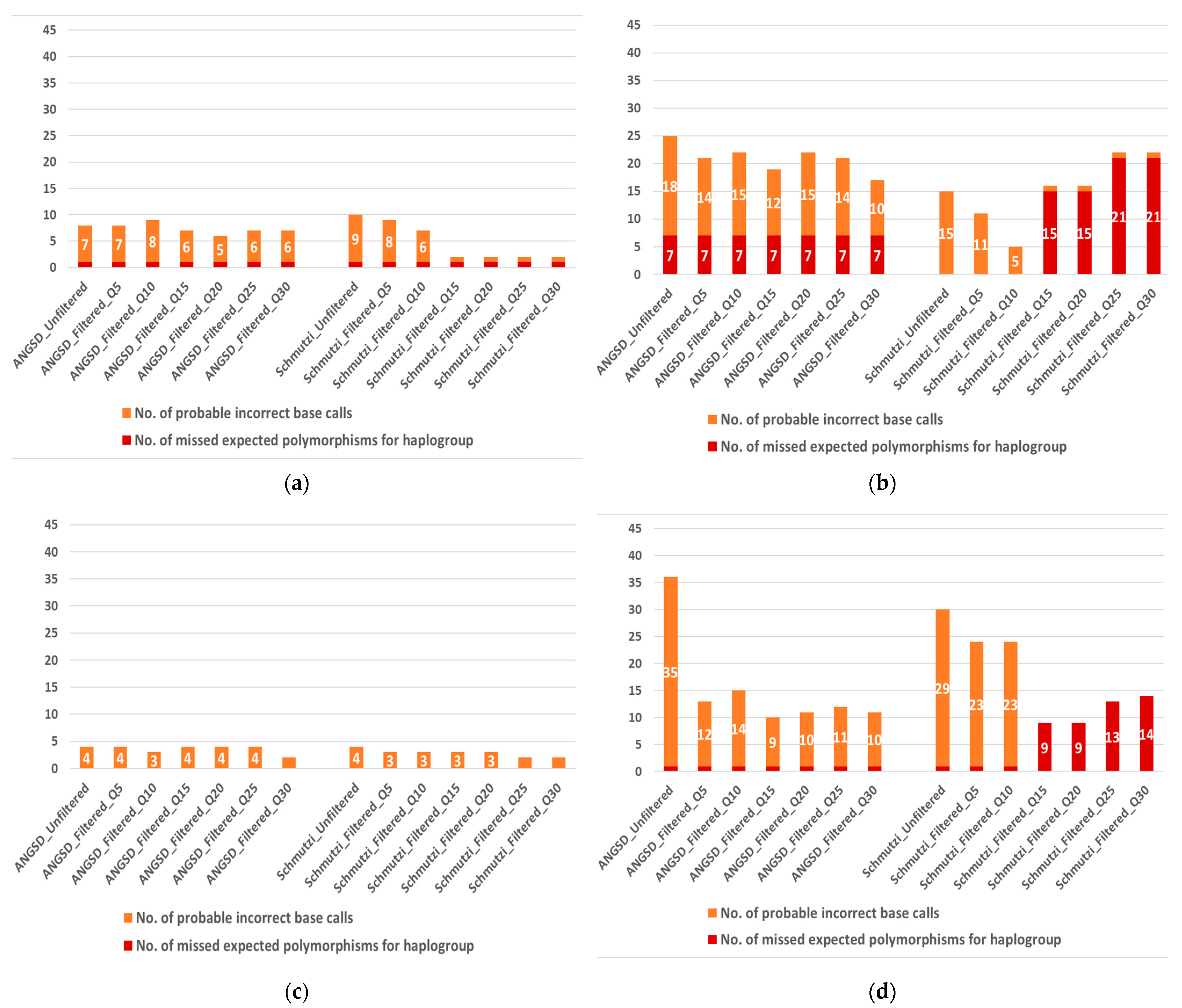
2.2. Quality-Improved Mitogenome Sequences through Filtering
2.2.1. Schmutzi Filtering
2.2.2. ANGSD Filtering
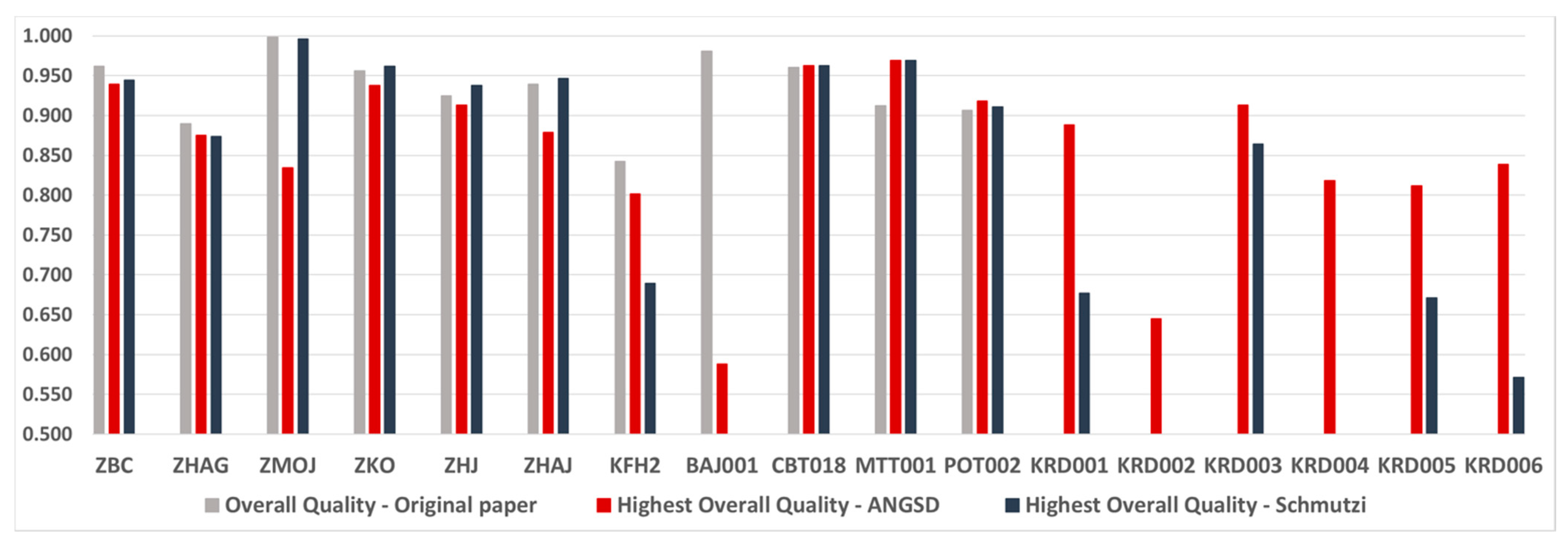
2.3. Mt Haplogroup Determination and Evaluation of Overall Sample Quality
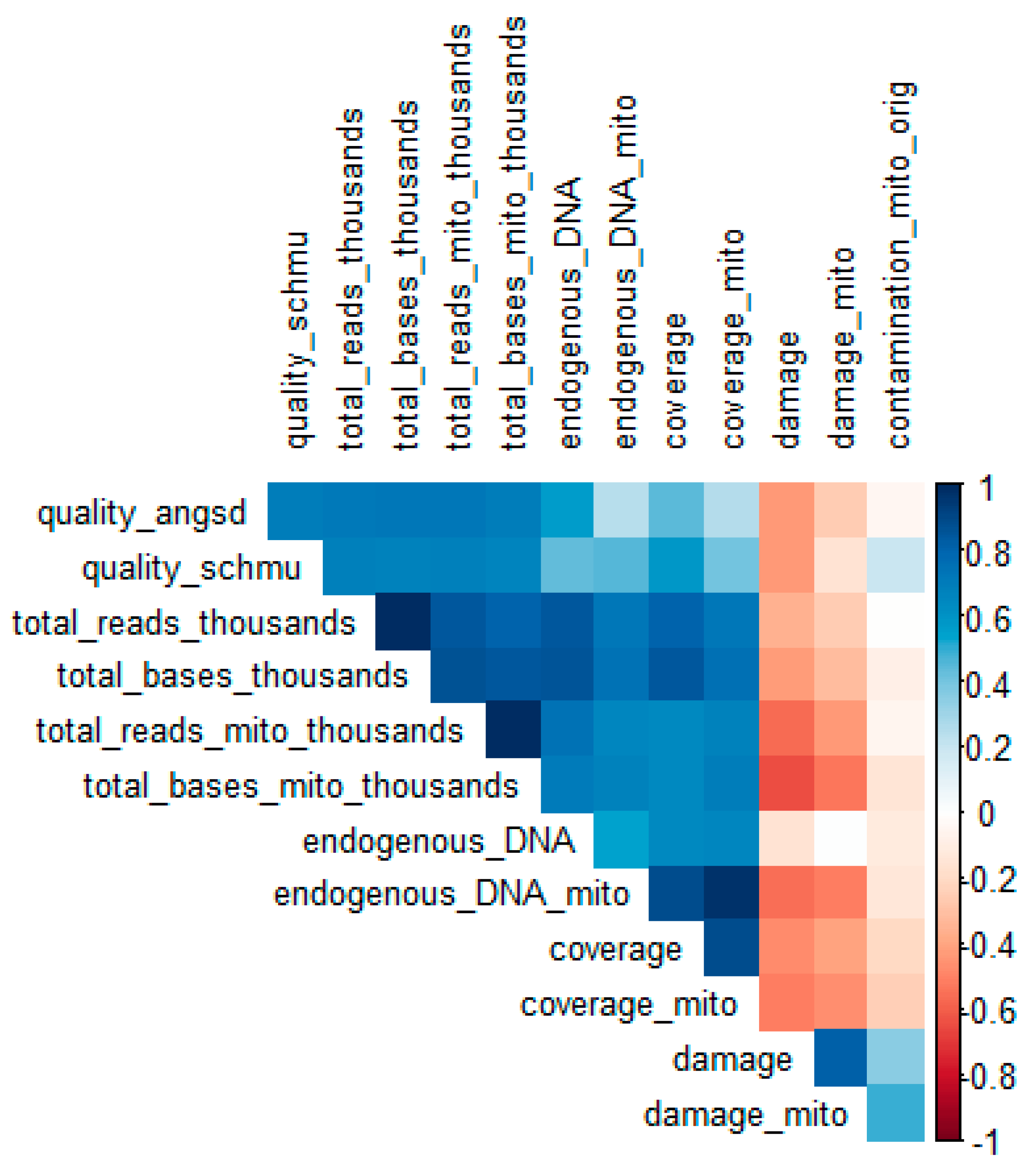
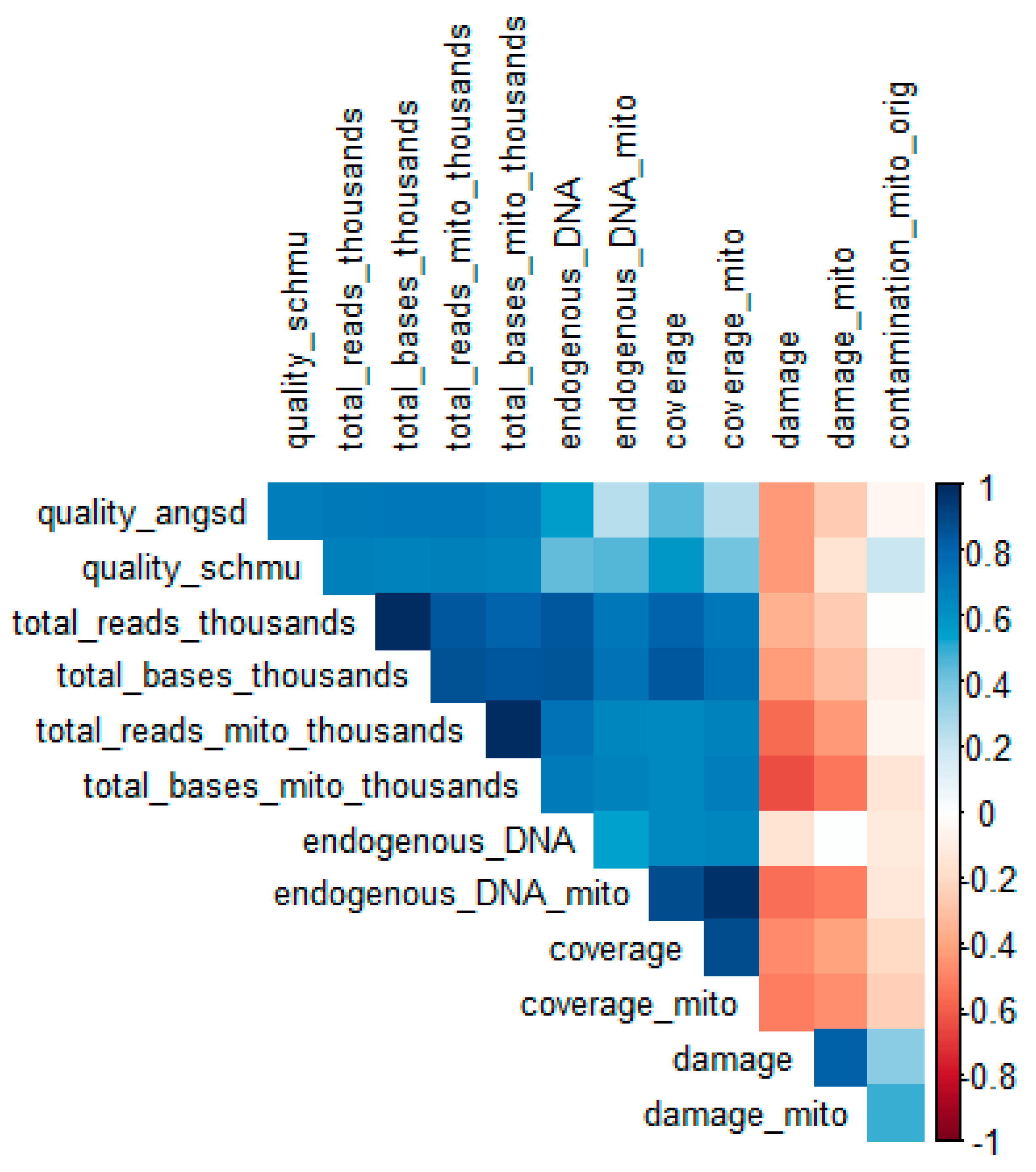
2.4. Predictors of Sample Quality Score
3. Discussion
3.1. Reconstructing Ancient Mitogenomes from Raw Genomic Data
3.2. Improving the Quality of Reconstructed Ancient Mitogenomes through Filtering
3.3. Newly Predicted mtDNA Haplogroups and Subclades
3.4. Implications for Downstream Ancient mtDNA Phylogenetic Analysis
4. Materials and Methods
4.1. Choice of Samples and Retrieval of Raw Genomic Data
4.2. Reconstructing Ancient Mitogenome Sequences from Raw Genomic Data
4.2.1. Schmutzi Pipeline
4.2.2. ANGSD Pipeline
4.3. Filtered Consensus Calling for Quality Improvement of Reconstructed Mitogenomes
4.3.1. Schmutzi Filtering
4.3.2. ANGSD Filtering
4.4. Determination of mtDNA Haplogroup Prediction Accuracy and Sample Quality
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Codes (Command Lines) for Reconstructing Mitogenome Sequences from Raw Genomic Data Using Two Alternative Tools (Schmutzi and ANGSD)
| Schmutzi Pipeline |
| ##STEP 1: Sample preparation using BWA and SAMtools## |
| #1a: Align FASTQ sample to reference fasta and create a SAM file |
| bwa aln MT_reference_file input_file.fastq > input_file.sai |
| bwa samse MT_reference_file input_file.sai input_file.fastq > output_file_MTaligned.sam |
| #1b (optional): Realignment to mtDNA reference by CircularMapper (automatic conversion to BAM) |
| java -jar realign-1.93.5.jar -e 500 -i output_file_MTaligned.sam -r MT_reference_file.fa |
| #1c: Sort BAM file (use aligned BAM file derived in previous step) |
| samtools sort output_file_MTaligned _CM_realigned.bam -o output_file_MTaligned _CM_realigned_sort.bam |
| #1d: MD-Tagging (use aligned and sorted BAM file derived in previous step) |
| samtools calmd -b output_file_MTaligned _CM_realigned_sort.bam MT_reference_file.fa > output_file_MTaligned _CM_realigned_sort_MD.bam |
| #1e: Index the MD-tagged BAM file |
| samtools index output_file_MTaligned _CM_realigned_sort_MD.bam |
| ##STEP 2: Estimate endogenous deamination using contDeam## |
| contDeam.pl --library double --out output_name output_file_MTaligned _CM_realigned_sort_MD.bam |
| ##STEP 3: Estimate contamination and call the (unfiltered) consensus sequence using schmutzi## |
| #3a: First run the iterative procedure without the prediction of the contaminant |
| schmutzi.pl --notusepredC --uselength --ref MT_reference_file.fa --out output_name_npred freqs output_name output_file_MTaligned _CM_realigned_sort_MD.bam |
| #3b: Then repeat the above with the prediction of the contaminant |
