
SARS-CoV-2 Pandemic Tracing in Italy Highlights Lineages with Mutational Burden in Growing Subsets

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results
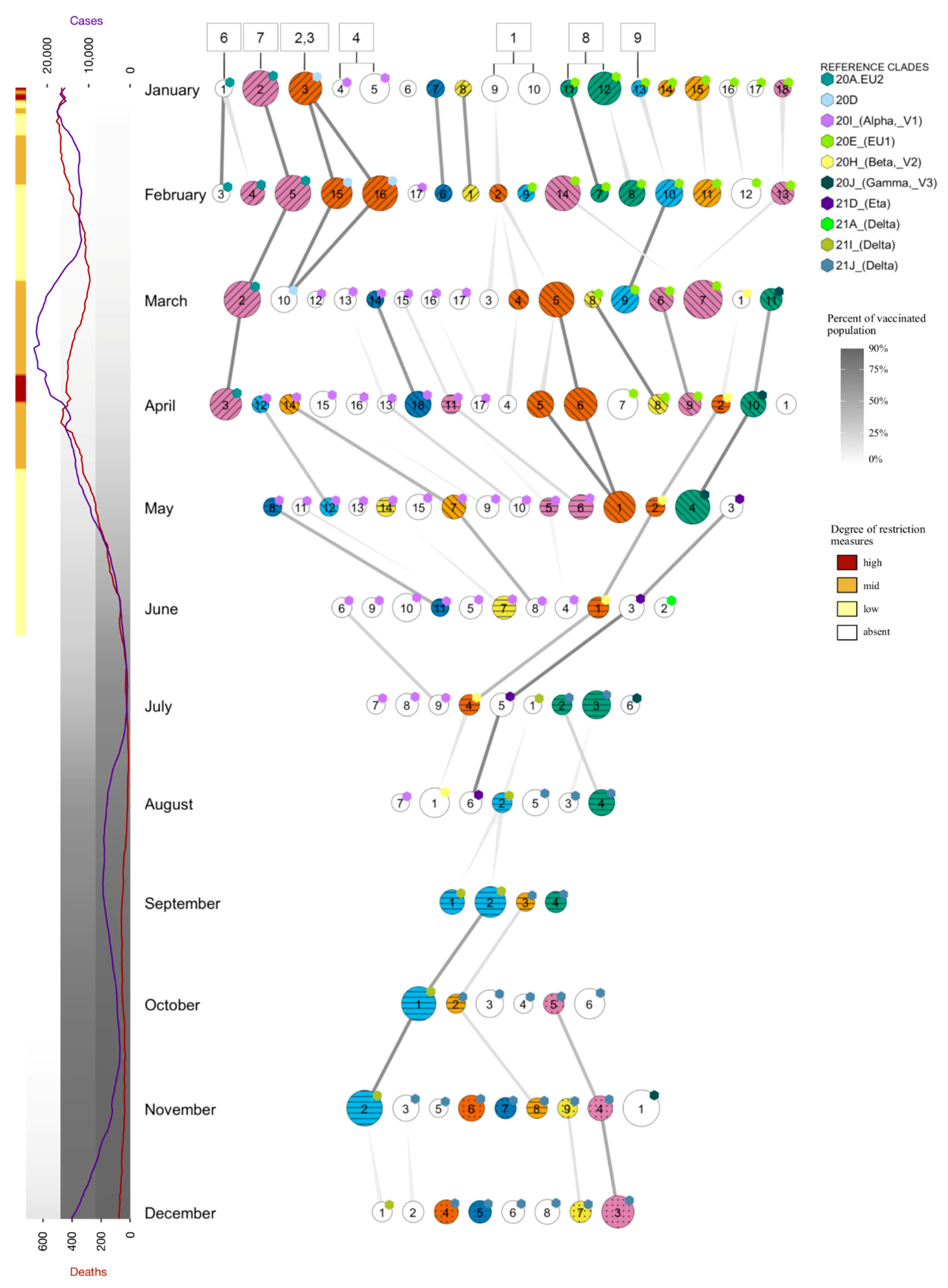
2.1. Epidemic Trend in Italy during the First Year
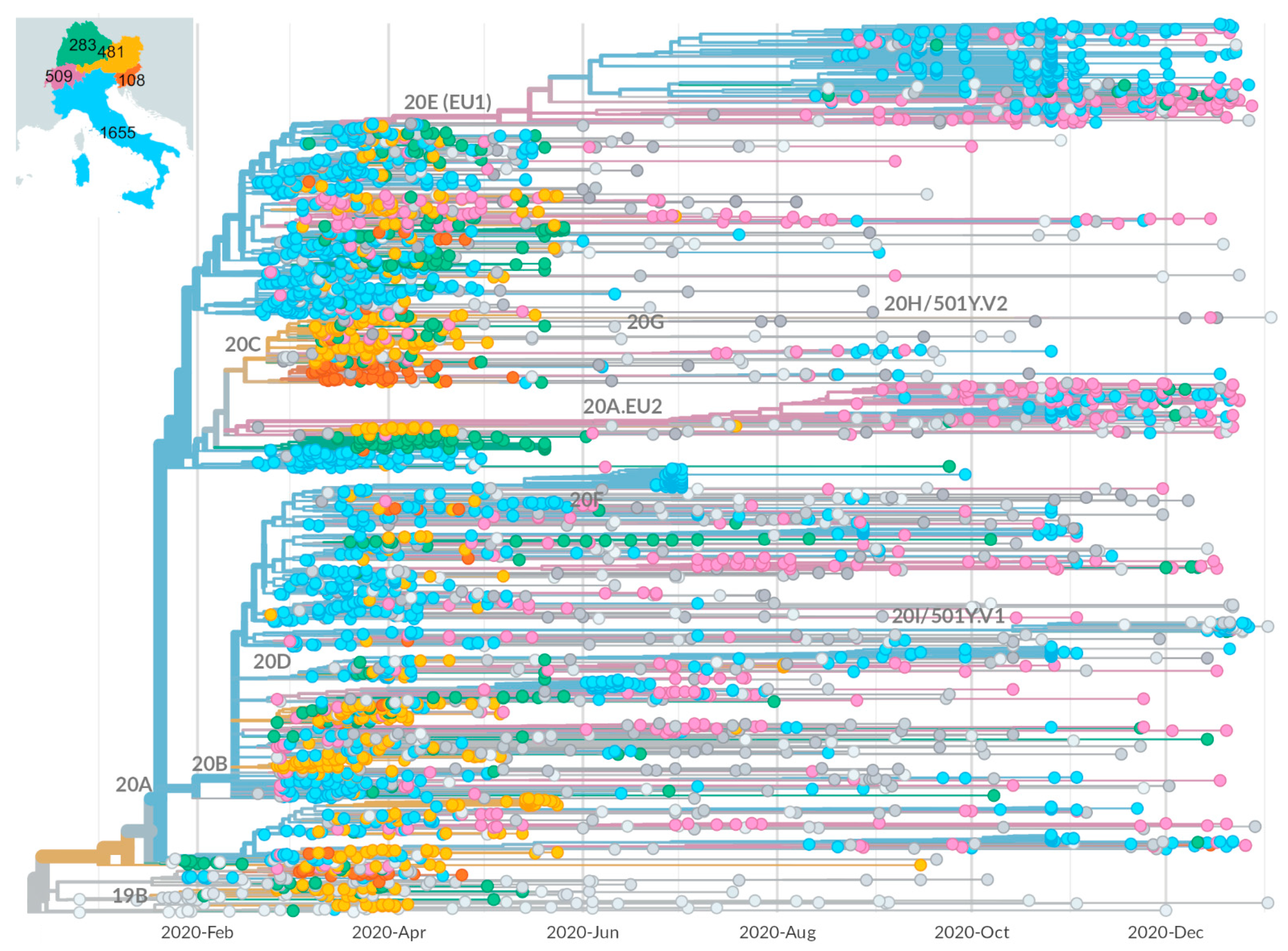
2.2. Identification of Growing Virus Subsets in Italy
2.3. Mutational Characterization of Growing Subsets
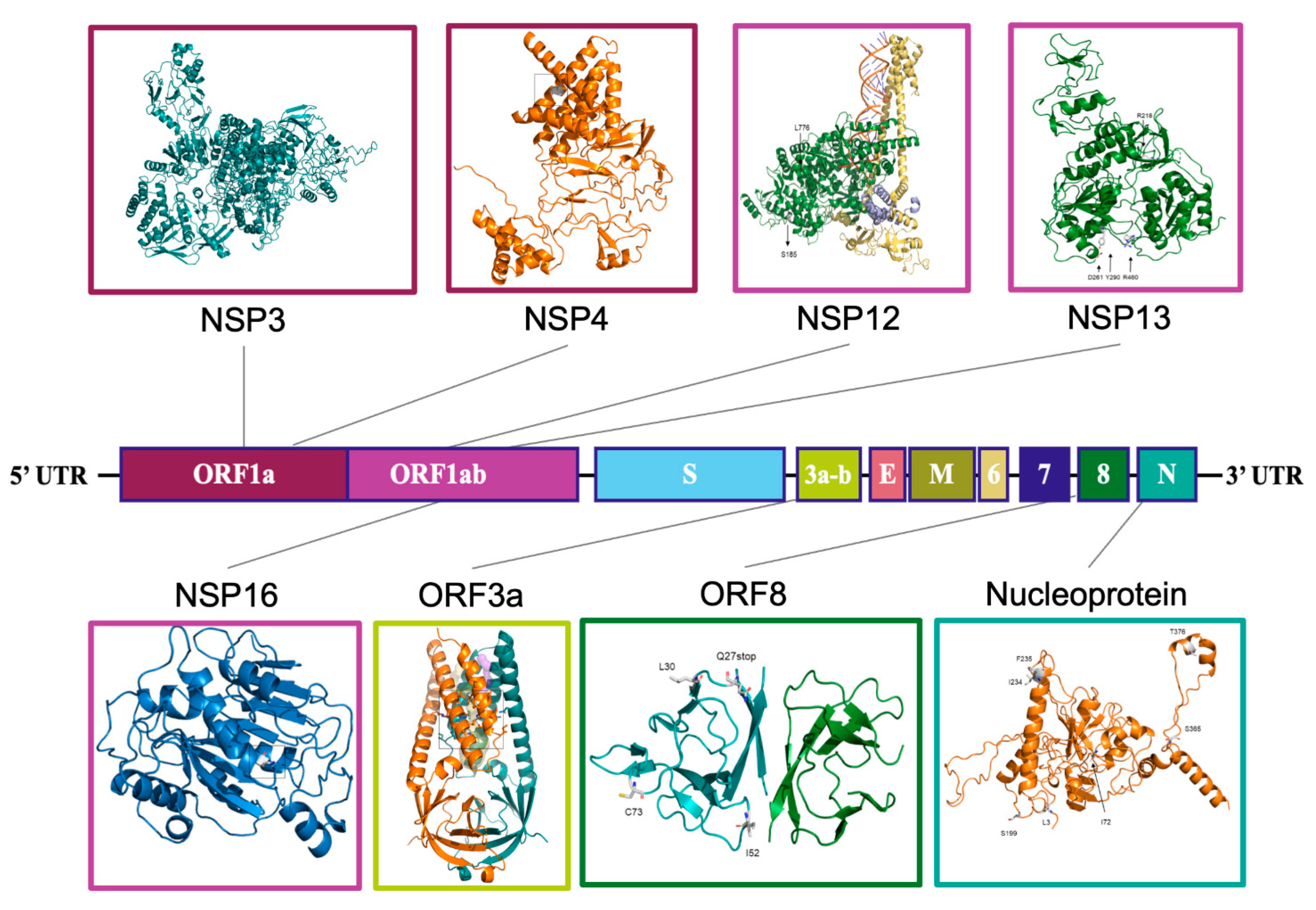
2.4. Structural Analysis
2.5. Tracing of Variants
3. Discussion
4. Materials and Methods
4.1. Sequence Datasets and Phylodynamic Analysis
4.2. Identification of Growing Virus Subsets
4.3. Detection of Mutated Sites
4.4. Impact of Mutations on Protein Conformation and Stability
4.5. Subset Relationships with Nextstrain Clades/Pango Lineages
4.6. Subset Tracing
- (a)
- full correspondence, if the product is 0.8 or more;
- (b)
- partial correspondence, if the product is less than 0.8, but at least one of the two ratios is more than 0.8;
- (c)
- no relationship, in the other cases.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Code Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kesheh, M.M.; Hosseini, P.; Soltani, S.; Zandi, M. An Overview on the Seven Pathogenic Human Coronaviruses. Rev. Med. Virol. 2022, 32, e2282. [Google Scholar] [CrossRef]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef]
- Singh, D.; Yi, S.V. On the Origin and Evolution of SARS-CoV-2. Exp. Mol. Med. 2021, 53, 537–547. [Google Scholar] [CrossRef]
- Sola, I.; Almazán, F.; Zúñiga, S.; Enjuanes, L. Continuous and Discontinuous RNA Synthesis in Coronaviruses. Annu. Rev. Virol. 2015, 2, 265–288. [Google Scholar] [CrossRef] [Green Version]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus Biology and Replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Ferrucci, V.; Kong, D.-Y.; Asadzadeh, F.; Marrone, L.; Boccia, A.; Siciliano, R.; Criscuolo, G.; Anastasio, C.; Quarantelli, F.; Comegna, M.; et al. Long-Chain Polyphosphates Impair SARS-CoV-2 Infection and Replication. Sci. Signal. 2021, 14, 690. [Google Scholar] [CrossRef]
