
The Importance of CXCL1 in Physiology and Noncancerous Diseases of Bone, Bone Marrow, Muscle and the Nervous System

 ,
,  , , and
, , and
Abstract
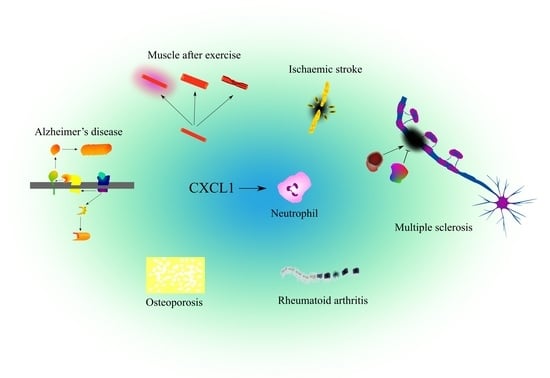
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
- CX3C chemokines (1 representative in humans),
- CXC chemokines (17 in humans),
- CC chemokines (26 in humans),
- XC chemokines (2 in humans).
2. Commentary on the Research Methodology
2.1. Search and Selection of Articles
2.2. The Lack of In Vivo Models for CXCL1 Functions
3. CXCL1 Action at the Single Cell Level
4. Cartilage and Bone Tissue
4.1. Bone, Fracture Healing, Osteoporosis
4.2. Bone Marrow
4.3. Rheumatoid Arthritis
5. Muscles
5.1. Muscle Physiology
5.2. Muscle, CXCL1 and Obesity
5.3. Tumor-Induced Muscle Wasting
6. The Nervous System
6.1. Prenatal Development of the Brain
6.2. Neurogenesis, Hippocampus and Neural Stem Cells
6.3. Addiction and Reward System
6.4. Alzheimer’s Disease
6.5. Epilepsy
6.6. Herpes Simplex Virus Type 1 (HSV-1) Encephalitis and Herpetic Stromal Keratitis (HSK)
6.7. Ischemic Stroke
6.8. Major Depression
6.9. Multiple Sclerosis
6.10. Neuromyelitis Optica
6.11. Neuropathic Pain and Sickness Behaviors
6.12. Prion Diseases
6.13. Tick-Borne Encephalitis (TBE) and Ticks
6.14. Traumatic Spinal Cord Injury
6.15. West Nile Fever
7. Directions of Further Research
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef] [PubMed]
- Do, H.T.T.; Lee, C.H.; Cho, J. Chemokines and their Receptors: Multifaceted Roles in Cancer Progression and Potential Value as Cancer Prognostic Markers. Cancers 2020, 12, 287. [Google Scholar] [CrossRef] [PubMed]
- Haskill, S.; Peace, A.; Morris, J.; Sporn, S.A.; Anisowicz, A.; Lee, S.W.; Smith, T.; Martin, G.; Ralph, P.; Sager, R. Identification of three related human GRO genes encoding cytokine functions. Proc. Natl. Acad. Sci. USA 1990, 87, 7732–7736. [Google Scholar] [CrossRef] [PubMed]
- Richmond, A.; Lawson, D.H.; Nixon, D.W.; Chawla, R.K. Characterization of autostimulatory and transforming growth factors from human melanoma cells. Cancer Res. 1985, 45, 6390–6394. [Google Scholar] [PubMed]
- Nomiyama, H.; Mera, A.; Ohneda, O.; Miura, R.; Suda, T.; Yoshie, O. Organization of the chemokine genes in the human and mouse major clusters of CC and CXC chemokines: Diversification between the two species. Genes Immun. 2001, 2, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Nomiyama, H.; Osada, N.; Yoshie, O. Systematic classification of vertebrate chemokines based on conserved synteny and evolutionary history. Genes Cells 2013, 18, 1–16. [Google Scholar] [CrossRef]
- Wuyts, A.; Proost, P.; Lenaerts, J.P.; Ben-Baruch, A.; Van Damme, J.; Wang, J.M. Differential usage of the CXC chemokine receptors 1 and 2 by interleukin-8, granulocyte chemotactic protein-2 and epithelial-cell-derived neutrophil attractant-78. Eur. J. Biochem. 1998, 255, 67–73. [Google Scholar] [CrossRef]
- Girbl, T.; Lenn, T.; Perez, L.; Rolas, L.; Barkaway, A.; Thiriot, A.; Del Fresno, C.; Lynam, E.; Hub, E.; Thelen, M.; et al. Distinct Compartmentalization of the Chemokines CXCL1 and CXCL2 and the Atypical Receptor ACKR1 Determine Discrete Stages of Neutrophil Diapedesis. Immunity 2018, 49, 1062–1076.e6. [Google Scholar] [CrossRef]
- Zagorski, J.; DeLarco, J.E. Rat CINC (cytokine-induced neutrophil chemoattractant) is the homolog of the human GRO proteins but is encoded by a single gene. Biochem. Biophys. Res. Commun. 1993, 190, 104–110. [Google Scholar] [CrossRef]
- Shibata, F.; Konishi, K.; Nakagawa, H. Identification of a common receptor for three types of rat cytokine-induced neutrophil chemoattractants (CINCs). Cytokine 2000, 12, 1368–1373. [Google Scholar] [CrossRef]
- Bozic, C.R.; Kolakowski, L.F., Jr.; Gerard, N.P.; Garcia-Rodriguez, C.; von Uexkull-Guldenband, C.; Conklyn, M.J.; Breslow, R.; Showell, H.J.; Gerard, C. Expression and biologic characterization of the murine chemokine KC. J. Immunol. 1995, 154, 6048–6057. [Google Scholar] [PubMed]
- Shea-Donohue, T.; Thomas, K.; Cody, M.J.; Zhao, A.; Detolla, L.J.; Kopydlowski, K.M.; Fukata, M.; Lira, S.A.; Vogel, S.N. Mice deficient in the CXCR2 ligand, CXCL1 (KC/GRO-alpha), exhibit increased susceptibility to dextran sodium sulfate (DSS)-induced colitis. Innate Immun. 2008, 14, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Patera, A.C.; Pong-Kennedy, A.; Deno, G.; Gonsiorek, W.; Manfra, D.J.; Vassileva, G.; Zeng, M.; Jackson, C.; Sullivan, L.; et al. Murine CXCR1 is a functional receptor for GCP-2/CXCL6 and interleukin-8/CXCL8. J. Biol. Chem. 2007, 282, 11658–11666. [Google Scholar] [CrossRef]
- Fox, S.E.; Lu, W.; Maheshwari, A.; Christensen, R.D.; Calhoun, D.A. The effects and comparative differences of neutrophil specific chemokines on neutrophil chemotaxis of the neonate. Cytokine 2005, 29, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.K.; Stojkov, D.; Feigelson, S.W.; Roncato, F.; Simon, H.U.; Yousefi, S.; Alon, R. Chemokine-triggered microtubule polymerization promotes neutrophil chemotaxis and invasion but not transendothelial migration. J. Leukoc. Biol. 2019, 105, 755–766. [Google Scholar] [CrossRef]
- Liu, L.; Li, M.; Spangler, L.C.; Spear, C.; Veenstra, M.; Darnall, L.; Chang, C.; Cotleur, A.C.; Ransohoff, R.M. Functional defect of peripheral neutrophils in mice with induced deletion of CXCR2. Genesis 2013, 51, 587–595. [Google Scholar] [CrossRef]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef]
- Geiser, T.; Dewald, B.; Ehrengruber, M.U.; Clark-Lewis, I.; Baggiolini, M. The interleukin-8-related chemotactic cytokines GRO alpha, GRO beta, and GRO gamma activate human neutrophil and basophil leukocytes. J. Biol. Chem. 1993, 268, 15419–15424. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Wang, G.; Westwood, J.A.; Pachynski, R.K.; Tiffany, H.L.; Marincola, F.M.; Wang, E.; Young, H.A.; Murphy, P.M.; Hwu, P. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum. Gene Ther. 2002, 13, 1971–1980. [Google Scholar] [CrossRef]
- Dunican, A.; Grutkoski, P.; Leuenroth, S.; Ayala, A.; Simms, H.H. Neutrophils regulate their own apoptosis via preservation of CXC receptors. J. Surg. Res. 2000, 90, 32–38. [Google Scholar] [CrossRef]
- Addison, C.L.; Daniel, T.O.; Burdick, M.D.; Liu, H.; Ehlert, J.E.; Xue, Y.Y.; Buechi, L.; Walz, A.; Richmond, A.; Strieter, R.M. The CXC chemokine receptor 2, CXCR2, is the putative receptor for ELR+ CXC chemokine-induced angiogenic activity. J. Immunol. 2000, 165, 5269–5277. [Google Scholar] [CrossRef] [PubMed]
- Keane, M.P.; Belperio, J.A.; Xue, Y.Y.; Burdick, M.D.; Strieter, R.M. Depletion of CXCR2 inhibits tumor growth and angiogenesis in a murine model of lung cancer. J. Immunol. 2004, 172, 2853–2860. [Google Scholar] [CrossRef] [PubMed]
- Moser, B.; Schumacher, C.; von Tscharner, V.; Clark-Lewis, I.; Baggiolini, M. Neutrophil-activating peptide 2 and gro/melanoma growth-stimulatory activity interact with neutrophil-activating peptide 1/interleukin 8 receptors on human neutrophils. J. Biol. Chem. 1991, 266, 10666–10671. [Google Scholar] [CrossRef]
- Loetscher, P.; Seitz, M.; Clark-Lewis, I.; Baggiolini, M.; Moser, B. Both interleukin-8 receptors independently mediate chemotaxis: Jurkat cells transfected with IL-8R1 or IL-8R2 migrate in response to IL-8, GRO alpha and NAP-2. FEBS Lett. 1994, 341, 187–192. [Google Scholar] [CrossRef]
