
A Non-Hazardous Deparaffinization Protocol Enables Quantitative Proteomics of Core Needle Biopsy-Sized Formalin-Fixed and Paraffin-Embedded (FFPE) Tissue Specimens

 ,
,  ,
,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
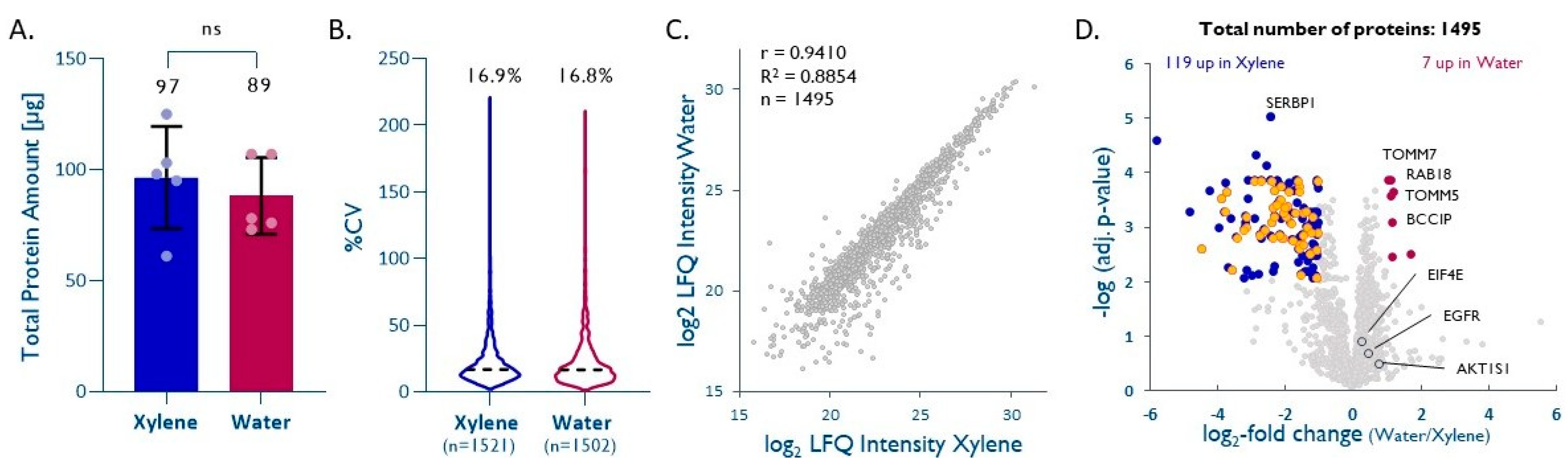
2.1. Water-Based Deparaffinization Competes with the Gold-Standard Xylene and Takes Only a Fraction of the Time
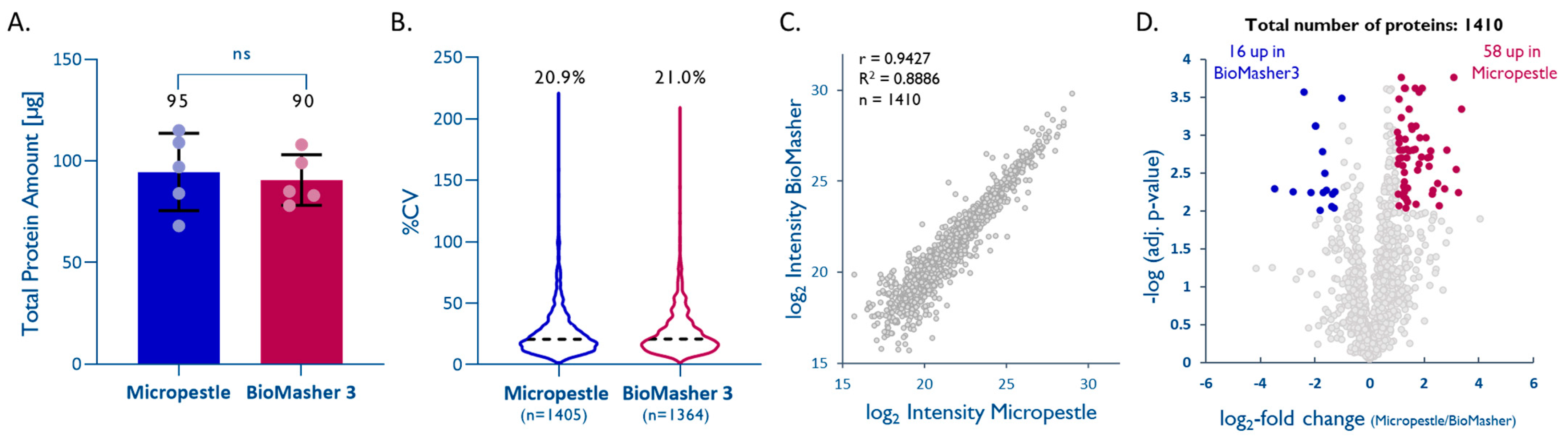
2.2. Efficient Tissue Homogenization Using Micropestles
2.3. Improved Protein Extraction with Sodium Deoxycholate (SDC)
2.4. PAC and STRAP Are Good Alternatives to FASP
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
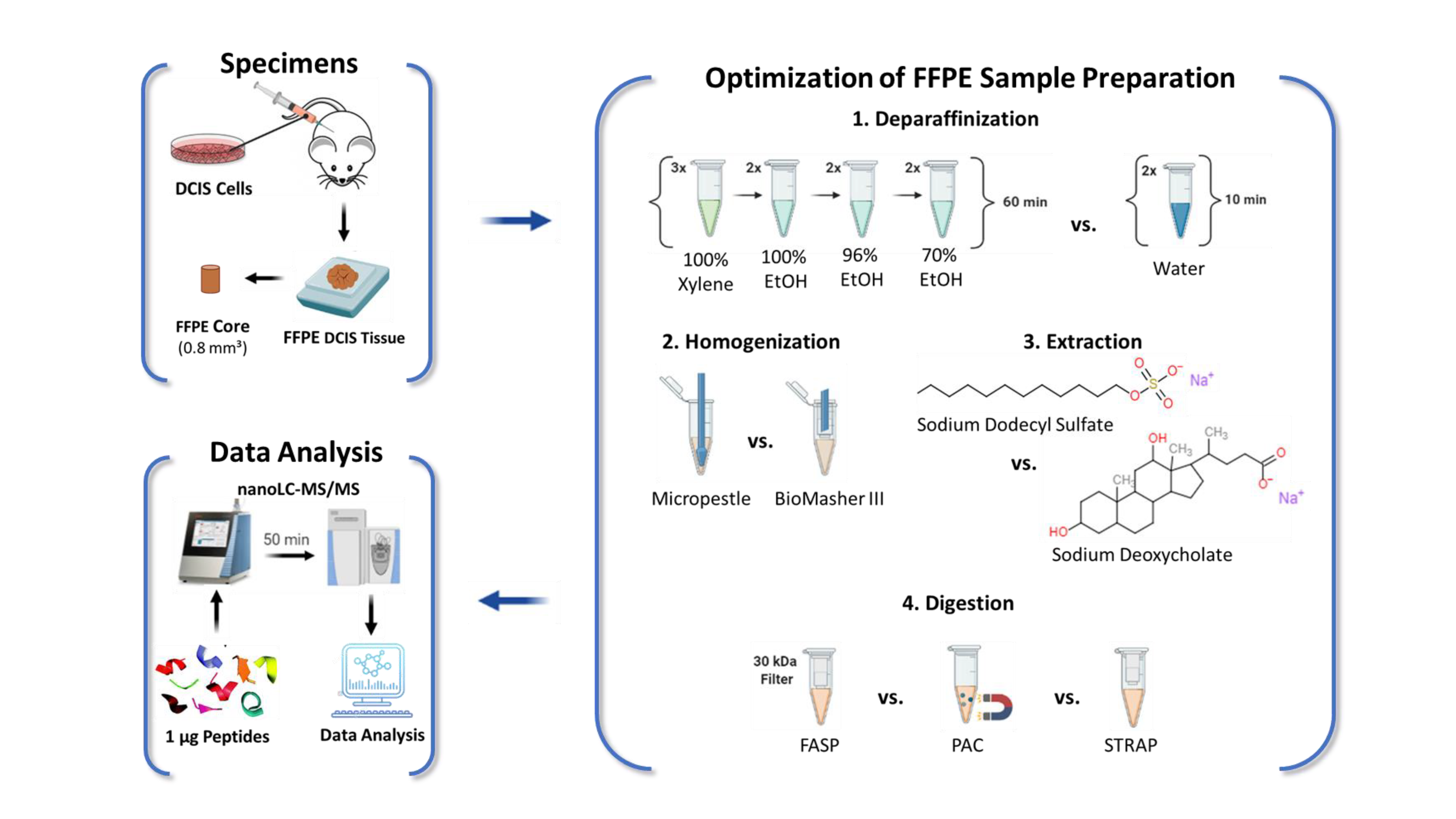
4.2. Source of Specimens
4.3. Sample Preparation of Core Needle Biopsy-Sized Specimens
4.3.1. Optimization of Deparaffinization
- (a)
- depX [54]: The samples were washed with 1 mL of 100% xylene and incubated for 10 min at room temperature (RT), followed by centrifugation at 14,000× g for 2 min and disposal of the supernatant, followed by another 2 repetitions. Then, the samples were washed twice each with 1 mL of 100%, 96%, and 70% ethanol, followed by incubation for 1 min at RT and centrifugation as above.
- (b)
- depW (modified from [14]): The samples were washed 2× with 500 µL of hot deionized water and incubated for 1 min at RT under vigorous vortex mixing. Each washing step was followed by centrifugation at 20,000× g for 5 min at 4 °C. The supernatant, containing paraffin either floating on the liquid surface or stuck to the wall of the tube (Figure 7), was discarded and the deparaffinized and rehydrated core was transferred to a clean LoBind Eppendorf tube.