- Zollo, M.; Ferrucci, V.; Izzo, B.; Quarantelli, F.; Domenico, C.D.; Comegna, M.; Paolillo, C.; Amato, F.; Siciliano, R.; Castaldo, G.; et al. SARS-CoV-2 Subgenomic N (sgN) Transcripts in Oro-Nasopharyngeal Swabs Correlate with the Highest Viral Load, as Evaluated by Five Different Molecular Methods. Diagn. Basel Switz. 2021, 11, 288. [Google Scholar] [CrossRef]
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.-L. Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Mol. Cell 2020, 79, 710–727. [Google Scholar] [CrossRef]
- Rochman, N.D.; Wolf, Y.I.; Faure, G.; Mutz, P.; Zhang, F.; Koonin, E.V. Ongoing Global and Regional Adaptive Evolution of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2021, 118, e2104241118. [Google Scholar] [CrossRef]
- Lai, A.; Bergna, A.; Caucci, S.; Clementi, N.; Vicenti, I.; Dragoni, F.; Cattelan, A.M.; Menzo, S.; Pan, A.; Callegaro, A.; et al. Molecular Tracing of SARS-CoV-2 in Italy in the First Three Months of the Epidemic. Viruses 2020, 12, 798. [Google Scholar] [CrossRef]
- Di Giallonardo, F.; Duchene, S.; Puglia, I.; Curini, V.; Profeta, F.; Cammà, C.; Marcacci, M.; Calistri, P.; Holmes, E.C.; Lorusso, A. Genomic Epidemiology of the First Wave of SARS-CoV-2 in Italy. Viruses 2020, 12, 1438. [Google Scholar] [CrossRef]
- Alteri, C.; Cento, V.; Piralla, A.; Costabile, V.; Tallarita, M.; Colagrossi, L.; Renica, S.; Giardina, F.; Novazzi, F.; Gaiarsa, S.; et al. Genomic Epidemiology of SARS-CoV-2 Reveals Multiple Lineages and Early Spread of SARS-CoV-2 Infections in Lombardy, Italy. Nat. Commun. 2021, 12, 434. [Google Scholar] [CrossRef]
- Faggioni, G.; Stefanelli, P.; Giordani, F.; Fillo, S.; Anselmo, A.; Vera Fain, V.; Fortunato, A.; Petralito, G.; Molinari, F.; Lo Presti, A.; et al. Identification and Characterization of SARS-CoV-2 Clusters in the EU/EEA in the First Pandemic Wave: Additional Elements to Trace the Route of the Virus. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2021, 96, 105108. [Google Scholar] [CrossRef]