- Ahuja, S.K.; Murphy, P.M. The CXC chemokines growth-regulated oncogene (GRO) alpha, GRObeta, GROgamma, neutrophil-activating peptide-2, and epithelial cell-derived neutrophil-activating peptide-78 are potent agonists for the type B, but not the type A, human interleukin-8 receptor. J. Biol. Chem. 1996, 271, 20545–20550. [Google Scholar] [CrossRef]
- Zhao, Y.; Mangalmurti, N.S.; Xiong, Z.; Prakash, B.; Guo, F.; Stolz, D.B.; Lee, J.S. Duffy antigen receptor for chemokines mediates chemokine endocytosis through a macropinocytosis-like process in endothelial cells. PLoS ONE 2011, 6, e29624. [Google Scholar] [CrossRef] [PubMed]
- Fukuma, N.; Akimitsu, N.; Hamamoto, H.; Kusuhara, H.; Sugiyama, Y.; Sekimizu, K. A role of the Duffy antigen for the maintenance of plasma chemokine concentrations. Biochem. Biophys. Res. Commun. 2003, 303, 137–139. [Google Scholar] [CrossRef]
- Dawson, T.C.; Lentsch, A.B.; Wang, Z.; Cowhig, J.E.; Rot, A.; Maeda, N.; Peiper, S.C. Exaggerated response to endotoxin in mice lacking the Duffy antigen/receptor for chemokines (DARC). Blood 2000, 96, 1681–1684. [Google Scholar] [CrossRef]
- Natoli, R.; Fernando, N.; Madigan, M.; Chu-Tan, J.A.; Valter, K.; Provis, J.; Rutar, M. Microglia-derived IL-1β promotes chemokine expression by Müller cells and RPE in focal retinal degeneration. Mol. Neurodegener. 2017, 12, 31. [Google Scholar] [CrossRef]
- Korbecki, J.; Barczak, K.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. CXCL1: Gene, Promoter, Regulation of Expression, mRNA Stability, Regulation of Activity in the Intercellular Space. Int. J. Mol. Sci. 2022, 23, 792. [Google Scholar] [CrossRef]
- Sun, D.; Novotny, M.; Bulek, K.; Liu, C.; Li, X.; Hamilton, T. Treatment with IL-17 prolongs the half-life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicing-regulatory factor SF2 (ASF). Nat. Immunol. 2011, 12, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Herjan, T.; Yao, P.; Qian, W.; Li, X.; Liu, C.; Bulek, K.; Sun, D.; Yang, W.P.; Zhu, J.; He, A.; et al. HuR is required for IL-17-induced Act1-mediated CXCL1 and CXCL5 mRNA stabilization. J. Immunol. 2013, 191, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Herjan, T.; Hong, L.; Bubenik, J.; Bulek, K.; Qian, W.; Liu, C.; Li, X.; Chen, X.; Yang, H.; Ouyang, S.; et al. IL-17-receptor-associated adaptor Act1 directly stabilizes mRNAs to mediate IL-17 inflammatory signaling. Nat. Immunol. 2018, 19, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, N.; Sakao, Y.; Hayashi, S.; Hadden, W.A., III; Harmon, C.L.; Miller, E.J. alpha-Chemokine growth factors for adenocarcinomas; a synthetic peptide inhibitor for alpha-chemokines inhibits the growth of adenocarcinoma cell lines. J. Cancer Res. Clin. Oncol. 2000, 126, 19–26. [Google Scholar] [CrossRef]
- Li, A.; Varney, M.L.; Singh, R.K. Constitutive expression of growth regulated oncogene (gro) in human colon carcinoma cells with different metastatic potential and its role in regulating their metastatic phenotype. Clin. Exp. Metastasis 2004, 21, 571–579. [Google Scholar] [CrossRef]
- Wang, B.; Hendricks, D.T.; Wamunyokoli, F.; Parker, M.I. A growth-related oncogene/CXC chemokine receptor 2 autocrine loop contributes to cellular proliferation in esophageal cancer. Cancer Res. 2006, 66, 3071–3077. [Google Scholar] [CrossRef]
- Bolitho, C.; Hahn, M.A.; Baxter, R.C.; Marsh, D.J. The chemokine CXCL1 induces proliferation in epithelial ovarian cancer cells by transactivation of the epidermal growth factor receptor. Endocr. Relat. Cancer 2010, 17, 929–940. [Google Scholar] [CrossRef]
- Robinson, S.; Tani, M.; Strieter, R.M.; Ransohoff, R.M.; Miller, R.H. The chemokine growth-regulated oncogene-alpha promotes spinal cord oligodendrocyte precursor proliferation. J. Neurosci. 1998, 18, 10457–10463. [Google Scholar] [CrossRef]
- Omari, K.M.; Lutz, S.E.; Santambrogio, L.; Lira, S.A.; Raine, C.S. Neuroprotection and remyelination after autoimmune demyelination in mice that inducibly overexpress CXCL1. Am. J. Pathol. 2009, 174, 164–176. [Google Scholar] [CrossRef]
- Cullen, S.P.; Henry, C.M.; Kearney, C.J.; Logue, S.E.; Feoktistova, M.; Tynan, G.A.; Lavelle, E.C.; Leverkus, M.; Martin, S.J. Fas/CD95-induced chemokines can serve as “find-me” signals for apoptotic cells. Mol. Cell 2013, 49, 1034–1048. [Google Scholar] [CrossRef]
- Eigenbrod, T.; Park, J.H.; Harder, J.; Iwakura, Y.; Núñez, G. Cutting edge: Critical role for mesothelial cells in necrosis-induced inflammation through the recognition of IL-1 alpha released from dying cells. J. Immunol. 2008, 181, 8194–8198. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef]
- Guo, H.; Liu, Z.; Xu, B.; Hu, H.; Wei, Z.; Liu, Q.; Zhang, X.; Ding, X.; Wang, Y.; Zhao, M.; et al. Chemokine receptor CXCR2 is transactivated by p53 and induces p38-mediated cellular senescence in response to DNA damage. Aging Cell 2013, 12, 1110–1121. [Google Scholar] [CrossRef]
- Alexander, E.; Hildebrand, D.G.; Kriebs, A.; Obermayer, K.; Manz, M.; Rothfuss, O.; Schulze-Osthoff, K.; Essmann, F. IκBζ is a regulator of the senescence-associated secretory phenotype in DNA damage- and oncogene-induced senescence. J. Cell Sci. 2013, 126, 3738–3745. [Google Scholar] [CrossRef]
- Lesina, M.; Wörmann, S.M.; Morton, J.; Diakopoulos, K.N.; Korneeva, O.; Wimmer, M.; Einwächter, H.; Sperveslage, J.; Demir, I.E.; Kehl, T.; et al. RelA regulates CXCL1/CXCR2-dependent oncogene-induced senescence in murine Kras-driven pancreatic carcinogenesis. J. Clin. Investig. 2016, 126, 2919–2932. [Google Scholar] [CrossRef]
- Yang, G.; Rosen, D.G.; Zhang, Z.; Bast, R.C., Jr.; Mills, G.B.; Colacino, J.A.; Mercado-Uribe, I.; Liu, J. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 16472–16477. [Google Scholar] [CrossRef]
- Cai, L.; Xu, S.; Piao, C.; Qiu, S.; Li, H.; Du, J. Adiponectin induces CXCL1 secretion from cancer cells and promotes tumor angiogenesis by inducing stromal fibroblast senescence. Mol. Carcinog. 2016, 55, 1796–1806. [Google Scholar] [CrossRef]
- Govey, P.M.; Jacobs, J.M.; Tilton, S.C.; Loiselle, A.E.; Zhang, Y.; Freeman, W.M.; Waters, K.M.; Karin, N.J.; Donahue, H.J. Integrative transcriptomic and proteomic analysis of osteocytic cells exposed to fluid flow reveals novel mechano-sensitive signaling pathways. J. Biomech. 2014, 47, 1838–1845. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dwivedi, A.; Kiely, P.A.; Hoey, D.A. Mechanically stimulated osteocytes promote the proliferation and migration of breast cancer cells via a potential CXCL1/2 mechanism. Biochem. Biophys. Res. Commun. 2021, 534, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Onan, D.; Allan, E.H.; Quinn, J.M.; Gooi, J.H.; Pompolo, S.; Sims, N.A.; Gillespie, M.T.; Martin, T.J. The chemokine Cxcl1 is a novel target gene of parathyroid hormone (PTH)/PTH-related protein in committed osteoblasts. Endocrinology 2009, 150, 2244–2253. [Google Scholar] [CrossRef] [PubMed]
- Hardaway, A.L.; Herroon, M.K.; Rajagurubandara, E.; Podgorski, I. Marrow adipocyte-derived CXCL1 and CXCL2 contribute to osteolysis in metastatic prostate cancer. Clin. Exp. Metastasis 2015, 32, 353–368. [Google Scholar] [CrossRef]
- Grassi, F.; Piacentini, A.; Cristino, S.; Toneguzzi, S.; Cavallo, C.; Facchini, A.; Lisignoli, G. Human osteoclasts express different CXC chemokines depending on cell culture substrate: Molecular and immunocytochemical evidence of high levels of CXCL10 and CXCL12. Histochem. Cell Biol. 2003, 120, 391–400. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, L.; Zhao, Z.; Lu, W.; Fan, J.; Gao, B.; Luo, Z.; Jie, Q.; Shi, X.; Yang, L. Cytokines CCL2 and CXCL1 may be potential novel predictors of early bone loss. Mol. Med. Rep. 2020, 22, 4716–4724. [Google Scholar] [CrossRef]