4.3.2. Optimization of Tissue Homogenization
4.3.3. Optimization of Protein Extraction
4.3.4. Optimization of Tryptic Digestion
- (a)
- FASP was performed as described above.
- (b)
- PAC was performed using amine microparticles (MagReSyn) based on Batth et al. [39]. For 20 µg of protein lysate, 12 µL of microparticle stock solution (20 µg/mL) were equilibrated with 100 µL of 70% ACN, briefly vortexed and placed on a magnetic rack to remove the supernatant. This step was repeated another two times. Next, the protein extracts were added to the beads and the sample was adjusted to a final concentration of 70% ACN, thoroughly vortexed and incubated for 10 min at RT without shaking. The following washing steps were performed on a magnetic rack without disturbing the protein/bead aggregate. The supernatants were discarded, and the beads were washed on the magnetic rack with 1 mL of 95% ACN for 10 s, followed by a wash with 1 mL of 70% ACN without disturbing the protein/bead aggregate. The tubes were removed from the magnetic rack, 100 µL of digestion buffer (1:20 (w/w) trypsin:protein in 0.2 M GuHCl, 50 mM AmBic, 2 mM CaCl2) were added and the samples were incubated at 37 °C for 3 h. After acidification with trifluoroacetic acid (TFA) to a final concentration of 2%, the tubes were placed on the magnetic rack for 1 min, followed by removal of the supernatant. To remove residual beads, the samples were centrifuged at 20,000× g for 10 min. The supernatants were dried under vacuum and reconstituted in 0.1% FA for nano-LC-MS/MS.
- (c)
- STRAP digestion was performed according to the manufacturer’s instructions [51]. Lysate corresponding to 20 µg of total protein was acidified to a final concentration of 1.2% phosphoric acid. SDS was added to a final concentration of 2% followed by a 7-fold dilution with STRAP binding buffer (90% methanol, 100 mM Tris-HCl, pH 7.1). The sample was loaded onto the STRAP and centrifuged at 4000× g for 1 min, followed by three washes with 150 µL binding buffer, with the spin-column being rotated by 180° between centrifugation steps. Then, 200 µL of STRAP digestion buffer, comprised of 1:10 (w/w) trypsin:protein in 0.2 M GuHCl, 50 mM AmBic, 2 mM CaCl2 were added to the STRAP, which was briefly spun on a benchtop centrifuge to assure saturation of the column material with the digestion buffer. The flow-through was loaded again on top of the column. The sample was incubated at 47 °C for 3 h. Peptides were eluted by sequential elution (1000xg, 1 min) using 40 µL of 50 mM AmBic, 40 µL of 0.1% FA, and 35 µL of 50% ACN, 0.1% FA. The collected peptide sample was dried under vacuum and reconstituted in 0.1% FA for nano-LC-MS/MS.