- Giovanetti, M.; Cella, E.; Benedetti, F.; Rife Magalis, B.; Fonseca, V.; Fabris, S.; Campisi, G.; Ciccozzi, A.; Angeletti, S.; Borsetti, A.; et al. SARS-CoV-2 Shifting Transmission Dynamics and Hidden Reservoirs Potentially Limit Efficacy of Public Health Interventions in Italy. Commun. Biol. 2021, 4, 489. [Google Scholar] [CrossRef]
- Di Giallonardo, F.; Puglia, I.; Curini, V.; Cammà, C.; Mangone, I.; Calistri, P.; Cobbin, J.C.A.; Holmes, E.C.; Lorusso, A. Emergence and Spread of SARS-CoV-2 Lineages B.1.1.7 and P.1 in Italy. Viruses 2021, 13, 794. [Google Scholar] [CrossRef]
- Lai, A.; Bergna, A.; Menzo, S.; Zehender, G.; Caucci, S.; Ghisetti, V.; Rizzo, F.; Maggi, F.; Cerutti, F.; Giurato, G.; et al. Circulating SARS-CoV-2 Variants in Italy, October 2020–March 2021. Virol. J. 2021, 18, 168. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinforma 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, Disease and Diplomacy: GISAID’s Innovative Contribution to Global Health. Glob. Chall. Hoboken NJ 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Shu, Y.; McCauley, J. GISAID: Global Initiative on Sharing All Influenza Data–from Vision to Reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Rangarajan, E.S.; Izard, T.; Farzan, M.; Choe, H. The D614G Mutation in the SARS-CoV-2 Spike Protein Reduces S1 Shedding and Increases Infectivity. BioRxiv 2020. [Google Scholar] [CrossRef]
- Groves, D.C.; Rowland-Jones, S.L.; Angyal, A. The D614G Mutations in the SARS-CoV-2 Spike Protein: Implications for Viral Infectivity, Disease Severity and Vaccine Design. Biochem. Biophys. Res. Commun. 2021, 538, 104–107. [Google Scholar] [CrossRef]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.D.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D.; et al. Spread of a SARS-CoV-2 Variant through Europe in the Summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef]
- Transmission of SARS-CoV-2 on Mink Farms between Humans and Mink and Back to Humans. Available online: https://www.science.org/doi/10.1126/science.abe5901 (accessed on 28 March 2022).