| schmutzi.pl --uselength --ref MT_reference_file.fa --out output_name_npred freqs output_name output_file_MTaligned _CM_realigned_sort_MD.bam |
| ##STEP 4: Apply filtering in the consensus sequence derived in the previous step using log2fasta## |
| #Use the log file created via running the schmutzi application to filter by base Q-score (in this case discards bases with Q-score below 10-repeat with different cut-offs) |
| log2fasta -q 10 output_name_npred_final_endo.log > output_name_filtered_schmu_q10.fasta |
| ANGSD Pipeline |
| ##STEP 1: Sample preparation using SAMtools## |
| #1a: check whether the input BAM file is sorted |
| samtools stats input_file.bam | grep “is sorted:” |
| #1b: create a BAM index file (BAI) |
| samtools index input_file.bam input_file.bai |
| #1c: Isolate the MT chromosome as a separate BAM file (this step will not be needed if the mitocapture BAM file is available) |
| samtools view -b input_file.bam MT > input_file_MT.bam |
| ##STEP 2: Call the consensus sequence using ANGSD (no filtering)## |
| #2a: Call the mitogenome consensus sequence from the MT BAM file (option 1: use the most common base) |
| angsd -out output_file -i input_file.bam -doFasta 2 -doCounts 1 |
| #2b (optional): Call the mitogenome consensus sequence from the MT BAM file (option 2: use the base with the highest effective depth-EBD) |
| angsd -out output_file -i input_file.bam -doFasta 3 |
| ##STEP 3: Call the consensus sequence using ANGSD (with filtering)## |
| #3a: Call the mitogenome consensus sequence from the MT BAM file (option 1) and filter by trimming the first/last 1 base of each read (in this case trims first/last base of each read-repeat by setting different n of bases to be trimmed) |
| angsd -out output_file -i input_file.bam -doFasta 2 -doCounts 1 -trim 1 |
| #3b: Call the mitogenome consensus sequence from the MT BAM file (option 1) and filter by trimming the first/last 1 base of each read and setting minimum base quality to 10 (in this case discards base quality below 10-repeat with different cut-offs) |
| angsd -out output_file -i input_file.bam -doFasta 2 -doCounts 1 -trim 1 -minQ 10 |
| HaploGrep |
| #mtDNA Haplogroup prediction using Haplogrep 2, based on the fasta sequence files derived in the above pipelines |
| haplogrep classify --extend-report --format fasta --in output_file.fasta --out output_file.txt |
References
- Handt, O.; Richards, M.; Trommsdorff, M.; Kilger, C.; Simanainen, J.; Georgiev, O.; Bauer, K.; Stone, A.; Hedges, R.; Schaffner, W.; et al. Molecular genetic analyses of the Tyrolean Ice Man. Science 1994, 264, 1775–1778. [Google Scholar] [CrossRef] [PubMed]