4.4. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gaffney, E.F.; Riegman, P.H.; Grizzle, W.E.; Watson, P.H. Factors that drive the increasing use of FFPE tissue in basic and translational cancer research. Biotech. Histochem. 2018, 93, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Foll, M.C.; Fahrner, M.; Oria, V.O.; Kuhs, M.; Biniossek, M.L.; Werner, M.; Bronsert, P.; Schilling, O. Reproducible proteomics sample preparation for single FFPE tissue slices using acid-labile surfactant and direct trypsinization. Clin. Proteom. 2018, 15, 11. [Google Scholar] [CrossRef] [PubMed]
- Maes, E.; Broeckx, V.; Mertens, I.; Sagaert, X.; Prenen, H.; Landuyt, B.; Schoofs, L. Analysis of the formalin-fixed paraffin-embedded tissue proteome: Pitfalls, challenges, and future prospectives. Amino Acids 2013, 45, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Maes, E.; Valkenborg, D.; Mertens, I.; Broeckx, V.; Baggerman, G.; Sagaert, X.; Landuyt, B.; Prenen, H.; Schoofs, L. Proteomic analysis of formalin-fixed paraffin-embedded colorectal cancer tissue using tandem mass tag protein labeling. Mol. Biosyst. 2013, 9, 2686–2695. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ralton, L.D.; Murray, G.I. The use of formalin fixed wax embedded tissue for proteomic analysis. J. Clin. Pathol. 2011, 64, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Seyhan, A.A.; Carini, C. Are innovation and new technologies in precision medicine paving a new era in patients centric care? J. Transl. Med. 2019, 17, 114. [Google Scholar] [CrossRef]
- Sobsey, C.A.; Ibrahim, S.; Richard, V.R.; Gaspar, V.; Mitsa, G.; Lacasse, V.; Zahedi, R.P.; Batist, G.; Borchers, C.H. Targeted and Untargeted Proteomics Approaches in Biomarker Development. Proteomics 2019, 20, e1900029. [Google Scholar] [CrossRef]
- Alvarez-Chaver, P.; De Chiara, L.; Martinez-Zorzano, V.S. Proteomic Profiling for Colorectal Cancer Biomarker Discovery. Methods Mol. Biol. 2018, 1765, 241–269. [Google Scholar] [CrossRef]
- Choi, C.H.; Chung, J.Y.; Kang, J.H.; Paik, E.S.; Lee, Y.Y.; Park, W.; Byeon, S.J.; Chung, E.J.; Kim, B.G.; Hewitt, S.M.; et al. Chemoradiotherapy response prediction model by proteomic expressional profiling in patients with locally advanced cervical cancer. Gynecol. Oncol. 2020, 157, 437–443. [Google Scholar] [CrossRef]
- Toomey, S.; Carr, A.; Mezynski, M.J.; Elamin, Y.; Rafee, S.; Cremona, M.; Morgan, C.; Madden, S.; Abdul-Jalil, K.I.; Gately, K.; et al. Identification and clinical impact of potentially actionable somatic oncogenic mutations in solid tumor samples. J. Transl. Med. 2020, 18, 99. [Google Scholar] [CrossRef]
- Faoláin, E.Ó.; Hunter, M.B.; Byrne, J.M.; Kelehan, P.; Lambkin, H.A.; Byrne, H.J.; Lyng, F.M. Raman spectroscopic evaluation of efficacy of current paraffin wax section dewaxing agents. J. Histochem. Cytochem. 2005, 53, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Kandyala, R.; Raghavendra, S.P.C.; Rajasekharan, S.T. Xylene: An overview of its health hazards and preventive measures. J. Oral Maxillofac. Pathol. 2010, 14, 1–5. [Google Scholar] [CrossRef] [PubMed]
- European Chemical Agency. Xylene (Last Updated: 16 April 2020). Available online: https://echa.europa.eu/brief-profile/-/briefprofile/100.014.124 (accessed on 11 August 2021).