- Kovtun, A.; Bergdolt, S.; Wiegner, R.; Radermacher, P.; Huber-Lang, M.; Ignatius, A. The crucial role of neutrophil granulocytes in bone fracture healing. Eur. Cells Mater. 2016, 32, 152–162. [Google Scholar] [CrossRef]
- Washam, C.L.; Byrum, S.D.; Leitzel, K.; Ali, S.M.; Tackett, A.J.; Gaddy, D.; Sundermann, S.E.; Lipton, A.; Suva, L.J. Identification of PTHrP(12-48) as a plasma biomarker associated with breast cancer bone metastasis. Cancer Epidemiol. Biomark. Prev. 2013, 22, 972–983. [Google Scholar] [CrossRef]
- Lee, Y.C.; Gajdosik, M.S.; Josic, D.; Clifton, J.G.; Logothetis, C.; Yu-Lee, L.Y.; Gallick, G.E.; Maity, S.N.; Lin, S.H. Secretome analysis of an osteogenic prostate tumor identifies complex signaling networks mediating cross-talk of cancer and stromal cells within the tumor microenvironment. Mol. Cell. Proteom. 2015, 14, 471–483. [Google Scholar] [CrossRef]
- Bhat, K.; Sarkissyan, M.; Wu, Y.; Vadgama, J.V. GROα overexpression drives cell migration and invasion in triple negative breast cancer cells. Oncol. Rep. 2017, 38, 21–30. [Google Scholar] [CrossRef]
- Sinclair, A.; Park, L.; Shah, M.; Drotar, M.; Calaminus, S.; Hopcroft, L.E.; Kinstrie, R.; Guitart, A.V.; Dunn, K.; Abraham, S.A.; et al. CXCR2 and CXCL4 regulate survival and self-renewal of hematopoietic stem/progenitor cells. Blood 2016, 128, 371–383. [Google Scholar] [CrossRef]
- Martin, C.; Burdon, P.C.; Bridger, G.; Gutierrez-Ramos, J.C.; Williams, T.J.; Rankin, S.M. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity 2003, 19, 583–593. [Google Scholar] [CrossRef]
- Ao, T.; Kikuta, J.; Sudo, T.; Uchida, Y.; Kobayashi, K.; Ishii, M. Local sympathetic neurons promote neutrophil egress from the bone marrow at the onset of acute inflammation. Int. Immunol. 2020, 32, 727–736. [Google Scholar] [CrossRef]
- Shi, H.; Han, X.; Sun, Y.; Shang, C.; Wei, M.; Ba, X.; Zeng, X. Chemokine (C-X-C motif) ligand 1 and CXCL2 produced by tumor promote the generation of monocytic myeloid-derived suppressor cells. Cancer Sci. 2018, 109, 3826–3839. [Google Scholar] [CrossRef]
- Han, X.; Shi, H.; Sun, Y.; Shang, C.; Luan, T.; Wang, D.; Ba, X.; Zeng, X. CXCR2 expression on granulocyte and macrophage progenitors under tumor conditions contributes to mo-MDSC generation via SAP18/ERK/STAT3. Cell Death Dis. 2019, 10, 598. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Skrzypkowska, M.; Stasiak, M.; Sakowska, J.; Chmiel, J.; Maciejewska, A.; Buciński, A.; Słomiński, B.; Trzonkowski, P.; Łuczkiewicz, P. Cytokines and chemokines multiplex analysis in patients with low disease activity rheumatoid arthritis. Rheumatol. Int. 2022, 42, 609–619. [Google Scholar] [CrossRef]
- Hogan, M.; Sherry, B.; Ritchlin, C.; Fabre, M.; Winchester, R.; Cerami, A.; Bucala, R. Differential expression of the small inducible cytokines GRO alpha and GRO beta by synovial fibroblasts in chronic arthritis: Possible role in growth regulation. Cytokine 1994, 6, 61–69. [Google Scholar] [CrossRef]
- Hou, S.M.; Chen, P.C.; Lin, C.M.; Fang, M.L.; Chi, M.C.; Liu, J.F. CXCL1 contributes to IL-6 expression in osteoarthritis and rheumatoid arthritis synovial fibroblasts by CXCR2, c-Raf, MAPK, and AP-1 pathway. Arthritis Res. Ther. 2020, 22, 251. [Google Scholar] [CrossRef]
- Borzi, R.M.; Mazzetti, I.; Macor, S.; Silvestri, T.; Bassi, A.; Cattini, L.; Facchini, A. Flow cytometric analysis of intracellular chemokines in chondrocytes in vivo: Constitutive expression and enhancement in osteoarthritis and rheumatoid arthritis. FEBS Lett. 1999, 455, 238–242. [Google Scholar] [CrossRef]
- König, A.; Krenn, V.; Toksoy, A.; Gerhard, N.; Gillitzer, R. Mig, GRO alpha and RANTES messenger RNA expression in lining layer, infiltrates and different leucocyte populations of synovial tissue from patients with rheumatoid arthritis, psoriatic arthritis and osteoarthritis. Virchows Arch. 2000, 436, 449–458. [Google Scholar]
- Bertazzolo, N.; Punzi, L.; Stefani, M.P.; Cesaro, G.; Pianon, M.; Finco, B.; Todesco, S. Interrelationships between interleukin (IL)-1, IL-6 and IL-8 in synovial fluid of various arthropathies. Agents Actions 1994, 41, 90–92. [Google Scholar] [CrossRef]
- Hussein, M.R.; Fathi, N.A.; El-Din, A.M.; Hassan, H.I.; Abdullah, F.; Al-Hakeem, E.; Backer, E.A. Alterations of the CD4+, CD8+ T cell subsets, interleukins-1beta, IL-10, IL-17, tumor necrosis factor-alpha and soluble intercellular adhesion molecule-1 in rheumatoid arthritis and osteoarthritis: Preliminary observations. Pathol. Oncol. Res. 2008, 14, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Sun, L.; Jiang, T.; Zhang, D.; He, D.; Nie, H. TNFα promotes Th17 cell differentiation through IL-6 and IL-1β produced by monocytes in rheumatoid arthritis. J. Immunol. Res. 2014, 2014, 385352. [Google Scholar] [CrossRef] [PubMed]
- Roşu, A.; Mărgăritescu, C.; Stepan, A.; Muşetescu, A.; Ene, M. IL-17 patterns in synovium, serum and synovial fluid from treatment-naïve, early rheumatoid arthritis patients. Rom. J. Morphol. Embryol. 2012, 53, 73–80. [Google Scholar] [PubMed]
- Kehlen, A.; Thiele, K.; Riemann, D.; Langner, J. Expression, modulation and signalling of IL-17 receptor in fibroblast-like synoviocytes of patients with rheumatoid arthritis. Clin. Exp. Immunol. 2002, 127, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Muraoka, S.; Kusunoki, N.; Masuoka, S.; Yamada, S.; Ogasawara, H.; Imai, T.; Akasaka, Y.; Tochigi, N.; Takahashi, H.; et al. Resistin upregulates chemokine production by fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Res. Ther. 2017, 19, 263. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, E.; Vitellozzi, R.; Fernandez, P.; Falcieri, E.; Battistelli, M.; Burattini, S.; Facchini, A.; Flamigni, F.; Santi, S.; Facchini, A.; et al. Chondrocyte hypertrophy and apoptosis induced by GROalpha require three-dimensional interaction with the extracellular matrix and a co-receptor role of chondroitin sulfate and are associated with the mitochondrial splicing variant of cathepsin B. J. Cell. Physiol. 2007, 210, 417–427. [Google Scholar] [CrossRef]
- Unemori, E.N.; Amento, E.P.; Bauer, E.A.; Horuk, R. Melanoma growth-stimulatory activity/GRO decreases collagen expression by human fibroblasts. Regulation by C-X-C but not C-C cytokines. J. Biol. Chem. 1993, 268, 1338–1342. [Google Scholar] [CrossRef]
- Favalli, E.G. Understanding the Role of Interleukin-6 (IL-6) in the Joint and Beyond: A Comprehensive Review of IL-6 Inhibition for the Management of Rheumatoid Arthritis. Rheumatol. Ther. 2020, 7, 473–516. [Google Scholar] [CrossRef]
- Coelho, F.M.; Pinho, V.; Amaral, F.A.; Sachs, D.; Costa, V.V.; Rodrigues, D.H.; Vieira, A.T.; Silva, T.A.; Souza, D.G.; Bertini, R.; et al. The chemokine receptors CXCR1/CXCR2 modulate antigen-induced arthritis by regulating adhesion of neutrophils to the synovial microvasculature. Arthritis Rheumatol. 2008, 58, 2329–2337. [Google Scholar] [CrossRef] [PubMed]
- Grespan, R.; Fukada, S.Y.; Lemos, H.P.; Vieira, S.M.; Napimoga, M.H.; Teixeira, M.M.; Fraser, A.R.; Liew, F.Y.; McInnes, I.B.; Cunha, F.Q. CXCR2-specific chemokines mediate leukotriene B4-dependent recruitment of neutrophils to inflamed joints in mice with antigen-induced arthritis. Arthritis Rheumatol. 2008, 58, 2030–2040. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, L.J.; Kaplan, M.J. Neutrophils in Rheumatoid Arthritis: Breaking Immune Tolerance and Fueling Disease. Trends Mol. Med. 2019, 25, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Nedachi, T.; Fujita, H.; Kanzaki, M. Contractile C2C12 myotube model for studying exercise-inducible responses in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1191–E1204. [Google Scholar] [CrossRef] [PubMed]