- Vernesi, C.; Caramelli, D.; Dupanloup, I.; Bertorelle, G.; Lari, M.; Cappellini, E.; Moggi-Cecchi, J.; Chiarelli, B.; Castrì, L.; Casoli, A.; et al. The Etruscans: A population-genetic study. Am. J. Hum. Genet. 2004, 74, 694–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampietro, M.L.; Caramelli, D.; Lao, O.; Calafell, F.; Comas, D.; Lari, M.; Agusti, B.; Bertranpetit, J.; Lalueza-Fox, C. The genetics of the pre-Roman Iberian Peninsula: A mtDNA study of ancient Iberians. Ann. Hum. Genet. 2005, 69, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Haak, W.; Forster, P.; Bramanti, B.; Matsumura, S.; Brandt, G.; Tanzer, M.; Villems, R.; Renfrew, C.; Gronenborn, D.; Alt, K.W.; et al. Ancient DNA from the first European farmers in 7500-year-old Neolithic sites. Science 2005, 310, 1016–1018. [Google Scholar] [CrossRef] [Green Version]
- Mathieson, I.; Lazaridis, I.; Rohland, N.; Mallick, S.; Patterson, N.; Roodenberg, S.A.; Harney, E.; Stewardson, K.; Fernandes, D.; Novak, M.; et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature 2015, 528, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Patterson, N.; Isakov, M.; Booth, T.; Büster, L.; Fischer, C.; Olalde, I.; Ringbauer, H.; Akbari, A.; Cheronet, O.; Bleasdale, M.; et al. Large-scale migration into Britain during the Middle to Late Bronze Age. Nature 2022, 601, 588–594. [Google Scholar] [CrossRef]
- Liu, Y.; Mao, X.; Krause, J.; Fu, Q. Insights into human history from the first decade of ancient human genomics. Science 2021, 373, 1479–1484. [Google Scholar] [CrossRef]
- Orlando, L.; Allaby, R.; Skoglund, P.; Der Sarkissian, C.; Stockhammer, P.W.; Ávila-Arcos, M.C.; Fu, Q.; Krause, J.; Willerslev, E.; Stone, A.C.; et al. Ancient DNA analysis. Nat. Rev. Methods Primers 2021, 1, 14. [Google Scholar] [CrossRef]
- Kivisild, T. Maternal ancestry and population history from whole mitochondrial genomes. Investig. Genet. 2015, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q.; Mittnik, A.; Johnson, P.L.; Bos, K.; Lari, M.; Bollongino, R.; Sun, C.; Giemsch, L.; Schmitz, R.; Burger, J.; et al. A revised timescale for human evolution based on ancient mitochondrial genomes. Curr. Biol. 2013, 23, 553–559. [Google Scholar] [CrossRef] [Green Version]
- Bramanti, B.; Thomas, M.G.; Haak, W.; Unterländer, M.; Jores, P.; Tambets, K.; Antanaitis-Jacobs, I.; Haidle, M.N.; Jankauskas, R.; Kind, C.; et al. Genetic discontinuity between local hunter-gatherers and central Europe’s first farmers. Science 2009, 326, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Haak, W.; Balanovsky, O.; Sanchez, J.J.; Koshel, S.; Zaporozhchenko, V.; Adler, C.J.; Der Sarkissian, C.S.; Brandt, G.; Schwarz, C.; Nicklisch, N.; et al. Ancient DNA from European early neolithic farmers reveals their near eastern affinities. PLoS Biol. 2010, 8, e1000536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, E.; Pérez-Pérez, A.; Gamba, C.; Prats, E.; Cuesta, P.; Anfruns, J.; Molist, M.; Arroyo-Pardo, E.; Turbón, D. Ancient DNA analysis of 8000 BC near eastern farmers supports an early neolithic pioneer maritime colonization of Mainland Europe through Cyprus and the Aegean Islands. PLoS Genet. 