
A Monoclonal Anti-HMGB1 Antibody Attenuates Neurodegeneration in an Experimental Animal Model of Glaucoma

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Chronic IOP Elevation by Episcleral Vein Occlusion
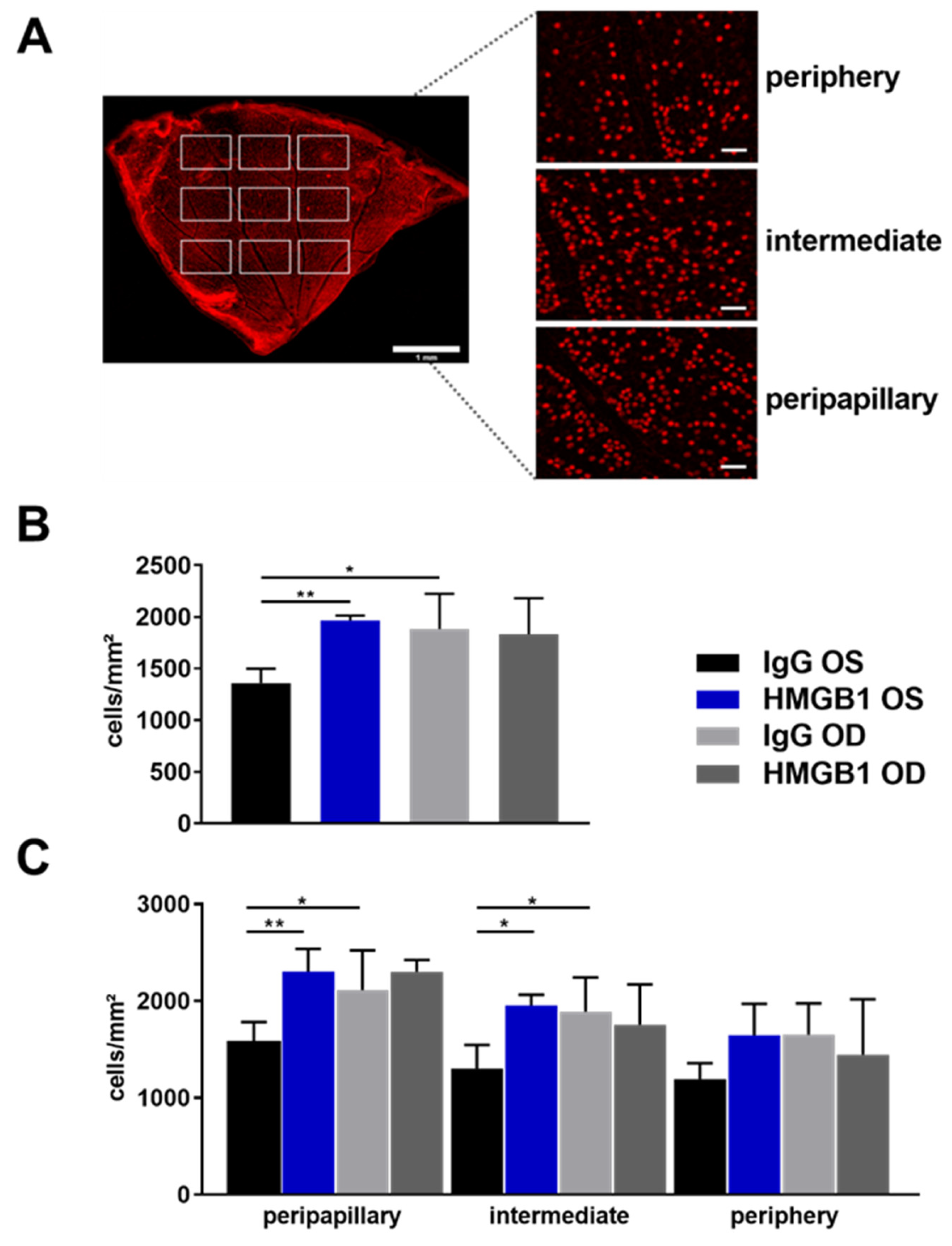
2.2. Anti-HMGB1 Antibodies Attenuate RGC Loss