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated Transmissibility and Impact of SARS-CoV-2 Lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef]
- Latif, A.A.L.; Mullen, J.L.; Alkuzweny, M.; Tsueng, G.; Cano, M.; Haag, E.; Zhou, J.; Zeller, M.; Hufbauer, E.; Matteson, N.; et al. B.1.258 Lineage Report. Available online: https://outbreak.info/situation-reports?pango=B.1.258 (accessed on 15 December 2021).
- Latif, A.A.L.; Mullen, J.L.; Alkuzweny, M.; Tsueng, G.; Cano, M.; Haag, E.; Zhou, J.; Zeller, M.; Hufbauer, E.; Matteson, N.; et al. B.1.1.39 Lineage Report. Available online: https://outbreak.info/situation-reports?pango=B.1.1.39 (accessed on 15 December 2021).
- Latif, A.A.L.; Mullen, J.L.; Alkuzweny, M.; Tsueng, G.; Cano, M.; Haag, E.; Zhou, J.; Zeller, M.; Hufbauer, E.; Matteson, N.; et al. B.1.1.177.33 Lineage Report. Available online: https://outbreak.info/situation-reports?pango=B.1.1.177.33 (accessed on 15 December 2021).
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Thomson, E.C.; Rosen, L.E.; Shepherd, J.G.; Spreafico, R.; da Silva Filipe, A.; Wojcechowskyj, J.A.; Davis, C.; Piccoli, L.; Pascall, D.J.; Dillen, J.; et al. Circulating SARS-CoV-2 Spike N439K Variants Maintain Fitness While Evading Antibody-Mediated Immunity. Cell 2021, 184, 1171–1187.e20. [Google Scholar] [CrossRef]
- CoVariants: 20I (Alpha, V1). Available online: https://covariants.org/variants/20I.Alpha.V1 (accessed on 29 March 2022).
- Chen, J.; Wang, R.; Wang, M.; Wei, G.-W. Mutations Strengthened SARS-CoV-2 Infectivity. J. Mol. Biol. 2020, 432, 5212–5226. [Google Scholar] [CrossRef]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.-M.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 Neutralizing Antibody Structures Inform Therapeutic Strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef]
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.; Muecksch, F.; Rutkowska, M.; Hoffmann, H.-H.; Michailidis, E.; et al. Escape from Neutralizing Antibodies by SARS-CoV-2 Spike Protein Variants. eLife 2020, 9, e61312. [Google Scholar] [CrossRef]
- Gaebler, C.; Wang, Z.; Lorenzi, J.C.C.; Muecksch, F.; Finkin, S.; Tokuyama, M.; Cho, A.; Jankovic, M.; Schaefer-Babajew, D.; Oliveira, T.Y.; et al. Evolution of Antibody Immunity to SARS-CoV-2. Nature 2021, 591, 639–644. [Google Scholar] [CrossRef]
- Liu, Z.; VanBlargan, L.A.; Bloyet, L.-M.; Rothlauf, P.W.; Chen, R.E.; Stumpf, S.; Zhao, H.; Errico, J.M.; Theel, E.S.; Liebeskind, M.J.; et al. Landscape Analysis of Escape Variants Identifies SARS-CoV-2 Spike Mutations That Attenuate Monoclonal and Serum Antibody Neutralization. BioRxiv 2021. [Google Scholar] [CrossRef]
- Latif, A.A.L.; Mullen, J.L.; Alkuzweny, M.; Tsueng, G.; Cano, M.; Haag, E.; Zhou, J.; Zeller, M.; Hufbauer, E.; Matteson, N.; et al. B.1.258.14 Lineage Report. Available online: https://outbreak.info/situation-reports?pango=B.1.258.14 (accessed on 15 December 2021).
- Latif, A.A.L.; Mullen, J.L.; Alkuzweny, M.; Tsueng, G.; Cano, M.; Haag, E.; Zhou, J.; Zeller, M.; Hufbauer, E.; Matteson, N.; et al. B.1.160 Lineage Report. Available online: https://outbreak.info/situation-reports?pango=B.1.160 (accessed on 15 December 2021).
- Latif, A.A.L.; Mullen, J.L.; Alkuzweny, M.; Tsueng, G.; Cano, M.; Haag, E.; Zhou, J.; Zeller, M.; Hufbauer, E.; Matteson, N.; et al. C.18 Lineage Report. Available online: https://outbreak.info/situation-reports?pango=C.18 (accessed on 15 December 2021).