- Mansour, A.G.; Khalil, P.A.; Bejjani, N.; Chatila, R.; Dagher-Hamalian, C.; Faour, W.H. An optimized xylene-free protein extraction method adapted to formalin-fixed paraffin embedded tissue sections for western blot analysis. Histol. Histopathol. 2017, 32, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Magdeldin, S.; Yamamoto, T. Toward deciphering proteomes of formalin-fixed paraffin-embedded (FFPE) tissues. Proteomics 2012, 12, 1045–1058. [Google Scholar] [CrossRef]
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Guo, Q.; Li, V.Z.; Nichol, J.N.; Huang, F.; Yang, W.; Preston, S.E.J.; Talat, Z.; Lefrere, H.; Yu, H.; Zhang, G.; et al. MNK1/NODAL Signaling Promotes Invasive Progression of Breast Ductal Carcinoma In Situ. Cancer Res. 2019, 79, 1646–1657. [Google Scholar] [CrossRef] [PubMed]
- Kalantari, N.; Bayani, M.; Ghaffari, T. Deparaffinization of formalin-fixed paraffin-embedded tissue blocks using hot water instead of xylene. Anal. Biochem. 2016, 507, 71–73. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ (accessed on 11 August 2021).
- Maertens, A.; Tran, V.P.; Maertens, M.; Kleensang, A.; Luechtefeld, T.H.; Hartung, T.; Paller, C.J. Functionally Enigmatic Genes in Cancer: Using TCGA Data to Map the Limitations of Annotations. Sci. Rep. 2020, 10, 4106. [Google Scholar] [CrossRef]
- Sekine, S.; Wang, C.; Sideris, D.P.; Bunker, E.; Zhang, Z.; Youle, R.J. Reciprocal roles of Tom7 and OMA1 during mitochondrial import and activation of PINK1. Mol. Cell 2019, 73, 1028–1043.e5. [Google Scholar] [CrossRef]
- Alnouti, Y. Bile Acid Sulfation: A Pathway of Bile Acid Elimination and Detoxification. Toxicol. Sci. 2009, 108, 225–246. [Google Scholar] [CrossRef]
- Zhong, K.; Chen, K.; Han, L.; Li, B. MicroRNA-30b/c inhibits non-small cell lung cancer cell proliferation by targeting Rab18. BMC Cancer 2014, 14, 703. [Google Scholar] [CrossRef]
- Meng, X.; Liu, J.; Shen, Z. Genomic structure of the human BCCIP gene and its expression in cancer. Gene 2003, 302, 139–146. [Google Scholar] [CrossRef]
- Fujikawa, T.; Miyata, S.-I.; Iwanami, T. Convenient detection of the citrus greening (huanglongbing) bacterium ‘Candidatus Liberibacter asiaticus’ by direct PCR from the midrib extract. PLoS ONE 2013, 8, e57011. [Google Scholar] [CrossRef] [PubMed]
- Alqaydi, M.; Roy, R. Quantitative and qualitative study of STR DNA from ethanol and formalin fixed tissues. Forensic Sci. Int. 2016, 262, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Nakashima, K.; Maruta, Y.; Kiriyama, T.; Sasaki, M.; Sugiyama, S.; Suzuki, K.; Fujisaki, H.; Sasaki, J.; Kaku-Ushiki, Y. Improved RNA extraction method using the BioMasher and BioMasher power-plus. J. Vet. Med. Sci. 2012, 74, 1561–1567. [Google Scholar] [CrossRef]
- Yamamoto, T.; Ushiki, Y.; Hara, S.; Hall, W.W.; Tsukagoshi-Nagai, H.; Yokoyama, T.; Tagawa, Y.; Sata, T.; Yamakawa, Y.; Kinoshita, N. An advantageous method utilizing new homogenizing device BioMasher and a sensitive ELISA to detect bovine spongiform encephalopathy accurately in brain tissue. J. Virol. Methods 2008, 149, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Muller, M.; Xu, B.; Yoshida, Y.; Horlacher, O.; Nikitin, F.; Garessus, S.; Magdeldin, S.; Kinoshita, N.; Fujinaka, H.; et al. Unrestricted modification search reveals lysine methylation as major modification induced by tissue formalin fixation and paraffin embedding. Proteomics 2015, 15, 2568–2579. [Google Scholar] [CrossRef]