- Nedachi, T.; Hatakeyama, H.; Kono, T.; Sato, M.; Kanzaki, M. Characterization of contraction-inducible CXC chemokines and their roles in C2C12 myocytes. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E866–E878. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.; Pilegaard, H.; Hansen, J.; Brandt, C.; Adser, H.; Hidalgo, J.; Olesen, J.; Pedersen, B.K.; Hojman, P. Exercise-induced liver chemokine CXCL-1 expression is linked to muscle-derived interleukin-6 expression. J. Physiol. 2011, 589, 1409–1420. [Google Scholar] [CrossRef]
- Pedersen, L.; Olsen, C.H.; Pedersen, B.K.; Hojman, P. Muscle-derived expression of the chemokine CXCL1 attenuates diet-induced obesity and improves fatty acid oxidation in the muscle. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E831–E840. [Google Scholar] [CrossRef]
- Farmawati, A.; Kitajima, Y.; Nedachi, T.; Sato, M.; Kanzaki, M.; Nagatomi, R. Characterization of contraction-induced IL-6 up-regulation using contractile C2C12 myotubes. Endocr. J. 2013, 60, 137–147. [Google Scholar] [CrossRef]
- Masuda, S.; Tanaka, M.; Inoue, T.; Ohue-Kitano, R.; Yamakage, H.; Muranaka, K.; Kusakabe, T.; Shimatsu, A.; Hasegawa, K.; Satoh-Asahara, N. Chemokine (C-X-C motif) ligand 1 is a myokine induced by palmitate and is required for myogenesis in mouse satellite cells. Acta Physiol. 2018, 222, e12975. [Google Scholar] [CrossRef]
- Iwasaki, S.; Miyake, M.; Hayashi, S.; Watanabe, H.; Nagasawa, Y.; Terada, S.; Watanabe, K.; Ohwada, S.; Kitazawa, H.; Rose, M.T.; et al. Effect of myostatin on chemokine expression in regenerating skeletal muscle cells. Cells Tissues Organs 2013, 198, 66–74. [Google Scholar] [CrossRef]
- Yang, M.; Wei, D.; Mo, C.; Zhang, J.; Wang, X.; Han, X.; Wang, Z.; Xiao, H. Saturated fatty acid palmitate-induced insulin resistance is accompanied with myotube loss and the impaired expression of health benefit myokine genes in C2C12 myotubes. Lipids Health Dis. 2013, 12, 104. [Google Scholar] [CrossRef] [PubMed]
- Chooi, Y.C.; Ding, C.; Magkos, F. The epidemiology of obesity. Metabolism 2019, 92, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Hogan, K.A.; Cho, D.S.; Arneson, P.C.; Samani, A.; Palines, P.; Yang, Y.; Doles, J.D. Tumor-derived cytokines impair myogenesis and alter the skeletal muscle immune microenvironment. Cytokine 2018, 107, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Callaway, C.S.; Delitto, A.E.; Patel, R.; Nosacka, R.L.; D’Lugos, A.C.; Delitto, D.; Deyhle, M.R.; Trevino, J.G.; Judge, S.M.; Judge, A.R. IL-8 Released from Human Pancreatic Cancer and Tumor-Associated Stromal Cells Signals through a CXCR2-ERK1/2 Axis to Induce Muscle Atrophy. Cancers 2019, 11, 1863. [Google Scholar] [CrossRef] [PubMed]
- Divella, R.; Daniele, A.; Savino, E.; Palma, F.; Bellizzi, A.; Giotta, F.; Simone, G.; Lioce, M.; Quaranta, M.; Paradiso, A.; et al. Circulating levels of transforming growth factor-βeta (TGF-β) and chemokine (C-X-C motif) ligand-1 (CXCL1) as predictors of distant seeding of circulating tumor cells in patients with metastatic breast cancer. Anticancer Res. 2013, 33, 1491–1497. [Google Scholar]
- Zhang, H.; Yue, J.; Jiang, Z.; Zhou, R.; Xie, R.; Xu, Y.; Wu, S. CAF-secreted CXCL1 conferred radioresistance by regulating DNA damage response in a ROS-dependent manner in esophageal squamous cell carcinoma. Cell Death Dis. 2017, 8, e2790. [Google Scholar] [CrossRef]
- Wang, Q.; Li, D.; Zhang, W.; Tang, B.; Li, Q.Q.; Li, L. Evaluation of proteomics-identified CCL18 and CXCL1 as circulating tumor markers for differential diagnosis between ovarian carcinomas and benign pelvic masses. Int. J. Biol. Markers 2011, 26, 262–273. [Google Scholar] [CrossRef]
- Mestas, J.; Burdick, M.D.; Reckamp, K.; Pantuck, A.; Figlin, R.A.; Strieter, R.M. The role of CXCR2/CXCR2 ligand biological axis in renal cell carcinoma. J. Immunol. 2005, 175, 5351–5357. [Google Scholar] [CrossRef]
- Bhardwaj, D.; Náger, M.; Camats, J.; David, M.; Benguria, A.; Dopazo, A.; Cantí, C.; Herreros, J. Chemokines induce axon outgrowth downstream of Hepatocyte Growth Factor and TCF/β-catenin signaling. Front. Cell. Neurosci. 2013, 7, 52. [Google Scholar] [CrossRef]
- Deftu, A.T.; Ciorescu, R.; Gheorghe, R.O.; Mihăilescu, D.; Ristoiu, V. CXCL1 and CXCL2 Inhibit the Axon Outgrowth in a Time- and Cell-Type-Dependent Manner in Adult Rat Dorsal Root Ganglia Neurons. Neurochem. Res. 2019, 44, 2215–2229. [Google Scholar] [CrossRef]
- Vora, P.; Pillai, P.; Mustapha, J.; Kowal, C.; Shaffer, S.; Bose, R.; Namaka, M.; Frost, E.E. CXCL1 regulation of oligodendrocyte progenitor cell migration is independent of calcium signaling. Exp. Neurol. 2012, 236, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Filipovic, R.; Zecevic, N. The effect of CXCL1 on human fetal oligodendrocyte progenitor cells. Glia 2008, 56, 1–15. [Google Scholar] [CrossRef]
- Tsai, H.H.; Frost, E.; To, V.; Robinson, S.; Ffrench-Constant, C.; Geertman, R.; Ransohoff, R.M.; Miller, R.H. The chemokine receptor CXCR2 controls positioning of oligodendrocyte precursors in developing spinal cord by arresting their migration. Cell 2002, 110, 373–383. [Google Scholar] [CrossRef]
- Huang, F.; Lan, Y.; Qin, L.; Dong, H.; Shi, H.; Wu, H.; Zou, Q.; Hu, Z.; Wu, X. Astragaloside IV Promotes Adult Neurogenesis in Hippocampal Dentate Gyrus of Mouse through CXCL1/CXCR2 Signaling. Molecules 2018, 23, 2178. [Google Scholar] [CrossRef]
- Zonis, S.; Breunig, J.J.; Mamelak, A.; Wawrowsky, K.; Bresee, C.; Ginzburg, N.; Chesnokova, V. Inflammation-induced Gro1 triggers senescence in neuronal progenitors: Effects of estradiol. J. Neuroinflamm. 2018, 15, 260. [Google Scholar] [CrossRef]
- Shang, Y.; Tian, L.; Chen, T.; Liu, X.; Zhang, J.; Liu, D.; Wei, J.; Fang, W.; Chen, Y.; Shang, D. CXCL1 promotes the proliferation of neural stem cells by stimulating the generation of reactive oxygen species in APP/PS1 mice. Biochem. Biophys. Res. Commun. 2019, 515, 201–206. [Google Scholar] [CrossRef]
- Gordon, R.J.; McGregor, A.L.; Connor, B. Chemokines direct neural progenitor cell migration following striatal cell loss. Mol. Cell. Neurosci. 2009, 41, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Saika, F.; Matsuzaki, S.; Kobayashi, D.; Kiguchi, N.; Kishioka, S. Chemokine CXCL1 is responsible for cocaine-induced reward in mice. Neuropsychopharmacol. Rep. 2018, 38, 145–148. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Craig-Schapiro, R.; Kuhn, M.; Xiong, C.; Pickering, E.H.; Liu, J.; Misko, T.P.; Perrin, R.J.; Bales, K.R.; Soares, H.; Fagan, A.M.; et al. Multiplexed immunoassay panel identifies novel CSF biomarkers for Alzheimer’s disease diagnosis and prognosis. PLoS ONE 2011, 6, e18850. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Hyman, B.T. GROalpha/KC, a chemokine receptor CXCR2 ligand, can be a potent trigger for neuronal ERK1/2 and PI-3 kinase pathways and for tau hyperphosphorylation-a role in Alzheimer’s disease? J. Neuroimmunol. 2002, 122, 55–64. [Google Scholar] [CrossRef]