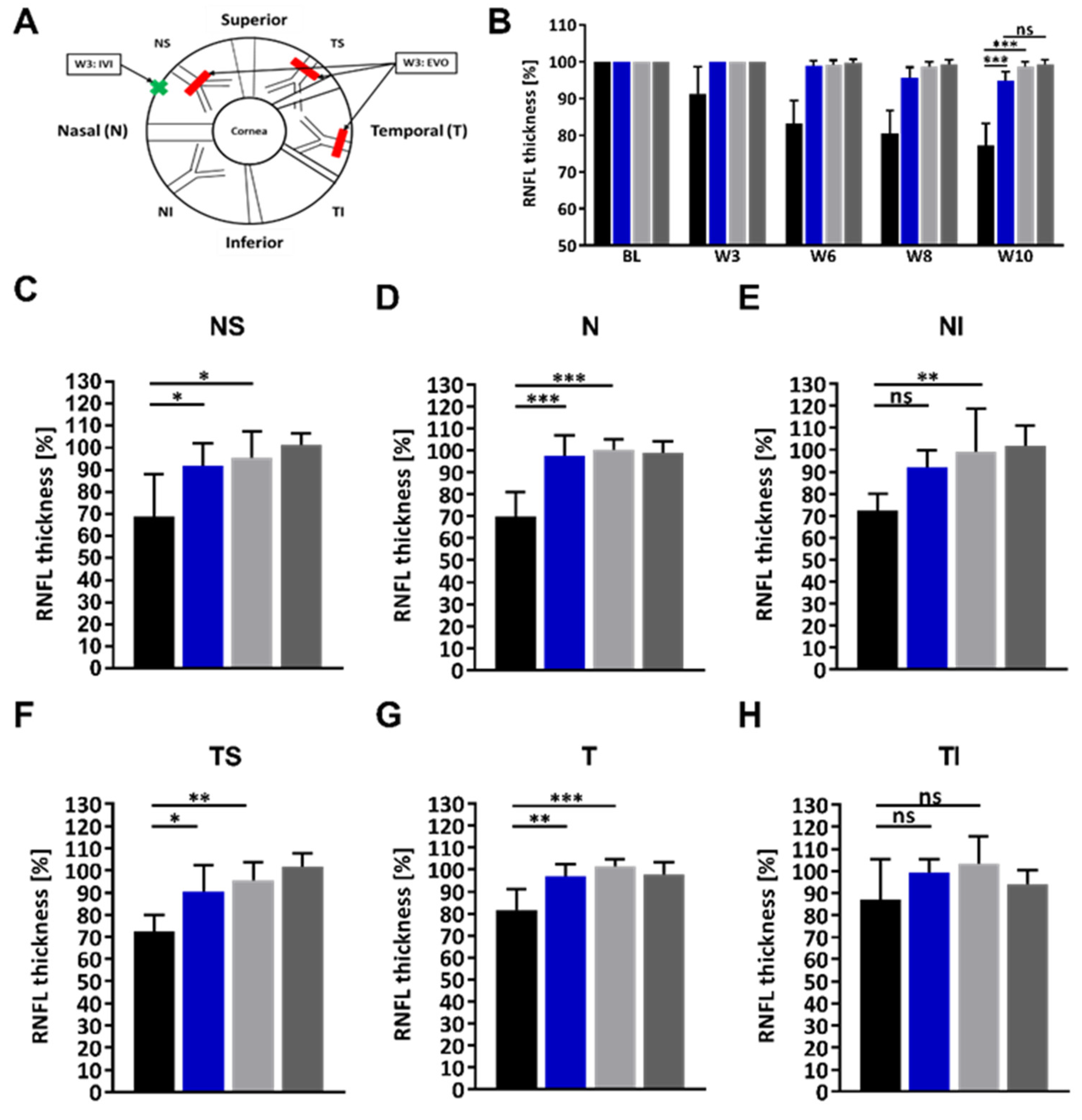
2.3. Administration of the HMGB1 Antibody Reduces the RNFLT Loss
2.4. Analysis of Retinal Functionality by Photopic Ganzfeld ERG