2014, 10, e1004401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posth, C.; Renaud, G.; Mittnik, A.; Drucker, D.G.; Rougier, H.; Cupillard, C.; Valentin, F.; Thevenet, C.; Furtwängler, A.; Wißing, C.; et al. Pleistocene mitochondrial genomes suggest a single major dispersal of non-Africans and a Late Glacial population turnover in Europe. Curr. Biol. 2016, 26, 827–833. [Google Scholar] [CrossRef] [Green Version]
- Pala, M.; Olivieri, A.; Achilli, A.; Accetturo, M.; Metspalu, E.; Reidla, M.; Tamm, E.; Karmin, M.; Reisberg, T.; Kashani, B.H.; et al. Mitochondrial DNA signals of late glacial recolonization of Europe from near eastern refugia. Am. J. Hum. Genet. 2012, 90, 915–924. [Google Scholar] [CrossRef] [Green Version]
- Vai, S.; Amorim, C.E.G.; Lari, M.; Caramelli, D. Kinship determination in archeological contexts through DNA analysis. Front. Ecol. Evol. 2020, 8, 83. [Google Scholar] [CrossRef] [Green Version]
- Fowler, C.; Olalde, I.; Cummings, V.; Armit, I.; Büster, L.; Cuthbert, S.; Rohland, N.; Cheronet, O.; Pinhasi, R.; Reich, D. A high-resolution picture of kinship practices in an Early Neolithic tomb. Nature 2022, 601, 584–587. [Google Scholar] [CrossRef]
- Dabney, J.; Meyer, M.; Pääbo, S. Ancient DNA damage. Cold Spring Harb. Perspect. Biol. 2013, 5, a012567. [Google Scholar] [CrossRef]
- Briggs, A.W.; Stenzel, U.; Johnson, P.L.; Green, R.E.; Kelso, J.; Prüfer, K.; Meyer, M.; Krause, J.; Ronan, M.T.; Lachmann, M.; et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl. Acad. Sci. USA 2007, 104, 14616–14621. [Google Scholar] [CrossRef] [Green Version]
- Peyrégne, S.; Prüfer, K. Present-Day DNA Contamination in Ancient DNA Datasets. Bioessays 2020, 42, e2000081. [Google Scholar] [CrossRef]
- Bendall, K.E.; Sykes, B.C. Length heteroplasmy in the first hypervariable segment of the human mtDNA control region. Am. J. Hum. Genet. 1995, 57, 248–256. [Google Scholar] [PubMed]
- Stewart, J.E.; Fisher, C.L.; Aagaard, P.J.; Wilson, M.R.; Isenberg, A.R.; Polanskey, D.; Pokorak, E.; DiZinno, J.A.; Budowle, B. Length variation in HV2 of the human mitochondrial DNA control region. J. Forensic Sci. 2001, 46, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Jónsson, H.; Ginolhac, A.; Schubert, M.; Johnson, P.L.; Orlando, L. Map Damage 2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 2013, 29, 1682–1684. [Google Scholar] [CrossRef] [PubMed]