- Zhang, X.F.; Zhao, Y.F.; Zhu, S.W.; Huang, W.J.; Luo, Y.; Chen, Q.Y.; Ge, L.J.; Li, R.S.; Wang, J.F.; Sun, M.; et al. CXCL1 Triggers Caspase-3 Dependent Tau Cleavage in Long-Term Neuronal Cultures and in the Hippocampus of Aged Mice: Implications in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 48, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, P.; Margenthaler, E.; Laporte, V.; Crawford, F.; Mullan, M. Novel role of CXCR2 in regulation of gamma-secretase activity. ACS Chem. Biol. 2008, 3, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, P.; Jin, C.; Broutin, P.; Berhane, B.; Reed, J.; Mullan, M. Structural optimization of a CXCR2-directed antagonist that indirectly inhibits gamma-secretase and reduces Abeta. Bioorg. Med. Chem. 2009, 17, 8102–8112. [Google Scholar] [CrossRef]
- Bakshi, P.; Margenthaler, E.; Reed, J.; Crawford, F.; Mullan, M. Depletion of CXCR2 inhibits γ-secretase activity and amyloid-β production in a murine model of Alzheimer’s disease. Cytokine 2011, 53, 163–169. [Google Scholar] [CrossRef]
- Zhang, K.; Tian, L.; Liu, L.; Feng, Y.; Dong, Y.B.; Li, B.; Shang, D.S.; Fang, W.G.; Cao, Y.P.; Chen, Y.H. CXCL1 contributes to β-amyloid-induced transendothelial migration of monocytes in Alzheimer’s disease. PLoS ONE 2013, 8, e72744. [Google Scholar] [CrossRef]
- Simard, A.R.; Rivest, S. Bone marrow stem cells have the ability to populate the entire central nervous system into fully differentiated parenchymal microglia. FASEB J. 2004, 18, 998–1000. [Google Scholar] [CrossRef]
- Thijs, R.D.; Surges, R.; O’Brien, T.J.; Sander, J.W. Epilepsy in adults. Lancet 2019, 393, 689–701. [Google Scholar] [CrossRef]
- Di Sapia, R.; Zimmer, T.S.; Kebede, V.; Balosso, S.; Ravizza, T.; Sorrentino, D.; Castillo, M.A.M.; Porcu, L.; Cattani, F.; Ruocco, A.; et al. CXCL1-CXCR1/2 signaling is induced in human temporal lobe epilepsy and contributes to seizures in a murine model of acquired epilepsy. Neurobiol. Dis. 2021, 158, 105468. [Google Scholar] [CrossRef]
- Liu, X.X.; Yang, L.; Shao, L.X.; He, Y.; Wu, G.; Bao, Y.H.; Lu, N.N.; Gong, D.M.; Lu, Y.P.; Cui, T.T.; et al. Endothelial Cdk5 deficit leads to the development of spontaneous epilepsy through CXCL1/CXCR2-mediated reactive astrogliosis. J. Exp. Med. 2020, 217, e20180992. [Google Scholar] [CrossRef]
- Kothur, K.; Bandodkar, S.; Wienholt, L.; Chu, S.; Pope, A.; Gill, D.; Dale, R.C. Etiology is the key determinant of neuroinflammation in epilepsy: Elevation of cerebrospinal fluid cytokines and chemokines in febrile infection-related epilepsy syndrome and febrile status epilepticus. Epilepsia 2019, 60, 1678–1688. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D. HSV-1 biology and life cycle. Methods Mol. Biol. 2014, 1144, 1–17. [Google Scholar] [PubMed]
- McQuillan, G.; Kruszon-Moran, D.; Flagg, E.W.; Paulose-Ram, R. Prevalence of Herpes Simplex Virus Type 1 and Type 2 in Persons Aged 14–49: United States, 2015–2016. In NCHS Data Brief No. 304; National Center for Health Statistics: Hyattsville, MD, USA, 2018; pp. 1–8. [Google Scholar]
- Marcocci, M.E.; Napoletani, G.; Protto, V.; Kolesova, O.; Piacentini, R.; Li Puma, D.D.; Lomonte, P.; Grassi, C.; Palamara, A.T.; De Chiara, G. Herpes Simplex Virus-1 in the Brain: The Dark Side of a Sneaky Infection. Trends Microbiol. 2020, 28, 808–820. [Google Scholar] [CrossRef]
- Vilela, M.C.; Mansur, D.S.; Lacerda-Queiroz, N.; Rodrigues, D.H.; Arantes, R.M.; Kroon, E.G.; Campos, M.A.; Teixeira, M.M.; Teixeira, A.L. Traffic of leukocytes in the central nervous system is associated with chemokine up-regulation in a severe model of herpes simplex encephalitis: An intravital microscopy study. Neurosci. Lett. 2008, 445, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Michael, B.D.; Bricio-Moreno, L.; Sorensen, E.W.; Miyabe, Y.; Lian, J.; Solomon, T.; Kurt-Jones, E.A.; Luster, A.D. Astrocyte- and Neuron-Derived CXCL1 Drives Neutrophil Transmigration and Blood-Brain Barrier Permeability in Viral Encephalitis. Cell. Rep. 2020, 32, 108150. [Google Scholar] [CrossRef]
- West, D.M.; Del Rosso, C.R.; Yin, X.T.; Stuart, P.M. CXCL1 but not IL-6 is required for recurrent herpetic stromal keratitis. J. Immunol. 2014, 192, 1762–1767. [Google Scholar] [CrossRef]
- Suryawanshi, A.; Veiga-Parga, T.; Reddy, P.B.; Rajasagi, N.K.; Rouse, B.T. IL-17A differentially regulates corneal vascular endothelial growth factor (VEGF)-A and soluble VEGF receptor 1 expression and promotes corneal angiogenesis after herpes simplex virus infection. J. Immunol. 2012, 188, 3434–3446. [Google Scholar] [CrossRef]
- Yan, X.T.; Tumpey, T.M.; Kunkel, S.L.; Oakes, J.E.; Lausch, R.N. Role of MIP-2 in neutrophil migration and tissue injury in the herpes simplex virus-1-infected cornea. Investig. Ophthalmol. Vis. Sci. 1998, 39, 1854–1862. [Google Scholar]
- Bryant-Hudson, K.M.; Carr, D.J. CXCL1-deficient mice are highly sensitive to pseudomonas aeruginosa but not herpes simplex virus type 1 corneal infection. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6785–6792. [Google Scholar] [CrossRef]
- Campbell, B.C.V.; Khatri, P. Stroke. Lancet 2020, 396, 129–142. [Google Scholar] [CrossRef]
- Park, H.J.; Yun, D.H.; Kim, S.K.; Chung, J.H.; Lee, J.S.; Park, H.K.; Chon, J.; Kim, D.H.; Yoo, S.D.; Kim, H.S. Association of CXCL1 promoter polymorphism with ischaemic stroke in Korean population. Int. J. Immunogenet. 2013, 40, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Losy, J.; Zaremba, J.; Skrobański, P. CXCL1 (GRO-alpha) chemokine in acute ischaemic stroke patients. Folia Neuropathol. 2005, 43, 97–102. [Google Scholar] [PubMed]
- Gelderblom, M.; Weymar, A.; Bernreuther, C.; Velden, J.; Arunachalam, P.; Steinbach, K.; Orthey, E.; Arumugam, T.V.; Leypoldt, F.; Simova, O.; et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood 2012, 120, 3793–3802. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.; Vakilian, A.; Mahmoodi, M.H.; Hassanshahi, G.; Falahati-Pour, S.K.; Dolatabadi, M.R.; Nadimi, A.E. Circulatory Levels of C-X-C Motif Chemokine Ligands 1, 9, and 10 Are Elevated in Patients with Ischemic Stroke. Eurasian J. Med. 2017, 49, 92–96. [Google Scholar] [CrossRef]
- Zhu, W.; Nan, Y.; Wang, S.; Liu, W. Bioinformatics Analysis of Gene Expression Profiles of Sex Differences in Ischemic Stroke. Biomed. Res. Int. 2019, 2019, 2478453. [Google Scholar] [CrossRef]
- Shi, Y.; Yi, Z.; Zhao, P.; Xu, Y.; Pan, P. MicroRNA-532-5p protects against cerebral ischemia-reperfusion injury by directly targeting CXCL1. Aging 2021, 13, 11528–11541. [Google Scholar] [CrossRef]
- Leng, J.; Liu, W.; Li, L.; Wei, F.Y.; Tian, M.; Liu, H.M.; Guo, W. MicroRNA-429/Cxcl1 Axis Protective Against Oxygen Glucose Deprivation/Reoxygenation-Induced Injury in Brain Microvascular Endothelial Cells. Dose Response 2020, 18, 1559325820913785. [Google Scholar] [CrossRef]
- Brait, V.H.; Rivera, J.; Broughton, B.R.; Lee, S.; Drummond, G.R.; Sobey, C.G. Chemokine-related gene expression in the brain following ischemic stroke: No role for CXCR2 in outcome. Brain Res. 2011, 1372, 169–179. [Google Scholar] [CrossRef]
- Ikegame, Y.; Yamashita, K.; Hayashi, S.; Yoshimura, S.; Nakashima, S.; Iwama, T. Neutrophil elastase inhibitor prevents ischemic brain damage via reduction of vasogenic edema. Hypertens. Res. 2010, 33, 703–707. [Google Scholar] [CrossRef]
- Malhi, G.S.; Mann, J.J. Depression. Lancet 2018, 392, 2299–2312. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, L.; Liu, Y.Z.; Shen, X.L.; Wu, T.Y.; Zhang, T.; Wang, W.; Wang, Y.X.; Jiang, C.L. NLRP3 Inflammasome Mediates Chronic Mild Stress-Induced Depression in Mice via Neuroinflammation. Int. J. Neuropsychopharmacol. 2015, 18, pyv006. [Google Scholar] [CrossRef] [PubMed]