- Sprung, R.W.; Brock, J.W.C.; Tanksley, J.P.; Li, M.; Washington, M.K.; Slebos, R.J.C.; Liebler, D.C. Equivalence of Protein Inventories Obtained from Formalin-fixed Paraffin-embedded and Frozen Tissue in Multidimensional Liquid Chromatography-Tandem Mass Spectrometry Shotgun Proteomic Analysis. Mol. Cell. Proteom. 2009, 8, 1988–1998. [Google Scholar] [CrossRef]
- Coscia, F.; Doll, S.; Bech, J.M.; Schweizer, L.; Mund, A.; Lengyel, E.; Lindebjerg, J.; Madsen, G.I.; Moreira, J.M.; Mann, M. A streamlined mass spectrometry–based proteomics workflow for large-scale FFPE tissue analysis. J. Pathol. 2020, 251, 100–112. [Google Scholar] [CrossRef]
- Metz, B.; Kersten, G.F.A.; Baart, G.J.E.; de Jong, A.; Meiring, H.; ten Hove, J.; van Steenbergen, M.J.; Hennink, W.E.; Crommelin, D.J.A.; Jiskoot, W. Identification of Formaldehyde-Induced Modifications in Proteins: Reactions with Insulin. Bioconjugate Chem. 2006, 17, 815–822. [Google Scholar] [CrossRef]
- Shi, S.R.; Liu, C.; Balgley, B.M.; Lee, C.; Taylor, C.R. Protein extraction from formalin-fixed, paraffin-embedded tissue sections: Quality evaluation by mass spectrometry. J. Histochem. Cytochem. 2006, 54, 739–743. [Google Scholar] [CrossRef]
- Proc, J.L.; Kuzyk, M.A.; Hardie, D.B.; Yang, J.; Smith, D.S.; Jackson, A.M.; Parker, C.E.; Borchers, C.H. A quantitative study of the effects of chaotropic agents, surfactants, and solvents on the digestion efficiency of human plasma proteins by trypsin. J. Proteome Res. 2010, 9, 5422–5437. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Lin, H.; Liu, Z.; Wang, K.; Yan, Y. Improvement of a sample preparation method assisted by sodium deoxycholate for mass-spectrometry-based shotgun membrane proteomics. J. Sep. Sci. 2014, 37, 3321–3329. [Google Scholar] [CrossRef] [PubMed]
- Erde, J.; Loo, R.R.; Loo, J.A. Enhanced FASP (eFASP) to increase proteome coverage and sample recovery for quantitative proteomic experiments. J. Proteome Res. 2014, 13, 1885–1895. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Manza, L.L.; Stamer, S.L.; Ham, A.J.; Codreanu, S.G.; Liebler, D.C. Sample preparation and digestion for proteomic analyses using spin filters. Proteomics 2005, 5, 1742–1745. [Google Scholar] [CrossRef]
- Batth, T.S.; Tollenaere, M.A.X.; Rüther, P.L.; Gonzalez-Franquesa, A.; Prabhakar, B.S.; Bekker-Jensen, S.H.; Deshmukh, A.S.; Olsen, J.V. Protein aggregation capture on microparticles enables multi-purpose proteomics sample preparation. Mol. Cell. Proteom. 2019, 18, 1027–1035. [Google Scholar] [CrossRef]
- Wilson, J.; Mayyappan, V.; Narducci, D.N.; Neely, B.; Laugharn, J.; Pappin, D. Universal Sample Processing of Multiple Sample Types for Reproducible Proteomic Sample Preparation. HUPO. 2018. Available online: https://cdn.shopify.com/s/files/1/0271/1964/8832/files/HUPO-ProtiFi-Covaris-poster-final.pdf (accessed on 11 August 2021).