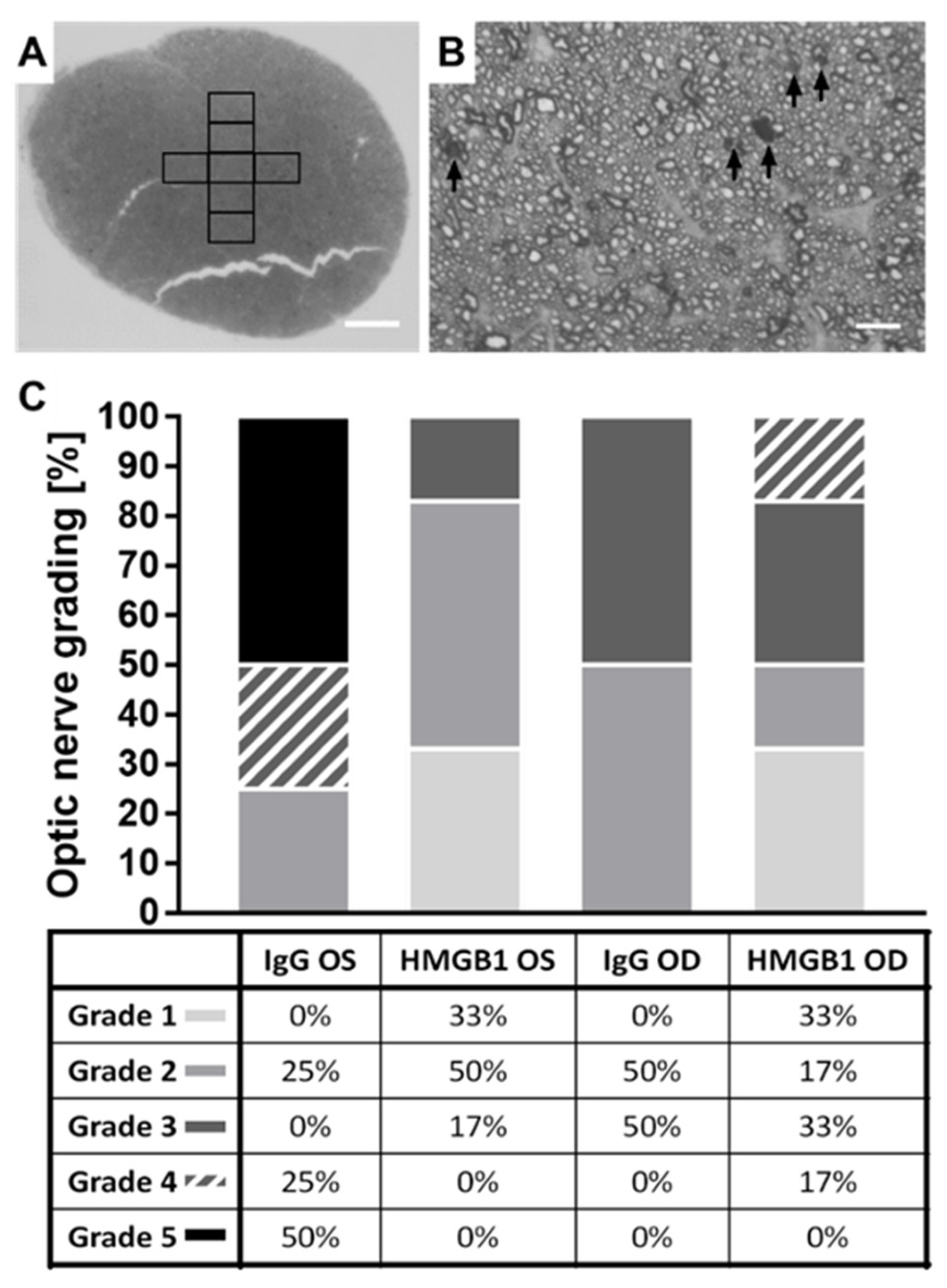
2.5. The HMGB1 Antibody Reduces the Grade of Optic Nerve (ON) Damage
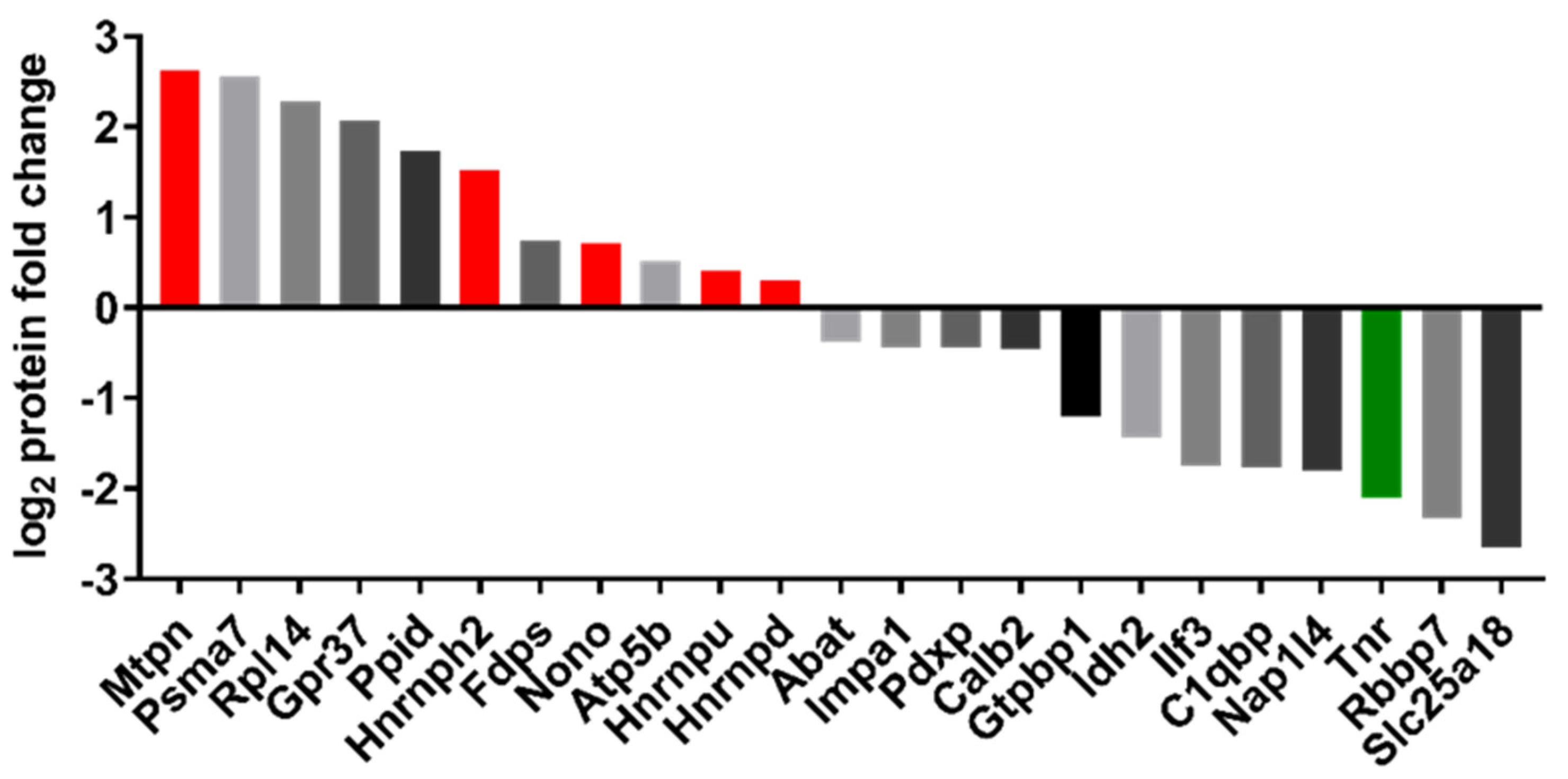