- Renaud, G.; Slon, V.; Duggan, A.T.; Kelso, J. Schmutzi: Estimation of contamination and endogenous mitochondrial consensus calling for ancient DNA. Genome Biol. 2015, 16, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neukamm, J.; Peltzer, A.; Nieselt, K. Damage Profiler: Fast damage pattern calculation for ancient DNA. Bioinformatics 2021, 37, 3652–3653. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuka, N.; Harney, É.; Mallick, S.; Mah, M.; Patterson, N.; Reich, D. ContamLD: Estimation of ancient nuclear DNA contamination using breakdown of linkage disequilibrium. Genome Biol. 2020, 21, 199. [Google Scholar] [CrossRef]
- Peyrégne, S.; Peter, B.M. AuthentiCT: A model of ancient DNA damage to estimate the proportion of present-day DNA contamination. Genome Biol. 2020, 21, 246. [Google Scholar] [CrossRef]
- Korneliussen, T.S.; Albrechtsen, A.; Nielsen, R. ANGSD: Analysis of next generation sequencing data. BMC Bioinform. 2014, 15, 356. [Google Scholar] [CrossRef] [Green Version]
- Feldman, M.; Fernández-Domínguez, E.; Reynolds, L.; Baird, D.; Pearson, J.; Hershkovitz, I.; May, H.; Goring-Morris, N.; Benz, M.; Gresky, J.; et al. Late Pleistocene human genome suggests a local origin for the first farmers of central Anatolia. Nat. Commun. 2019, 10, 1218. [Google Scholar] [CrossRef] [Green Version]
- Skourtanioti, E.; Erdal, Y.S.; Frangipane, M.; Restelli, F.B.; Yener, K.A.; Pinnock, F.; Matthiae, P.; Özbal, R.; Schoop, U.; Guliyev, F.; et al. Genomic history of neolithic to bronze age Anatolia, northern Levant, and southern Caucasus. Cell 2020, 181, 1158–1175.e28. [Google Scholar] [CrossRef]
- Weissensteiner, H.; Pacher, D.; Kloss-Brandstätter, A.; Forer, L.; Specht, G.; Bandelt, H.; Kronenberg, F.; Salas, A.; Schönherr, S. HaploGrep 2: Mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic. Acids. Res. 2016, 44, W58–W63. [Google Scholar] [CrossRef] [PubMed]
- Van Oven, M.; Kayser, M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum. Mutat. 2009, 30, E386–E394. [Google Scholar] [CrossRef] [PubMed]
- Feldman, M.; Master, D.M.; Bianco, R.A.; Burri, M.; Stockhammer, P.W.; Mittnik, A.; Aja, A.J.; Jeong, C.; Krause, J. Ancient DNA sheds light on the genetic origins of early Iron Age Philistines. Sci. Adv. 2019, 5, eaax0061. [Google Scholar] [CrossRef] [Green Version]
- Furtwängler, A.; Rohrlach, A.B.; Lamnidis, T.C.; Papac, L.; Neumann, G.U.; Siebke, I.; Reiter, E.; Steuri, N.; Hald, J.; Denaire, A. Ancient genomes reveal social and genetic structure of Late Neolithic Switzerland. Nat. Commun. 2020, 11, 1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Ning, C.; Scott, A.; Fu, Q.; Bjørn, R.; Li, W.; Wei, D.; Wang, W.; Fan, L.; Abuduresule, I.; et al. The genomic origins of the Bronze Age Tarim Basin mummies. Nature 2021, 599, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Chyleński, M.; Ehler, E.; Somel, M.; Yaka, R.; Krzewińska, M.; Dabert, M.; Juras, A.; Marciniak, A. Ancient mitochondrial genomes reveal the absence of maternal kinship in the burials of Çatalhöyük people and their genetic affinities. Genes 2019, 10, 207. [Google Scholar] [CrossRef] [Green Version]