- Brejová, B.; Boršová, K.; Hodorová, V.; Čabanová, V.; Reizigová, L.; Paul, E.D.; Čekan, P.; Klempa, B.; Nosek, J.; Vinař, T. A SARS-CoV-2 Mutant from B.1.258 Lineage with ∆H69/∆V70 Deletion in the Spike Protein Circulating in Central Europe in the Fall 2020. Virus Genes 2021, 57, 556–560. [Google Scholar] [CrossRef]
- Latif, A.A.L.; Mullen, J.L.; Alkuzweny, M.; Tsueng, G.; Cano, M.; Haag, E.; Zhou, J.; Zeller, M.; Hufbauer, E.; Matteson, N.; et al. B.1.177.75 Lineage Report. Available online: https://outbreak.info/situation-reports?pango=B.1.177.75 (accessed on 15 December 2021).
- Latif, A.A.L.; Mullen, J.L.; Alkuzweny, M.; Tsueng, G.; Cano, M.; Haag, E.; Zhou, J.; Zeller, M.; Hufbauer, E.; Matteson, N.; et al. B.1.177.83 Lineage Report. Available online: https://outbreak.info/situation-reports?pango=B.1.177.83 (accessed on 15 December 2021).
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 Variant of Concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Naveca, F.G.; Nascimento, V.; de Souza, V.C.; de Lima Corado, A.; Nascimento, F.; Silva, G.; Costa, Á.; Duarte, D.; Pessoa, K.; Mejía, M.; et al. COVID-19 in Amazonas, Brazil, Was Driven by the Persistence of Endemic Lineages and P.1 Emergence. Nat. Med. 2021, 27, 1230–1238. [Google Scholar] [CrossRef]
- O’Toole, Á.; Hill, V.; Pybus, O.; Watts, A.; Bogoch, I.; Khan, K.; Messina, J.; The COVID; Genomics UK. Tracking the International Spread of SARS-CoV-2 Lineages B.1.1.7 and B.1.351/501Y-V2 with Grinch [Version 2; Peer Review: 3 Approved]. Wellcome Open Res. 2021, 6, 121. [Google Scholar] [CrossRef]
- Latif, A.A.L.; Mullen, J.L.; Alkuzweny, M.; Tsueng, G.; Cano, M.; Haag, E.; Zhou, J.; Zeller, M.; Hufbauer, E.; Matteson, N.; et al. B.1.617.2 Lineage Report. Available online: https://outbreak.info/situation-reports?pango=B.1.617.2 (accessed on 15 December 2021).
- COVID-19 Italian Data; Presidenza del Consiglio dei Ministri-Dipartimento della Protezione Civile. Available online: https://github.com/pcm-dpc/COVID-19 (accessed on 22 March 2022).
- COVID-19 Opendata Vaccini; Developers Italia. Available online: https://github.com/italia/covid19-opendata-vaccini (accessed on 23 March 2022).
- Ragonnet-Cronin, M.; Hodcroft, E.; Hué, S.; Fearnhill, E.; Delpech, V.; Brown, A.J.L.; Lycett, S. Automated Analysis of Phylogenetic Clusters. BMC Bioinform. 2013, 14, 317. [Google Scholar] [CrossRef] [Green Version]
- Prosperi, M.C.F.; Ciccozzi, M.; Fanti, I.; Saladini, F.; Pecorari, M.; Borghi, V.; Di Giambenedetto, S.; Bruzzone, B.; Capetti, A.; Vivarelli, A.; et al. A Novel Methodology for Large-Scale Phylogeny Partition. Nat. Commun. 2011, 2, 321. [Google Scholar] [CrossRef] [Green Version]
- Capozzi, L.; Bianco, A.; Del Sambro, L.; Simone, D.; Lippolis, A.; Notarnicola, M.; Pesole, G.; Pace, L.; Galante, D.; Parisi, A. Genomic Surveillance of Circulating SARS-CoV-2 in South East Italy: A One-Year Retrospective Genetic Study. Viruses 2021, 13, 731. [Google Scholar] [CrossRef]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef]
- Meng, B.; Kemp, S.A.; Papa, G.; Datir, R.; Ferreira, I.A.T.M.; Marelli, S.; Harvey, W.T.; Lytras, S.; Mohamed, A.; Gallo, G.; et al. Recurrent Emergence of SARS-CoV-2 Spike Deletion H69/V70 and Its Role in the Alpha Variant B.1.1.7. Cell Rep. 2021, 35, 109292. [Google Scholar] [CrossRef]
- McCarthy, K.R.; Rennick, L.J.; Nambulli, S.; Robinson-McCarthy, L.R.; Bain, W.G.; Haidar, G.; Duprex, W.P. Recurrent Deletions in the SARS-CoV-2 Spike Glycoprotein Drive Antibody Escape. Science 2021, 371, 1139–1142. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in Adults with Severe COVID-19: A Randomised, Double-Blind, Placebo-Controlled, Multicentre Trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Ferrucci, V.; De Antonellis, P.; Quarantelli, F.; Asadzadeh, F.; Bibbò, F.; Siciliano, R.; Sorice, C.; Pisano, I.; Izzo, B.; Di Domenico, C.; et al. Loss of Detection of SgN Precedes Viral Abridged Replication in COVID19-Affected Patients—A Target for SARS-CoV-2 Propagation. Int. J. Mol. Sci. 2022, 23, 1941. [Google Scholar] [CrossRef]
- Yang, M.; He, S.; Chen, X.; Huang, Z.; Zhou, Z.; Zhou, Z.; Chen, Q.; Chen, S.; Kang, S. Structural Insight Into the SARS-CoV-2 Nucleocapsid Protein C-Terminal Domain Reveals a Novel Recognition Mechanism for Viral Transcriptional Regulatory Sequences. Front. Chem. 2020, 8, 624765. [Google Scholar] [CrossRef]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 Mutation Hot Spots Include a Novel RNA-Dependent-RNA Polymerase Variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef] [Green Version]
- Latif, A.A.L.; Mullen, J.L.; Alkuzweny, M.; Tsueng, G.; Cano, M.; Haag, E.; Zhou, J.; Zeller, M.; Hufbauer, E.; Matteson, N.; et al. Italy Variant Report. Available online: https://outbreak.info/location-reports?loc=ITA (accessed on 15 December 2021).