- Müller, T.; Kalxdorf, M.; Longuespée, R.; Kazdal, D.N.; Stenzinger, A.; Krijgsveld, J. Automated sample preparation with SP3 for low-input clinical proteomics. Mol. Syst. Biol. 2020, 16, e9111. [Google Scholar] [CrossRef]
- Buczak, K.; Ori, A.; Kirkpatrick, J.M.; Holzer, K.; Dauch, D.; Roessler, S.; Endris, V.; Lasitschka, F.; Parca, L.; Schmidt, A.; et al. Spatial tissue proteomics quantifies inter- and intra-tumor heterogeneity in hepatocellular carcinoma. Mol. Cell. Proteom. 2018, 17, 810–825. [Google Scholar] [CrossRef]
- Schweizer, L.; Coscia, F.; Müller, J.; Doll, S.; Wierer, M.; Mann, M. AFA-sonication Followed by Modified Protein Aggregation Capture (APAC) Enables Direct, Reproducible and Non-toxic Sample Preparation of FFPE Tissue for Mass Spectrometrybased Proteomics. Covaris Appl. Note-M020141. Available online: https://d24ci5y4j5ezt1.cloudfront.net/wp/wp-content/uploads/2020/06/M020141.pdf (accessed on 11 August 2021).
- Hughes, C.S.; Moggridge, S.; Müller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 2019, 14, 68–85. [Google Scholar] [CrossRef]
- Stoychev, S.; Govender, I.; Naicker, P.; Gerber, I.; Jordaan, J.; Pauw, M.; Tabb, D.; Arribas Diez, I.; Norregaard Jensen, O.; Baath, T.; et al. Development of a fully automated high throughput magnetic workflow for phosphoproteome profiling. HUPO. 2019. Available online: https://resynbio.com/wp-content/uploads/2020/01/HUPO_2019_Poster_SS_Phos_coupled_clean-up.pdf (accessed on 11 August 2021).
- Tape, C.J.; Worboys, J.D.; Sinclair, J.; Gourlay, R.; Vogt, J.; McMahon, K.M.; Trost, M.; Lauffenburger, D.A.; Lamont, D.J.; Jørgensen, C. Reproducible automated phosphopeptide enrichment using magnetic TiO2 and Ti-IMAC. Anal. Chem. 2014, 86, 10296–10302. [Google Scholar] [CrossRef]
- Martínez-Val, A.; Bekker-Jensen, D.B.; Steigerwald, S.; Stoychev, S.; Gerber, I.; Jordaan, J.; Bache, N.; Olsen, J.V. Fast and reproducible phosphoproteomics using MagReSyn Amine and Ti-IMAC HP magnetic beads and the Evosep One. Tech. Note. Available online: https://www.evosep.com/wp-content/uploads/2020/03/Phospho-app-note-Evosep-Resyn-A5layout-v5-lores.pdf (accessed on 11 August 2021).
- Leutert, M.; Rodriguez-Mias, R.A.; Fukuda, N.K.; Villén, J. R2-P2 rapid-robotic phosphoproteomics enables multidimensional cell signaling studies. Mol. Syst. Biol. 2019, 15, e9021. [Google Scholar] [CrossRef] [PubMed]
- Nolte, H.; MacVicar, T.D.; Tellkamp, F.; Krüger, M. Instant Clue: A Software Suite for Interactive Data Visualization and Analysis. Sci. Rep. 2018, 8, 12648. [Google Scholar] [CrossRef] [PubMed]
- The Human Proteome Atlas. Available online: https://www.proteinatlas.org (accessed on 23 February 2022).