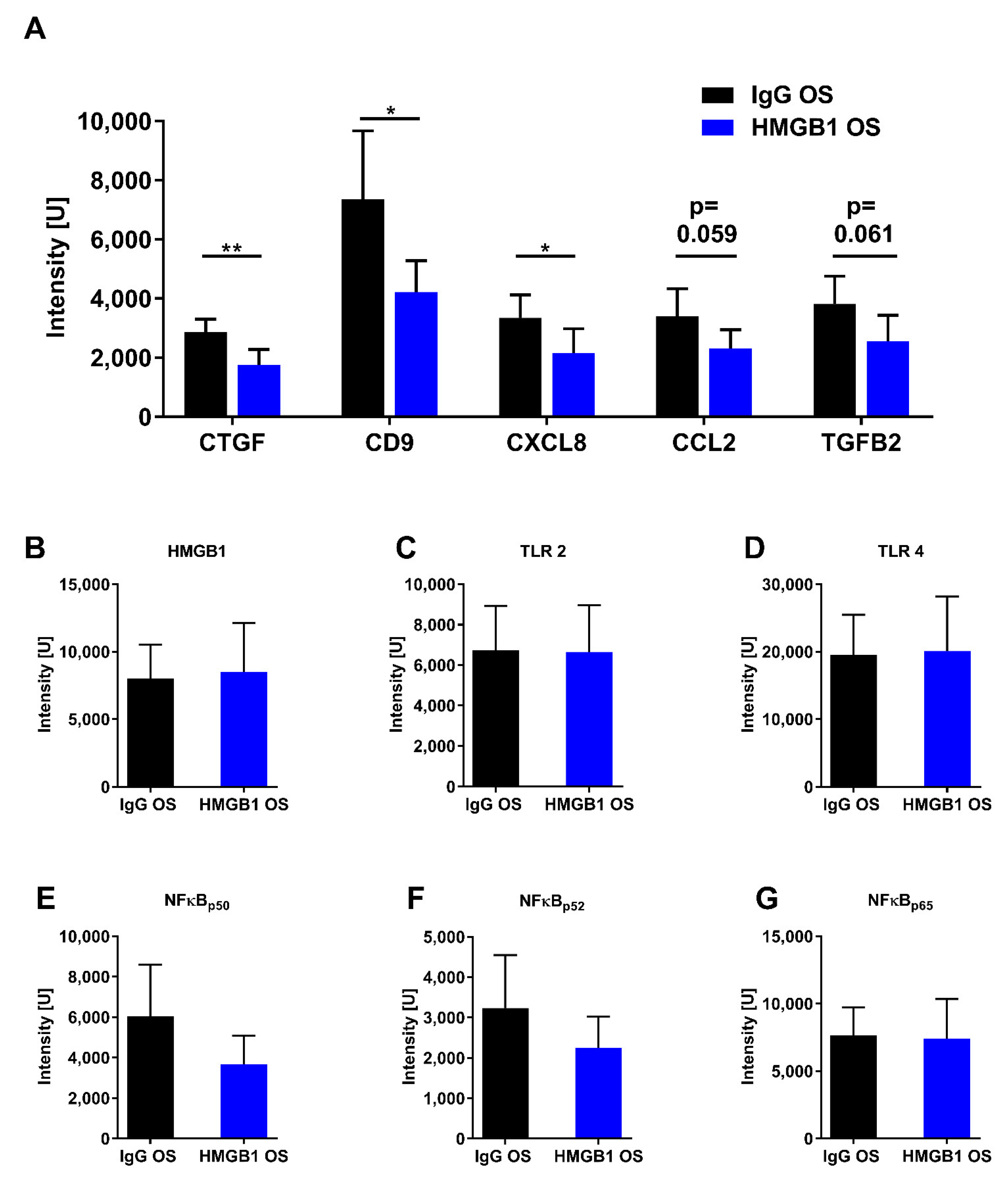
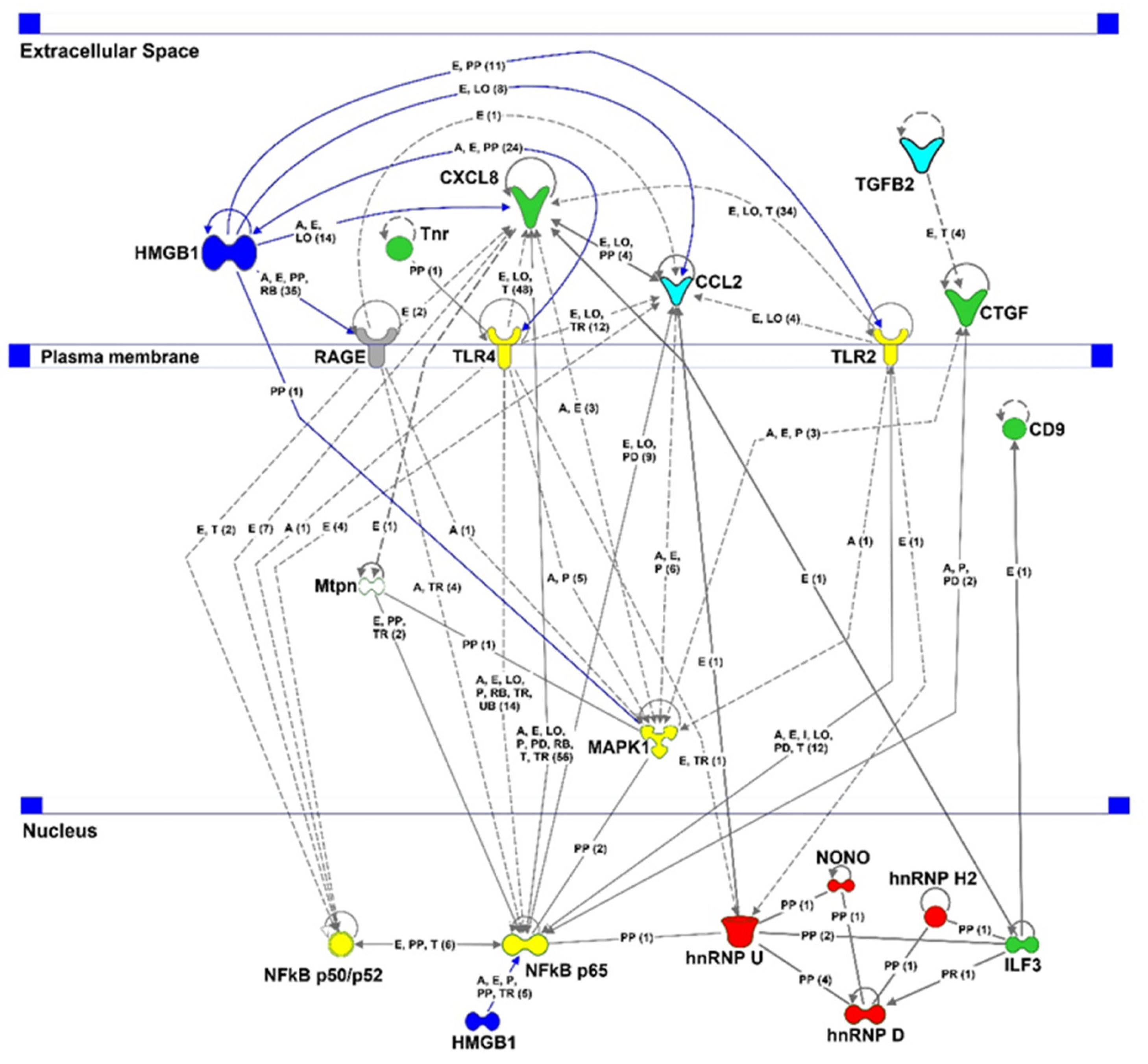
2.6. Proteomic Analysis
3. Discussion
4. Materials and Methods