- Brandt, G.; Haak, W.; Adler, C.J.; Roth, C.; Szécsényi-Nagy, A.; Karimnia, S.; Möller-Rieker, S.; Meller, H.; Ganslmeier, R.; Friederich, S.; et al. Ancient DNA reveals key stages in the formation of central European mitochondrial genetic diversity. Science 2013, 342, 257–261. [Google Scholar] [CrossRef] [Green Version]
- Szécsényi-Nagy, A.; Brandt, G.; Haak, W.; Keerl, V.; Jakucs, J.; Möller-Rieker, S.; Köhler, K.; Mende, B.G.; Oross, K.; Marton, T.; et al. Tracing the genetic origin of Europe’s first farmers reveals insights into their social organization. Proc. R. Soc. B Biol. Sci. 2015, 282, 20150339. [Google Scholar] [CrossRef] [Green Version]
- Olalde, I.; Brace, S.; Allentoft, M.E.; Armit, I.; Kristiansen, K.; Booth, T.; Rohland, N.; Mallick, S.; Szécsényi-Nagy, A.; Mittnik, A.; et al. The Beaker phenomenon and the genomic transformation of northwest Europe. Nature 2018, 555, 190–196. [Google Scholar] [CrossRef] [Green Version]
- Haak, W.; Lazaridis, I.; Patterson, N.; Rohland, N.; Mallick, S.; Llamas, B.; Brandt, G.; Nordenfelt, S.; Harney, E.; Stewardson, K.; et al. Massive migration from the steppe was a source for Indo-European languages in Europe. Nature 2015, 522, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q.; Posth, C.; Hajdinjak, M.; Petr, M.; Mallick, S.; Fernandes, D.; Furtwängler, A.; Haak, W.; Meyer, M.; Mittnik, A.; et al. The genetic history of ice age Europe. Nature 2016, 534, 200–205. [Google Scholar] [CrossRef] [Green Version]
- Lazaridis, I.; Nadel, D.; Rollefson, G.; Merrett, D.C.; Rohland, N.; Mallick, S.; Fernandes, D.; Novak, M.; Gamarra, B.; Sirak, K.; et al. Genomic insights into the origin of farming in the ancient Near East. Nature 2016, 536, 419–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Yeh, H.; Popov, A.N.; Zhang, H.; Matsumura, H.; Sirak, K.; Cheronet, O.; Kovalev, A.; Rohland, N.; Kim, A.M.; et al. Genomic insights into the formation of human populations in East Asia. Nature 2021, 591, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Hofmanová, Z.; Kreutzer, S.; Hellenthal, G.; Sell, C.; Diekmann, Y.; Díez-del-Molino, D.; Van Dorp, L.; López, S.; Kousathanas, A.; Link, V.; et al. Early farmers from across Europe directly descended from Neolithic Aegeans. Proc. Natl. Acad. Sci. USA 2016, 113, 6886–6891. [Google Scholar] [CrossRef] [Green Version]
- Schuenemann, V.J.; Peltzer, A.; Welte, B.; Van Pelt, W.P.; Molak, M.; Wang, C.; Furtwängler, A.; Urban, C.; Reiter, E.; Nieselt, K.; et al. Ancient Egyptian mummy genomes suggest an increase of Sub-Saharan African ancestry in post-Roman periods. Nat. Commun. 2017, 8, 15694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juras, A.; Chyleński, M.; Ehler, E.; Malmström, H.; Żurkiewicz, D.; Włodarczak, P.; Wilk, S.; Peška, J.; Fojtík, P.; Králík, M.; et al. Mitochondrial genomes reveal an east to west cline of steppe ancestry in Corded Ware populations. Sci. Rep. 2018, 8, 11603. [Google Scholar] [CrossRef]