- NCOV, Nextstrain Build for Novel Coronavirus SARS-CoV-2. Available online: https://github.com/nextstrain/ncov (accessed on 29 March 2022).
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-Likelihood Phylodynamic Analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef]
- Auspice, Web App for Visualizing Pathogen Evolution. Available online: https://github.com/nextstrain/auspice (accessed on 29 March 2022).
- Nextrain Exclusion List. Available online: https://github.com/nextstrain/ncov/blob/c52debc343e0792e3600963dc13fe94e57315c16/defaults/exclude.txt (accessed on 29 March 2022).
- European Centre for Disease Prevention and Control. Available online: https://www.ecdc.europa.eu/en (accessed on 29 March 2022).
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade Assignment, Mutation Calling and Quality Control for Viral Genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological Macromolecular Structures Enabling Research and Education in Fundamental Biology, Biomedicine, Biotechnology and Energy. Nucleic Acids Res. 2019, 47, D464–D474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein Structure and Function Prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger, LLC. The PyMOL Molecular Graphics System; Version 1.8; Schrödinger, LLC.: New York, NY, USA, 2015. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, C.H.; Pires, D.E.; Ascher, D.B. DynaMut: Predicting the Impact of Mutations on Protein Conformation, Flexibility and Stability. Nucleic Acids Res. 2018, 46, W350–W355. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Subset | Date | Internal Subsets | # Viral Seqs | Score | Skew | Origin | # Sources | Sex | Clade | Related Pango Lineage | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Italy | Others | Div. | Cent. | F | M | U | Parent | Is | Name | Rel. | |||||||
| 1 | 4/2020 | 61 | 11.32 | −0.84 | 41 (67%) | 20 (33%) | 6 | 10 | 18 | 31 | 12 | 20A | B.1.258 | ovr | |||
| 2−3 | 2 | 6/2020 | 47 | 7.21 | −0.68 | 47 (100%) | 0 (0%) | 3 | 5 | 23 | 24 | 0 | 20D | C.18 | not | ||
| 3 | 4/2020 | 2 | 91 | 14.33 | −0.93 | 56 (62%) | 35 (38%) | 8 | 12 | 34 | 34 | 23 | 20D | B.1.1.1 | par | ||
| 4 | 10/2020 | 43 | 54.79 | −3.07 | 25 (58%) | 18 (42%) | 6 | 12 | 18 | 16 | 9 | 20B | 20I/501Y.V1 | B.1.1.7 | ovr | ||
| 5 | 3/2020 | 45 | 6.65 | −0.66 | 4 (9%) | 41 (91%) | 3 | 3 | 7 | 6 | 32 | 20B | B.1.1.39 | ovr | |||
| 6−7 | 6 | 4/2020 | 31 | 7.77 | −0.77 | 20 (65%) | 11 (35%) | 4 | 7 | 6 | 17 | 8 | 20A.EU2 | B.1.160 | par | ||
| 7 | 6/2020 | 6 | 164 | 8.66 | −0.56 | 68 (41%) | 96 (59%) | 6 | 12 | 29 | 56 | 79 | 20A | 20A.EU2 | B.1.160 | ovr | |
| 8 | 6/2020 | 52 | 20.53 | −1.33 | 27 (52%) | 25 (48%) | 5 | 8 | 19 | 13 | 20 | 20E (EU1) | B.1.177 | par | |||
| 9 | 7/2020 | 33 | 5.81 | −0.66 | 22 (67%) | 11 (33%) | 5 | 6 | 9 | 16 | 8 | 20E (EU1) | B.1.177 | par | |||
| 10 | 8/2020 | 49 | 13.77 | −1 | 47 (96%) | 2 (4%) | 4 | 5 | 13 | 34 | 2 | 20E (EU1) | B.1.177.33 | not | |||
| Variant | Variant (ORF1ab Peptide) | Nucleotide | Subset | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |||
| 5UTR: C241T | - | 241 | C | C | C | C | C | C | C | C | C | C |
| ORF1ab: V60V | leader: V60V | 445 | C | C | C | |||||||
| ORF1ab: S216S | nsp2: S36S | 913 | B | |||||||||
| ORF1ab: F924F | nsp3: F106F | 3037 | C | C | C | C | C | C | C | C | C | C |
| ORF1ab: T1001I | nsp3: T183I | 3267 | B | |||||||||
| ORF1ab: T1246I | nsp3: T428I | 4002 | C | C | ||||||||
| ORF1ab: T1426T | nsp3: T608T | 4543 | C | B | ||||||||
| ORF1ab: Y1635Y | nsp3: Y817Y | 5170 | B | |||||||||
| ORF1ab: A1708D | nsp3: A890D | 5388 | B | |||||||||
| ORF1ab: T1788T | nsp3: T970T | 5629 | C | B | ||||||||
| ORF1ab: F1907F | nsp3: F1089F | 5986 | B | |||||||||
| ORF1ab: T2007T | nsp3: T1189T | 6286 | C | C | C | |||||||
| ORF1ab: I2230T | nsp3: I1412T | 6954 | B | |||||||||
| ORF1ab: T2274I | nsp3: T1456I | 7086 | B | |||||||||
| ORF1ab: I2501T | nsp3: I1683T | 7767 | B | |||||||||
| ORF1ab: Y2594Y | nsp3: Y1776Y | 8047 | B | |||||||||