- Song, A.Q.; Gao, B.; Fan, J.J.; Zhu, Y.J.; Zhou, J.; Wang, Y.L.; Xu, L.Z.; Wu, W.N. NLRP1 inflammasome contributes to chronic stress-induced depressive-like behaviors in mice. J. Neuroinflamm. 2020, 17, 178. [Google Scholar] [CrossRef] [PubMed]
- Pandey, G.N.; Rizavi, H.S.; Bhaumik, R.; Zhang, H. Chemokines gene expression in the prefrontal cortex of depressed suicide victims and normal control subjects. Brain Behav. Immun. 2021, 94, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Bot, M.; Chan, M.K.; Jansen, R.; Lamers, F.; Vogelzangs, N.; Steiner, J.; Leweke, F.M.; Rothermundt, M.; Cooper, J.; Bahn, S.; et al. Serum proteomic profiling of major depressive disorder. Transl. Psychiatry 2015, 5, e599. [Google Scholar] [CrossRef]
- Fanelli, G.; Benedetti, F.; Wang, S.M.; Lee, S.J.; Jun, T.Y.; Masand, P.S.; Patkar, A.A.; Han, C.; Serretti, A.; Pae, C.U.; et al. Reduced CXCL1/GRO chemokine plasma levels are a possible biomarker of elderly depression. J. Affect. Disord. 2019, 249, 410–417. [Google Scholar] [CrossRef]
- Walss-Bass, C.; Suchting, R.; Olvera, R.L.; Williamson, D.E. Inflammatory markers as predictors of depression and anxiety in adolescents: Statistical model building with component-wise gradient boosting. J. Affect. Disord. 2018, 234, 276–281. [Google Scholar] [CrossRef]
- Lee, K.S.; Chung, J.H.; Lee, K.H.; Shin, M.J.; Oh, B.H.; Lee, S.H.; Hong, C.H. Simultaneous measurement of 23 plasma cytokines in late-life depression. Neurol. Sci. 2009, 30, 435–438. [Google Scholar] [CrossRef]
- Leighton, S.P.; Nerurkar, L.; Krishnadas, R.; Johnman, C.; Graham, G.J.; Cavanagh, J. Chemokines in depression in health and in inflammatory illness: A systematic review and meta-analysis. Mol. Psychiatry 2018, 23, 48–58. [Google Scholar] [CrossRef]
- Omari, K.M.; John, G.R.; Sealfon, S.C.; Raine, C.S. CXC chemokine receptors on human oligodendrocytes: Implications for multiple sclerosis. Brain 2005, 128, 1003–1015. [Google Scholar] [CrossRef]
- Karim, H.; Kim, S.H.; Lapato, A.S.; Yasui, N.; Katzenellenbogen, J.A.; Tiwari-Woodruff, S.K. Increase in chemokine CXCL1 by ERβ ligand treatment is a key mediator in promoting axon myelination. Proc. Natl. Acad. Sci. USA 2018, 115, 6291–6296. [Google Scholar] [CrossRef]
- Howard, J.; Trevick, S.; Younger, D.S. Epidemiology of Multiple Sclerosis. Neurol. Clin. 2016, 34, 919–939. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.; Smith, T.W. An update on immunopathogenesis, diagnosis, and treatment of multiple sclerosis. Brain Behav. 2015, 5, e00362. [Google Scholar] [CrossRef] [PubMed]
- Kerstetter, A.E.; Padovani-Claudio, D.A.; Bai, L.; Miller, R.H. Inhibition of CXCR2 signaling promotes recovery in models of multiple sclerosis. Exp. Neurol. 2009, 220, 44–56. [Google Scholar] [CrossRef]
- Rumble, J.M.; Huber, A.K.; Krishnamoorthy, G.; Srinivasan, A.; Giles, D.A.; Zhang, X.; Wang, L.; Segal, B.M. Neutrophil-related factors as biomarkers in EAE and MS. J. Exp. Med. 2015, 212, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Dulamea, A.O. Role of Oligodendrocyte Dysfunction in Demyelination, Remyelination and Neurodegeneration in Multiple Sclerosis. Adv. Exp. Med. Biol. 2017, 958, 91–127. [Google Scholar] [PubMed]
- Burman, J.; Svensson, E.; Fransson, M.; Loskog, A.S.; Zetterberg, H.; Raininko, R.; Svenningsson, A.; Fagius, J.; Mangsbo, S.M. The cerebrospinal fluid cytokine signature of multiple sclerosis: A homogenous response that does not conform to the Th1/Th2/Th17 convention. J. Neuroimmunol. 2014, 277, 153–159. [Google Scholar] [CrossRef]
- Khaibullin, T.; Ivanova, V.; Martynova, E.; Cherepnev, G.; Khabirov, F.; Granatov, E.; Rizvanov, A.; Khaiboullina, S. Elevated Levels of Proinflammatory Cytokines in Cerebrospinal Fluid of Multiple Sclerosis Patients. Front. Immunol. 2017, 8, 531. [Google Scholar] [CrossRef]
- Lepennetier, G.; Hracsko, Z.; Unger, M.; Van Griensven, M.; Grummel, V.; Krumbholz, M.; Berthele, A.; Hemmer, B.; Kowarik, M.C. Cytokine and immune cell profiling in the cerebrospinal fluid of patients with neuro-inflammatory diseases. J. Neuroinflamm. 2019, 16, 219. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, J.; Wang, Z.; Wang, Y.; Zheng, D.; Wang, H.; Peng, Y. The CSF Levels of Neutrophil-Related Chemokines in Patients with Neuromyelitis Optica. Ann. Clin. Transl. Neurol. 2020, 7, 1245–1251. [Google Scholar] [CrossRef]
- Kostic, M.; Dzopalic, T.; Zivanovic, S.; Zivkovic, N.; Cvetanovic, A.; Stojanovic, I.; Vojinovic, S.; Marjanovic, G.; Savic, V.; Colic, M. IL-17 and glutamate excitotoxicity in the pathogenesis of multiple sclerosis. Scand. J. Immunol. 2014, 79, 181–186. [Google Scholar] [CrossRef]
- Wojkowska, D.W.; Szpakowski, P.; Ksiazek-Winiarek, D.; Leszczynski, M.; Glabinski, A. Interactions between neutrophils, Th17 cells, and chemokines during the initiation of experimental model of multiple sclerosis. Mediat. Inflamm. 2014, 2014, 590409. [Google Scholar] [CrossRef] [PubMed]
- Filipovic, R.; Jakovcevski, I.; Zecevic, N. GRO-alpha and CXCR2 in the human fetal brain and multiple sclerosis lesions. Dev. Neurosci. 2003, 25, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Carlson, T.; Kroenke, M.; Rao, P.; Lane, T.E.; Segal, B. The Th17-ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J. Exp. Med. 2008, 205, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Simmons, S.B.; Liggitt, D.; Goverman, J.M. Cytokine-regulated neutrophil recruitment is required for brain but not spinal cord inflammation during experimental autoimmune encephalomyelitis. J. Immunol. 2014, 193, 555–563. [Google Scholar] [CrossRef]
- Kang, Z.; Wang, C.; Zepp, J.; Wu, L.; Sun, K.; Zhao, J.; Chandrasekharan, U.; DiCorleto, P.E.; Trapp, B.D.; Ransohoff, R.M.; et al. Act1 mediates IL-17-induced EAE pathogenesis selectively in NG2+ glial cells. Nat. Neurosci. 2013, 16, 1401–1408. [Google Scholar] [CrossRef]
- Miller, N.M.; Wang, J.; Tan, Y.; Dittel, B.N. Anti-inflammatory mechanisms of IFN-γ studied in experimental autoimmune encephalomyelitis reveal neutrophils as a potential target in multiple sclerosis. Front. Neurosci. 2015, 9, 287. [Google Scholar] [CrossRef]
- Zehntner, S.P.; Brickman, C.; Bourbonnière, L.; Remington, L.; Caruso, M.; Owens, T. Neutrophils that infiltrate the central nervous system regulate T cell responses. J. Immunol. 2005, 174, 5124–5131. [Google Scholar] [CrossRef]
- Tirotta, E.; Kirby, L.A.; Hatch, M.N.; Lane, T.E. IFN-γ-induced apoptosis of human embryonic stem cell derived oligodendrocyte progenitor cells is restricted by CXCR2 signaling. Stem Cell Res. 2012, 9, 208–217. [Google Scholar] [CrossRef]
- Tirotta, E.; Ransohoff, R.M.; Lane, T.E. CXCR2 signaling protects oligodendrocyte progenitor cells from IFN-γ/CXCL10-mediated apoptosis. Glia 2011, 59, 1518–1528. [Google Scholar] [CrossRef]
- Grist, J.J.; Marro, B.S.; Skinner, D.D.; Syage, A.R.; Worne, C.; Doty, D.J.; Fujinami, R.S.; Lane, T.E. Induced CNS expression of CXCL1 augments neurologic disease in a murine model of multiple sclerosis via enhanced neutrophil recruitment. Eur. J. Immunol. 2018, 48, 1199–1210. [Google Scholar] [CrossRef]
- De Bondt, M.; Hellings, N.; Opdenakker, G.; Struyf, S. Neutrophils: Underestimated Players in the Pathogenesis of Multiple Sclerosis (MS). Int. J. Mol. Sci. 2020, 21, 4558. [Google Scholar] [CrossRef]