- Marchione, D.M.; Ilieva, I.; Devins, K.; Sharpe, D.; Pappin, D.J.; Garcia, B.A.; Wilson, J.P.; Wojcik, J.B. HYPERsol: High-Quality Data from Archival FFPE Tissue for Clinical Proteomics. J. Proteome Res. 2020, 19, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Sielaff, M.; Kuharev, J.; Bohn, T.; Hahlbrock, J.; Bopp, T.; Tenzer, S.; Distler, U. Evaluation of FASP, SP3, and iST Protocols for Proteomic Sample Preparation in the Low Microgram Range. J. Proteome Res. 2017, 16, 4060–4072. [Google Scholar] [CrossRef]
- Wiśniewski, J.R. Filter Aided Sample Preparation—A tutorial. Anal. Chim. Acta 2019, 1090, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Boellner, S.; Becker, K.F. Reverse Phase Protein Arrays-Quantitative Assessment of Multiple Biomarkers in Biopsies for Clinical Use. Microarrays 2015, 4, 98–114. [Google Scholar] [CrossRef] [PubMed]
- Shema, G.; Nguyen, M.T.N.; Solari, F.A.; Loroch, S.; Venne, A.S.; Kollipara, L.; Sickmann, A.; Verhelst, S.H.L.; Zahedi, R.P. Simple, scalable, and ultrasensitive tip-based identification of protease substrates. Mol. Cell. Proteom. 2018, 17, 826–834. [Google Scholar] [CrossRef]
- Kollipara, L.; Zahedi, R.P. Protein carbamylation: In vivo modification or in vitro artefact? Proteomics 2013, 13, 941–944. [Google Scholar] [CrossRef]
- Tanca, A.; Abbondio, M.; Pisanu, S.; Pagnozzi, D.; Uzzau, S.; Addis, M.F. Critical comparison of sample preparation strategies for shotgun proteomic analysis of formalin-fixed, paraffin-embedded samples: Insights from liver tissue. Clin. Proteom. 2014, 11, 28. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Rios, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef]
- Ibrahim, S.; Lan, C.; Chabot, C.; Mitsa, G.; Buchanan, M.; Aguilar-Mahecha, A.; Elchebly, M.; Poetz, O.; Spatz, A.; Basik, M.; et al. Precise quantitation of PTEN by immuno-MRM: A tool to resolve the breast cancer biomarker controversy. Anal. Chem. 2021, 93, 10816–10824. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitsa, G.; Guo, Q.; Goncalves, C.; Preston, S.E.J.; Lacasse, V.; Aguilar-Mahecha, A.; Benlimame, N.; Basik, M.; Spatz, A.; Batist, G.; et al. A Non-Hazardous Deparaffinization Protocol Enables Quantitative Proteomics of Core Needle Biopsy-Sized Formalin-Fixed and Paraffin-Embedded (FFPE) Tissue Specimens. Int. J. Mol. Sci. 2022, 23, 4443. https://doi.org/10.3390/ijms23084443
Mitsa G, Guo Q, Goncalves C, Preston SEJ, Lacasse V, Aguilar-Mahecha A, Benlimame N, Basik M, Spatz A, Batist G, et al. A Non-Hazardous Deparaffinization Protocol Enables Quantitative Proteomics of Core Needle Biopsy-Sized Formalin-Fixed and Paraffin-Embedded (FFPE) Tissue Specimens. International Journal of Molecular Sciences. 2022; 23(8):4443. https://doi.org/10.3390/ijms23084443
Chicago/Turabian StyleMitsa, Georgia, Qianyu Guo, Christophe Goncalves, Samuel E. J. Preston, Vincent Lacasse, Adriana Aguilar-Mahecha, Naciba Benlimame, Mark Basik, Alan Spatz, Gerald Batist, and et al. 2022. "A Non-Hazardous Deparaffinization Protocol Enables Quantitative Proteomics of Core Needle Biopsy-Sized Formalin-Fixed and Paraffin-Embedded (FFPE) Tissue Specimens" International Journal of Molecular Sciences 23, no. 8: 4443. https://doi.org/10.3390/ijms23084443
APA StyleMitsa, G., Guo, Q., Goncalves, C., Preston, S. E. J., Lacasse, V., Aguilar-Mahecha, A., Benlimame, N., Basik, M., Spatz, A., Batist, G., Miller, W. H., Jr., del Rincon, S. V., Zahedi, R. P., & Borchers, C. H. (2022). A Non-Hazardous Deparaffinization Protocol Enables Quantitative Proteomics of Core Needle Biopsy-Sized Formalin-Fixed and Paraffin-Embedded (FFPE) Tissue Specimens. International Journal of Molecular Sciences, 23(8), 4443. https://doi.org/10.3390/ijms23084443