- Ning, C.; Zheng, H.; Zhang, F.; Wu, S.; Li, C.; Zhao, Y.; Xu, Y.; Wei, D.; Wu, Y.; Gao, S.; et al. Ancient Mitochondrial Genomes Reveal Extensive Genetic Influence of the Steppe Pastoralists in Western Xinjiang. Front. Genet. 2021, 12, 740167. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Peltzer, A.; Jäger, G.; Herbig, A.; Seitz, A.; Kniep, C.; Krause, J.; Nieselt, K. EAGER: Efficient ancient genome reconstruction. Genome Biol. 2016, 17, 60. [Google Scholar] [CrossRef] [Green Version]
- Ewing, B.; Green, P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998, 8, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-calling of automated sequencer traces usingPhred. I. Accuracy assessment. Genome Res. 1998, 8, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, M.; Ginolhac, A.; Lindgreen, S.; Thompson, J.F.; Al-Rasheid, K.A.; Willerslev, E.; Krogh, A.; Orlando, L. Improving ancient DNA read mapping against modern reference genomes. BMC Genom. 2012, 13, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | Sample | Site | Location and Era | Sex | Estimated Date | Total Read Count | Total Base Count | Endogenous DNA (%) | Mean Coverage (Fold) | Damage (5′ Terminal) | Mt Contamination | Μt Haplogroup (Original Study) | Mt Haplogroup (Current Study) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [29] | ZBC | Pinarbași | Anatolia Epipaleolithic | M | 13642–13073 cal BCE | 10,036,367 | 638,989,878 | 2.42 | 2.90 | 0.09 | 0.01 (0.00–0.02) | K2b | K2b2 |
| [29] | ZHAG | Boncuklu | Anatolia Aceramic Neolithic | F | 8300–7800 BCE | 8,127,772 | 488,875,951 | 2.14 | 1.48 | 0.11 | 0.01 (0.00–0.02) | N1a1a1 | N1a1a1 |
| [29] | ZMOJ | Boncuklu | Anatolia Aceramic Neolithic | M | 8300–7800 BCE | 3,513,636 | 176,572,167 | 1.03 | 0.80 | 0.22 | 0.03 (0.02–0.04) | K1a | K1a19 |
| [29] | ZKO | Boncuklu | Anatolia Aceramic Neolithic | M | 8300–7800 BCE | 4,594,498 | 257,455,118 | 1.51 | 0.90 | 0.16 | 0.01 (0.00–0.02) | U3 | U3b |
| [29] | ZHJ | Boncuklu | Anatolia Aceramic Neolithic | F | 8300–7800 BCE | 5,063,868 | 290,873,189 | 2.36 | 0.76 | 0.09 | 0.01 (0.00–0.02) | U3 | U3b |
| [29] | ZHAJ | Boncuklu | Anatolia Aceramic Neolithic | F | 8269–8210 cal BCE | 3,848,792 | 216,286,212 | 1.48 | 0.69 | 0.16 | 0.03 (0.02–0.04) | U3 | U3b |
| [29] | KFH2 | Kfar HaHoresh | Levant Early Neolithic (PPNB) | F | 7712–7589 cal BCE | 1,315,907 | 58,245,650 | 0.20 | 0.16 | 0.26 | 0.06 (0.04–0.08) | N1a1b | I6 |
| [29] | BAJ001 | Ba’ja | Levant Early Neolithic (PPNB) | F | 7027–6685 cal BCE | 2,038,720 | 104,654,562 | 5.51 | 0.75 | 0.28 | 0.01 (0.00–0.02) | N1b1a | N1b1a |