- Khaw, Y.M.; Cunningham, C.; Tierney, A.; Sivaguru, M.; Inoue, M. Neutrophil-selective deletion of Cxcr2 protects against CNS neurodegeneration in a mouse model of multiple sclerosis. J. Neuroinflamm. 2020, 17, 49. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, E.; Zanghì, A.; Romano, A.; Sciandra, M.; Palumbo, G.A.M.; Patti, F. The Neutrophil-to-Lymphocyte Ratio is Related to Disease Activity in Relapsing Remitting Multiple Sclerosis. Cells 2019, 8, 1114. [Google Scholar] [CrossRef] [PubMed]
- Hemond, C.C.; Glanz, B.I.; Bakshi, R.; Chitnis, T.; Healy, B.C. The neutrophil-to-lymphocyte and monocyte-to-lymphocyte ratios are independently associated with neurological disability and brain atrophy in multiple sclerosis. BMC Neurol. 2019, 19, 23. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Paul, F.; Weinshenker, B.G.; Levy, M.; Kim, H.J.; Wildemann, B. Neuromyelitis optica. Nat. Rev. Dis. Primers 2020, 6, 85. [Google Scholar] [CrossRef] [PubMed]
- Howe, C.L.; Kaptzan, T.; Magaña, S.M.; Ayers-Ringler, J.R.; LaFrance-Corey, R.G.; Lucchinetti, C.F. Neuromyelitis optica IgG stimulates an immunological response in rat astrocyte cultures. Glia 2014, 62, 692–708. [Google Scholar] [CrossRef]
- Jones, M.V.; Levy, M. Effect of CXCR2 Inhibition on Behavioral Outcomes and Pathology in Rat Model of Neuromyelitis Optica. J. Immunol. Res. 2018, 2018, 9034695. [Google Scholar] [CrossRef]
- Saadoun, S.; Waters, P.; MacDonald, C.; Bell, B.A.; Vincent, A.; Verkman, A.S.; Papadopoulos, M.C. Neutrophil protease inhibition reduces neuromyelitis optica-immunoglobulin G-induced damage in mouse brain. Ann. Neurol. 2012, 71, 323–333. [Google Scholar] [CrossRef]
- Zhang, Z.J.; Cao, D.L.; Zhang, X.; Ji, R.R.; Gao, Y.J. Chemokine contribution to neuropathic pain: Respective induction of CXCL1 and CXCR2 in spinal cord astrocytes and neurons. Pain 2013, 154, 2185–2197. [Google Scholar] [CrossRef]
- Cao, D.L.; Zhang, Z.J.; Xie, R.G.; Jiang, B.C.; Ji, R.R.; Gao, Y.J. Chemokine CXCL1 enhances inflammatory pain and increases NMDA receptor activity and COX-2 expression in spinal cord neurons via activation of CXCR2. Exp. Neurol. 2014, 261, 328–336. [Google Scholar] [CrossRef]
- Manjavachi, M.N.; Costa, R.; Quintão, N.L.; Calixto, J.B. The role of keratinocyte-derived chemokine (KC) on hyperalgesia caused by peripheral nerve injury in mice. Neuropharmacology 2014, 79, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Cunha, T.M.; Verri, W.A., Jr.; Silva, J.S.; Poole, S.; Cunha, F.Q.; Ferreira, S.H. A cascade of cytokines mediates mechanical inflammatory hypernociception in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 1755–1760. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Zhou, Y.; Wang, M.; Qian, C.; Wang, C.; Tang, J.; Cai, Z.; Dai, W.; Zhu, X. Pharmacological inhibition of CXCR2 alleviates neuropathic pain by inactivating microglia in a rat L5 spinal nerve ligation model. Am. J. Transl. Res. 2020, 12, 3803–3812. [Google Scholar]
- Liang, D.Y.; Shi, X.; Liu, P.; Sun, Y.; Sahbaie, P.; Li, W.W.; Yeomans, D.C.; Clark, J.D. The Chemokine Receptor CXCR2 Supports Nociceptive Sensitization after Traumatic Brain Injury. Mol. Pain 2017, 13, 1744806917730212. [Google Scholar] [CrossRef]
- Manjavachi, M.N.; Quintão, N.L.; Campos, M.M.; Deschamps, I.K.; Yunes, R.A.; Nunes, R.J.; Leal, P.C.; Calixto, J.B. The effects of the selective and non-peptide CXCR2 receptor antagonist SB225002 on acute and long-lasting models of nociception in mice. Eur. J. Pain 2010, 14, 23–31. [Google Scholar] [CrossRef]
- Moraes, T.R.; Elisei, L.S.; Malta, I.H.; Galdino, G. Participation of CXCL1 in the glial cells during neuropathic pain. Eur. J. Pharmacol. 2020, 875, 173039. [Google Scholar] [CrossRef]
- Dong, F.; Du, Y.R.; Xie, W.; Strong, J.A.; He, X.J.; Zhang, J.M. Increased function of the TRPV1 channel in small sensory neurons after local inflammation or in vitro exposure to the pro-inflammatory cytokine GRO/KC. Neurosci. Bull. 2012, 28, 155–164. [Google Scholar] [CrossRef]
- Wang, J.G.; Strong, J.A.; Xie, W.; Yang, R.H.; Coyle, D.E.; Wick, D.M.; Dorsey, E.D.; Zhang, J.M. The chemokine CXCL1/growth related oncogene increases sodium currents and neuronal excitability in small diameter sensory neurons. Mol. Pain 2008, 4, 38. [Google Scholar] [CrossRef]
- Yang, R.H.; Strong, J.A.; Zhang, J.M. NF-kappaB mediated enhancement of potassium currents by the chemokine CXCL1/growth related oncogene in small diameter rat sensory neurons. Mol. Pain 2009, 5, 26. [Google Scholar] [CrossRef]
- Qin, X.; Wan, Y.; Wang, X. CCL2 and CXCL1 trigger calcitonin gene-related peptide release by exciting primary nociceptive neurons. J. Neurosci. Res. 2005, 82, 51–62. [Google Scholar] [CrossRef]
- Cao, L.; Malon, J.T. Anti-nociceptive Role of CXCL1 in a Murine Model of Peripheral Nerve Injury-induced Neuropathic Pain. Neuroscience 2018, 372, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Harada, Y.; Zhang, J.; Imari, K.; Yamasaki, R.; Ni, J.; Wu, Z.; Yamamoto, K.; Kira, J.I.; Nakanishi, H.; Hayashi, Y. Cathepsin E in neutrophils contributes to the generation of neuropathic pain in experimental autoimmune encephalomyelitis. Pain 2019, 160, 2050–2062. [Google Scholar] [CrossRef] [PubMed]
- Rittner, H.L.; Labuz, D.; Schaefer, M.; Mousa, S.A.; Schulz, S.; Schäfer, M.; Stein, C.; Brack, A. Pain control by CXCR2 ligands through Ca2+-regulated release of opioid peptides from polymorphonuclear cells. FASEB J. 2006, 20, 2627–2629. [Google Scholar] [CrossRef] [PubMed]
- Carreira, E.U.; Carregaro, V.; Teixeira, M.M.; Moriconi, A.; Aramini, A.; Verri, W.A., Jr.; Ferreira, S.H.; Cunha, F.Q.; Cunha, T.M. Neutrophils recruited by CXCR1/2 signalling mediate post-incisional pain. Eur. J. Pain 2013, 17, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.J.; Meier, U.; Mardiguian, S.; Jiang, Y.; Littleton, E.T.; Bristow, A.; Relton, J.; Connor, T.J.; Anthony, D.C. Sickness behaviour is induced by a peripheral CXC-chemokine also expressed in multiple sclerosis and EAE. Brain Behav. Immun. 2010, 24, 738–746. [Google Scholar] [CrossRef]
- Sigurdson, C.J.; Bartz, J.C.; Glatzel, M. Cellular and Molecular Mechanisms of Prion Disease. Annu. Rev. Pathol. 2019, 14, 497–516. [Google Scholar] [CrossRef]
- Carroll, J.A.; Chesebro, B. Neuroinflammation, Microglia, and Cell-Association during Prion Disease. Viruses 2019, 11, 65. [Google Scholar] [CrossRef]
- Tribouillard-Tanvier, D.; Striebel, J.F.; Peterson, K.E.; Chesebro, B. Analysis of protein levels of 24 cytokines in scrapie agent-infected brain and glial cell cultures from mice differing in prion protein expression levels. J. Virol. 2009, 83, 11244–11253. [Google Scholar] [CrossRef]
- Tribouillard-Tanvier, D.; Race, B.; Striebel, J.F.; Carroll, J.A.; Phillips, K.; Chesebro, B. Early cytokine elevation, PrPres deposition, and gliosis in mouse scrapie: No effect on disease by deletion of cytokine genes IL-12p40 and IL-12p35. J. Virol. 2012, 86, 10377–10383. [Google Scholar] [CrossRef]
- Hennessy, E.; Griffin, É.W.; Cunningham, C. Astrocytes Are Primed by Chronic Neurodegeneration to Produce Exaggerated Chemokine and Cell Infiltration Responses to Acute Stimulation with the Cytokines IL-1β and TNF-α. J. Neurosci. 2015, 35, 8411–8422. [Google Scholar] [CrossRef]
- Miragliotta, G.; Fumarulo, R.; Fumarola, D. Inhibition of neutrophil functions by scrapie prion protein: Description of some inhibitory properties. Acta Virol. 1990, 34, 517–522. [Google Scholar] [PubMed]