| ORF1ab: S2625S | nsp3: S1807S | 8140 | b | |||||||||
| ORF1ab: S2839S | nsp4: S76S | 8782 | ||||||||||
| ORF1ab: V2955V | nsp4: V192V | 9130 | B | |||||||||
| ORF1ab: M3087I | nsp4: M324I | 9526 | C | B | ||||||||
| ORF1ab: G3278S | 3C-like proteinase: G15S | 10,097 | C | C | ||||||||
| ORF1ab: F3329F | 3C-like proteinase: F66F | 10,252 | B | b | ||||||||
| ORF1ab: A3623S | nsp6: A54S | 11,132 | B | |||||||||
| ORF1ab: S3675- | nsp6: S106- | 11,288–11,290 | B | |||||||||
| ORF1ab: G3676- | nsp6: G107- | 11,291–11,293 | B | |||||||||
| ORF1ab: F3677- | nsp6: F108- | 11,294–11,296 | B | |||||||||
| ORF1ab: Y3744Y | nsp6: Y175Y | 11,497 | C | C | ||||||||
| ORF1ab: D3897D | nsp7: D38D | 11,956 | B | b | ||||||||
| ORF1ab: Y4424Y | RdRp: Y32Y | 13,536 | C | C | ||||||||
| ORF1ab: A4577S | RdRp: A185S | 13,993 | C | B | ||||||||
| ORF1ab: P4715L | RdRp: P323L | 14,408 | C | C | C | C | C | C | C | C | C | C |
| ORF1ab: P4804P | RdRp: P412P | 14,676 | B | |||||||||
| ORF1ab: H5005H | RdRp: H613H | 15,279 | B | |||||||||
| ORF1ab: V5168L | RdRp: V776L | 15,766 | C | B | ||||||||
| ORF1ab: L5283L | RdRp: L891L | 16,111 | b | |||||||||
| ORF1ab: T5304T | RdRp: T912T | 16,176 | B | |||||||||
| ORF1ab: K5542R | helicase: K218R | 16,889 | C | B | ||||||||
| ORF1ab: E5585D | helicase: E261D | 17,019 | C | B | ||||||||
| ORF1ab: H5614Y | helicase: H290Y | 17,104 | B | |||||||||
| ORF1ab: K5784R | helicase: K460R | 17,615 | B | |||||||||
| ORF1ab: L6205L | 3′-to-5′ exon.: L280L | 18,877 | C | B | ||||||||
| ORF1ab: L6668L | endoRNAse: L216L | 20,268 | B | |||||||||
| ORF1ab: K6958K | 2′-o-MT: K160K | 21,138 | B | b | ||||||||
| ORF1ab: S6964A | 2′-o-MT: S166A | 21,154 | B | b | ||||||||
| ORF1ab: A6997A | 2′-o-MT: A199A | 21,255 | C | C | C | |||||||
| S: H69- | - | 21,767–21,769 | B | |||||||||
| S: V70- | - | 21,770 | B | |||||||||
| S: Y144- | - | 21,992–21,993 | B | |||||||||
| S: A222V | - | 22,227 | C | C | C | |||||||
| S: N439K | - | 22,879 | B | |||||||||
| S: S477N | - | 22,992 | C | B | ||||||||
| S: N501Y | - | 23,063 | B | |||||||||
| S: A570D | - | 23,271 | B | |||||||||
| S: D614G | - | 23,403 | C | C | C | C | C | C | C | C | C | C |
| S: G639S | - | 23,477 | B | |||||||||
| S: Q675H | - | 23,587 | b | |||||||||
| S: P681H | - | 23,604 | B | |||||||||
| S: T716I | - | 23,709 | B | |||||||||
| S: T723T | - | 23,731 | C | C | ||||||||
| S: S982A | - | 24,506 | B | |||||||||
| S: D1118H | - | 24,914 | B | |||||||||
| ORF3a: Q57H | - | 25,563 | C | B | ||||||||
| ORF3a: L106L | - | 25,710 | C | B | ||||||||
| M: Y71Y | - | 26,735 | C | B | ||||||||
| M: L93L | - | 26,801 | C | C | C | |||||||
| M: I118I | - | 26,876 | C | B | ||||||||
| ORF6: L40L | - | 27,319 | B | |||||||||
| ORF7b: A15A | - | 27,800 | B | |||||||||
| ORF8: H17H | - | 27,944 | B | |||||||||
| ORF8: Q27 * | - | 27,972 | B | |||||||||
| ORF8: P30L | - | 27,982 | b | |||||||||
| ORF8: R52I | - | 28,048 | B | |||||||||
| ORF8: Y73C | - | 28,111 | B | |||||||||
| N: D3L | - | 28,280–28,282 | B | |||||||||
| N: V72I | - | 28,487 | B | |||||||||
| N: P199S | - | 28,868 | B | |||||||||
| N: R203K | - | 28,881–28,882 | C | C | C | C | ||||||
| N: G204R | - | 28,883 | C | C | C | C | ||||||
| N: A220V | - | 28,932 | C | C | C | |||||||
| N: M234I | - | 28,975 | C | B | ||||||||
| N: S235F | - | 28,977 | B | |||||||||
| N: P365S | - | 29,366 | B | |||||||||
| N: A376T | - | 29,399 | C | B | ||||||||
| ORF10: V30L | - | 29,645 | C | C | C | |||||||
| 3UTR: G32T | - | 29,706 | b | |||||||||
| 3UTR: G60C | - | 29,734 | B | |||||||||
| Gene | Protein/Peptide | Nucleotide | AA Change | Interactions | Stability (ΔΔG) | Structure | |
|---|---|---|---|---|---|---|---|
| S1 | ORF1ab | nsp3 | 7767 | I1683T | Decreased hydrophobic interactions to V1678, L1685 | I-Tasser model | |
| helicase | 17,104 | H290Y | Hbond to peptide bond of E261. Stacking with F262 | Stabilizing ++ (+2.790 kcal/mol) | 5RL9 | ||