| [30] | CBT018 | Boğazköy-Büyükkaya | C./N. Anatolia 1 Early Chalcolithic | F | 7576–7463 cal BCE | 3,814,623 | 222,533,552 | 5.19 | 0.51 | 0.14 | 0.01 (0.00–0.03) | X2 | X2 |
| [30] | MTT001 | Mentesh Tepe | Caucasus lowlands Late Neolithic | F | 7679–7594 cal BCE | 11,887,457 | 582,094,002 | 14.08 | 0.55 | 0.21 | 0.03 (0.01–0.05) | U7 | U7b |
| [30] | POT002 | Polutepe | Caucasus lowlands Late Neolithic | F | 7458–7323 cal BCE | 8,339,513 | 392,934,499 | 6.63 | 0.41 | 0.31 | 0.02 (0.00–0.04) | H13a2b | H13a2b |
| [30] | KRD001 | Tell Kurdu | S. Anatolia/N. Levant 2 Early Chalcolithic | M | 7670–7590 cal BCE | 678,068 | 37,097,407 | 0.19 | 0.20 | 0.22 | 0.01 (0.00–0.03) | n/a 3 | P5 |
| [30] | KRD002 | Tell Kurdu | S. Anatolia/N. Levant 2 Middle Chalcolithic | M | 6955–6799 cal BCE | 520,046 | 25,733,234 | 0.26 | 0.15 | 0.27 | 0.01 (0.00–0.03) | n/a 3 | N1a1a |
| [30] | KRD003 | Tell Kurdu | S. Anatolia/N. Levant 2 Early Chalcolithic | M | 7656–7572 cal BCE | 1,646,820 | 83,050,541 | 0.74 | 0.35 | 0.26 | 0.03 (0.00–0.11) | n/a 3 | U3b |
| [30] | KRD004 | Tell Kurdu | S. Anatolia/N. Levant 2 Levant Early Chalcolithic | F | 7664–7582 cal BCE | 236,781 | 12,662,467 | 0.22 | 0.06 | 0.21 | 0.02 (0.00–0.06) | n/a 3 | F1a1a1*2 |
| [30] | KRD005 | Tell Kurdu | S. Anatolia/N. Levant 2 Early Chalcolithic | M | 7706–7614 cal BCE | 132,930 | 7,367,352 | 0.13 | 0.03 | 0.18 | 0.01 (0.00–0.03) | n/a 3 | U8b |
| [30] | KRD006 | Tell Kurdu | S. Anatolia/N. Levant 2 Early Chalcolithic | F | 7750–7350 BCE | 1,060,964 | 60,413,143 | 1.15 | 0.21 | 0.19 | 0.03 (0.01–0.05) | n/a 3 | H20a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heraclides, A.; Fernández-Domínguez, E. Mitochondrial DNA Consensus Calling and Quality Filtering for Constructing Ancient Human Mitogenomes: Comparison of Two Widely Applied Methods. Int. J. Mol. Sci. 2022, 23, 4651. https://doi.org/10.3390/ijms23094651
Heraclides A, Fernández-Domínguez E. Mitochondrial DNA Consensus Calling and Quality Filtering for Constructing Ancient Human Mitogenomes: Comparison of Two Widely Applied Methods. International Journal of Molecular Sciences. 2022; 23(9):4651. https://doi.org/10.3390/ijms23094651
Chicago/Turabian StyleHeraclides, Alexandros, and Eva Fernández-Domínguez. 2022. "Mitochondrial DNA Consensus Calling and Quality Filtering for Constructing Ancient Human Mitogenomes: Comparison of Two Widely Applied Methods" International Journal of Molecular Sciences 23, no. 9: 4651. https://doi.org/10.3390/ijms23094651
APA StyleHeraclides, A., & Fernández-Domínguez, E. (2022). Mitochondrial DNA Consensus Calling and Quality Filtering for Constructing Ancient Human Mitogenomes: Comparison of Two Widely Applied Methods. International Journal of Molecular Sciences, 23(9), 4651. https://doi.org/10.3390/ijms23094651