- Ruzek, D.; Avšič Županc, T.; Borde, J.; Chrdle, A.; Eyer, L.; Karganova, G.; Kholodilov, I.; Knap, N.; Kozlovskaya, L.; Matveev, A.; et al. Tick-borne encephalitis in Europe and Russia: Review of pathogenesis, clinical features, therapy, and vaccines. Antivir. Res. 2019, 164, 23–51. [Google Scholar] [CrossRef] [PubMed]
- Glatz, M.; Means, T.; Haas, J.; Steere, A.C.; Müllegger, R.R. Characterization of the early local immune response to Ixodes ricinus tick bites in human skin. Exp. Dermatol. 2017, 26, 263–269. [Google Scholar] [CrossRef]
- Kazimírová, M.; Štibrániová, I. Tick salivary compounds: Their role in modulation of host defences and pathogen transmission. Front. Cell. Infect. Microbiol. 2013, 3, 43. [Google Scholar] [CrossRef]
- Déruaz, M.; Frauenschuh, A.; Alessandri, A.L.; Dias, J.M.; Coelho, F.M.; Russo, R.C.; Ferreira, B.R.; Graham, G.J.; Shaw, J.P.; Wells, T.N.; et al. Ticks produce highly selective chemokine binding proteins with antiinflammatory activity. J. Exp. Med. 2008, 205, 2019–2031. [Google Scholar] [CrossRef] [PubMed]
- Hayward, J.; Sanchez, J.; Perry, A.; Huang, C.; Rodriguez Valle, M.; Canals, M.; Payne, R.J.; Stone, M.J. Ticks from diverse genera encode chemokine-inhibitory evasin proteins. J. Biol. Chem. 2017, 292, 15670–15680. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.W.; Deruaz, M.; Lynch, C.; Davies, G.; Singh, K.; Alenazi, Y.; Eaton, J.R.O.; Kawamura, A.; Shaw, J.; Proudfoot, A.E.I.; et al. A knottin scaffold directs the CXC-chemokine-binding specificity of tick evasins. J. Biol. Chem. 2019, 294, 11199–11212. [Google Scholar] [CrossRef]
- Denisov, S.S.; Heinzmann, A.C.A.; Vajen, T.; Vries, M.H.M.; Megens, R.T.A.; Suylen, D.; Koenen, R.R.; Post, M.J.; Ippel, J.H.; Hackeng, T.M.; et al. Tick Saliva Protein Evasin-3 Allows for Visualization of Inflammation in Arteries through Interactions with CXC-Type Chemokines Deposited on Activated Endothelium. Bioconjug. Chem. 2020, 31, 948–955. [Google Scholar] [CrossRef]
- Pokorna Formanova, P.; Palus, M.; Salat, J.; Hönig, V.; Stefanik, M.; Svoboda, P.; Ruzek, D. Changes in cytokine and chemokine profiles in mouse serum and brain, and in human neural cells, upon tick-borne encephalitis virus infection. J. Neuroinflamm. 2019, 16, 205. [Google Scholar] [CrossRef]
- Grygorczuk, S.; Świerzbińska, R.; Kondrusik, M.; Dunaj, J.; Czupryna, P.; Moniuszko, A.; Siemieniako, A.; Pancewicz, S. The intrathecal expression and pathogenetic role of Th17 cytokines and CXCR2-binding chemokines in tick-borne encephalitis. J. Neuroinflamm. 2018, 15, 115. [Google Scholar] [CrossRef]
- Rabinstein, A.A. Traumatic Spinal Cord Injury. Continuum 2018, 24, 551–566. [Google Scholar] [PubMed]
- Hassanshahi, G.; Amin, M.; Shunmugavel, A.; Vazirinejad, R.; Vakilian, A.; Sanji, M.; Shamsizadeh, A.; RafatPanah, H.; Poor, N.M.; Moosavi, S.R.; et al. Temporal expression profile of CXC chemokines in serum of patients with spinal cord injury. Neurochem. Int. 2013, 63, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Yates, A.G.; Jogia, T.; Gillespie, E.R.; Couch, Y.; Ruitenberg, M.J.; Anthony, D.C. Acute IL-1RA treatment suppresses the peripheral and central inflammatory response to spinal cord injury. J. Neuroinflamm. 2021, 18, 15. [Google Scholar] [CrossRef] [PubMed]
- Ellman, D.G.; Lund, M.C.; Nissen, M.; Nielsen, P.S.; Sørensen, C.; Lester, E.B.; Thougaard, E.; Jørgensen, L.H.; Nedospasov, S.A.; Andersen, D.C.; et al. Conditional Ablation of Myeloid TNF Improves Functional Outcome and Decreases Lesion Size after Spinal Cord Injury in Mice. Cells 2020, 9, 2407. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Kimura, A.; Kimura, H.; Ohmori, T.; Takahashi, M.; Takeshita, K. Deletion of inflammasome adaptor protein ASC enhances functional recovery after spinal cord injury in mice. J. Orthop. Sci. 2021, 26, 487–493. [Google Scholar] [CrossRef]
- Tonai, T.; Shiba, K.; Taketani, Y.; Ohmoto, Y.; Murata, K.; Muraguchi, M.; Ohsaki, H.; Takeda, E.; Nishisho, T. A neutrophil elastase inhibitor (ONO-5046) reduces neurologic damage after spinal cord injury in rats. J. Neurochem. 2001, 78, 1064–1072. [Google Scholar] [CrossRef]
- Habarugira, G.; Suen, W.W.; Hobson-Peters, J.; Hall, R.A.; Bielefeldt-Ohmann, H. West Nile Virus: An Update on Pathobiology, Epidemiology, Diagnostics, Control and “One Health” Implications. Pathogens 2020, 9, 589. [Google Scholar] [CrossRef]
- Garcia, M.; Alout, H.; Diop, F.; Damour, A.; Bengue, M.; Weill, M.; Missé, D.; Lévêque, N.; Bodet, C. Innate Immune Response of Primary Human Keratinocytes to West Nile Virus Infection and Its Modulation by Mosquito Saliva. Front. Cell. Infect. Microbiol. 2018, 8, 387. [Google Scholar] [CrossRef]
- Bai, F.; Kong, K.F.; Dai, J.; Qian, F.; Zhang, L.; Brown, C.R.; Fikrig, E.; Montgomery, R.R. A paradoxical role for neutrophils in the pathogenesis of West Nile virus. J. Infect. Dis. 2010, 202, 1804–1812. [Google Scholar] [CrossRef]
- Paul, A.M.; Acharya, D.; Duty, L.; Thompson, E.A.; Le, L.; Stokic, D.S.; Leis, A.A.; Bai, F. Osteopontin facilitates West Nile virus neuroinvasion via neutrophil “Trojan horse” transport. Sci. Rep. 2017, 7, 4722. [Google Scholar] [CrossRef]
- Cheeran, M.C.; Hu, S.; Sheng, W.S.; Rashid, A.; Peterson, P.K.; Lokensgard, J.R. Differential responses of human brain cells to West Nile virus infection. J. Neurovirol. 2005, 11, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Hunsperger, E.; Roehrig, J.T. Characterization of West Nile viral replication and maturation in peripheral neurons in culture. J. Neurovirol. 2005, 11, 11–22. [Google Scholar] [CrossRef]
- Quick, E.D.; Leser, J.S.; Clarke, P.; Tyler, K.L. Activation of intrinsic immune responses and microglial phagocytosis in an ex vivo spinal cord slice culture model of West Nile virus infection. J. Virol. 2014, 88, 13005–13014. [Google Scholar] [CrossRef] [PubMed]
- Bréhin, A.C.; Mouriès, J.; Frenkiel, M.P.; Dadaglio, G.; Desprès, P.; Lafon, M.; Couderc, T. Dynamics of immune cell recruitment during West Nile encephalitis and identification of a new CD19+B220−BST-2+ leukocyte population. J. Immunol. 2008, 180, 6760–6767. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korbecki, J.; Gąssowska-Dobrowolska, M.; Wójcik, J.; Szatkowska, I.; Barczak, K.; Chlubek, M.; Baranowska-Bosiacka, I. The Importance of CXCL1 in Physiology and Noncancerous Diseases of Bone, Bone Marrow, Muscle and the Nervous System. Int. J. Mol. Sci. 2022, 23, 4205. https://doi.org/10.3390/ijms23084205
Korbecki J, Gąssowska-Dobrowolska M, Wójcik J, Szatkowska I, Barczak K, Chlubek M, Baranowska-Bosiacka I. The Importance of CXCL1 in Physiology and Noncancerous Diseases of Bone, Bone Marrow, Muscle and the Nervous System. International Journal of Molecular Sciences. 2022; 23(8):4205. https://doi.org/10.3390/ijms23084205
Chicago/Turabian StyleKorbecki, Jan, Magdalena Gąssowska-Dobrowolska, Jerzy Wójcik, Iwona Szatkowska, Katarzyna Barczak, Mikołaj Chlubek, and Irena Baranowska-Bosiacka. 2022. "The Importance of CXCL1 in Physiology and Noncancerous Diseases of Bone, Bone Marrow, Muscle and the Nervous System" International Journal of Molecular Sciences 23, no. 8: 4205. https://doi.org/10.3390/ijms23084205
APA StyleKorbecki, J., Gąssowska-Dobrowolska, M., Wójcik, J., Szatkowska, I., Barczak, K., Chlubek, M., & Baranowska-Bosiacka, I. (2022). The Importance of CXCL1 in Physiology and Noncancerous Diseases of Bone, Bone Marrow, Muscle and the Nervous System. International Journal of Molecular Sciences, 23(8), 4205. https://doi.org/10.3390/ijms23084205