| S | spike | 22,879 | N439K | ||||
| S2–3 | ORF1ab | 2’-o-ribose methyl transferase | 21,154 | S166A | Hydrophobic interactions to L59 and L126 (H bond to N210 abolished) | Stabilizing + (+0.425 kcal/mol) | 6W75 |
| S4 | ORF1ab | nsp3 | 3267 | T183I | Remove a OH group. No polar interaction to Q185 or Q180. | I-Tasser model | |
| 5388 | A890D | No specific interaction. At the N-terminal of an a-helix | |||||
| 6954 | I1412T | Smaller side chain. No specific interaction | |||||
| nsp6 | 11,288–11,290 | S106- | |||||
| 11,291–11,293 | G107- | ||||||
| 11,294–11,296 | F108- | ||||||
| helicase | 17,615 | K460R | Hbond to Y457 and electrostatic interaction to D458. Contact to F437 | Stabilizing ++ (+1.254 kcal/mol) | 5RL9 | ||
| S | spike | 21,767–21,769 | H69- | ||||
| 21,770 | V70- | ||||||
| 21,992–21,993 | Y144- | ||||||
| 23,063 | N501Y | ||||||
| 23,271 | A570D | ||||||
| 23,604 | P681H | ||||||
| 23,709 | T716I | ||||||
| 24,506 | S982A | Loss of an inter-protomer H-bond between the S982 and T547 side chains D1118H: S2 | |||||
| 24,914 | D1118H | ||||||
| ORF8 | ORF8 | 27,972 | Q27 * | 7JX6 | |||
| 28,048 | R52I | Solvent exposed. Contact to S54 | |||||
| 28,111 | Y73C | Solvent exposed | |||||
| N | nucleocapside | 28,280–28,282 | D3L | Hydrophobic interactions to V324 | |||
| 28,977 | S235F | Exposed at the N-terminus of an a-helix | |||||
| S5 | N | nucleocapside | 28,487 | V72I | Increases hydrophobic interactions | ||
| 28,868 | P199S | H-bond to S197 | |||||
| S7 | ORF1ab | nsp4 | 9526 | M324I | Hydrophobic interactions to L321, L323 of the opposite helix | I-Tasser model | |
| RdRp | 13,993 | A185S | Add H-bond to V182 and N213 peptide bond | 6YYT | |||
| 15,766 | V776L | Increases hydrophobic interactions to H752, F753 and Y748 | |||||
| helicase | 16,889 | K218R | Exposed to the solvent | 5RL9 | |||
| 17,019 | E261D | Hbond to S259 (H to Y324 and H290 are abolished) | |||||
| S | spike | 22,992 | S477N | ||||
| ORF3a | ORF3a | 25,563 | Q57H | Contact to His57 from the other subunit. Wall of the central pore. Interacts to Lys61 | Stabilizing ++ (+1.620 kcal/mol) | 6XDC | |
| N | nucleocapside | 28,975 | M234I | C-terminal of an a-helix | |||
| 29,399 | A376T | Potential Hbond to K374 | |||||
| S8 | ORF1ab | nsp6 | 11,132 | A3623S | |||
| S | spike | 23,587 | Q675H | ||||
| ORF8 | ORF8 | 27,982 | P30L | Solvent exposed | Stabilizing ++ (+1.620 kcal/mol) | 7JX6 | |
| S10 | ORF1ab | nsp3 | 7086 | T1456I | Exposed in a loop at the C-terminal of an a-helix. Removes polar interaction to N1457 | I-Tasser model | |
| S | spike | 23,477 | G639S | ||||
| N | nucleocapside | 29,366 | P365S | Exposed in a loop. Increases local flexibility? |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boccia, A.; Tufano, R.; Ferrucci, V.; Sepe, L.; Bianchi, M.; Pascarella, S.; Zollo, M.; Paolella, G. SARS-CoV-2 Pandemic Tracing in Italy Highlights Lineages with Mutational Burden in Growing Subsets. Int. J. Mol. Sci. 2022, 23, 4155. https://doi.org/10.3390/ijms23084155
Boccia A, Tufano R, Ferrucci V, Sepe L, Bianchi M, Pascarella S, Zollo M, Paolella G. SARS-CoV-2 Pandemic Tracing in Italy Highlights Lineages with Mutational Burden in Growing Subsets. International Journal of Molecular Sciences. 2022; 23(8):4155. https://doi.org/10.3390/ijms23084155
Chicago/Turabian StyleBoccia, Angelo, Rossella Tufano, Veronica Ferrucci, Leandra Sepe, Martina Bianchi, Stefano Pascarella, Massimo Zollo, and Giovanni Paolella. 2022. "SARS-CoV-2 Pandemic Tracing in Italy Highlights Lineages with Mutational Burden in Growing Subsets" International Journal of Molecular Sciences 23, no. 8: 4155. https://doi.org/10.3390/ijms23084155
APA StyleBoccia, A., Tufano, R., Ferrucci, V., Sepe, L., Bianchi, M., Pascarella, S., Zollo, M., & Paolella, G. (2022). SARS-CoV-2 Pandemic Tracing in Italy Highlights Lineages with Mutational Burden in Growing Subsets. International Journal of Molecular Sciences, 23(8), 4155. https://doi.org/10.3390/ijms23084155