4.1. Animals and Anesthesia
4.2. Experimental Design
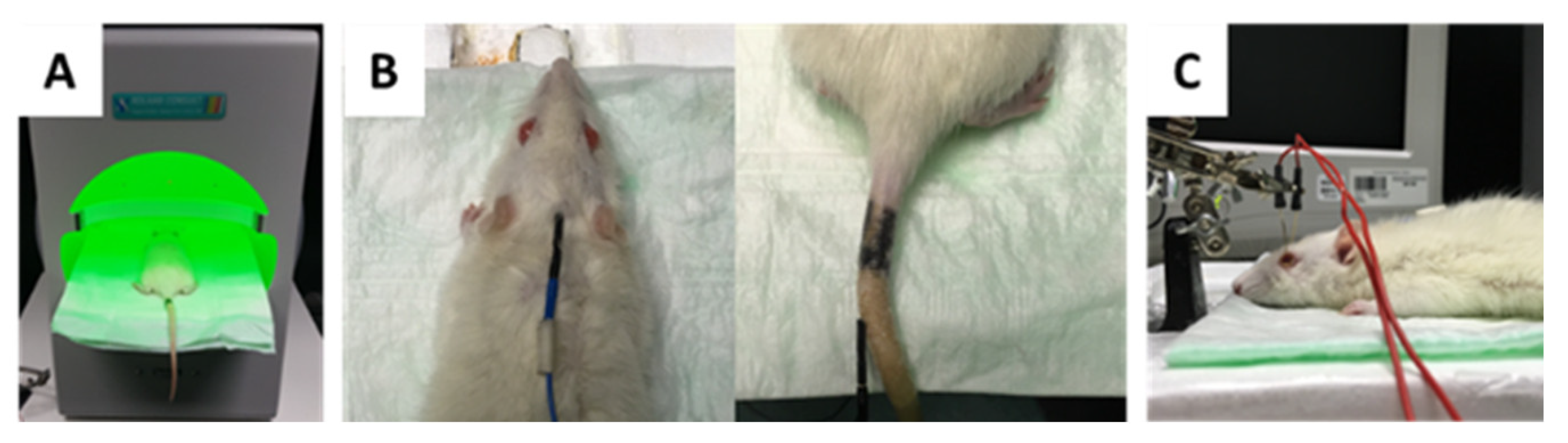
4.3. Intraocular Pressure (IOP) Monitoring by Rebound Tonometry
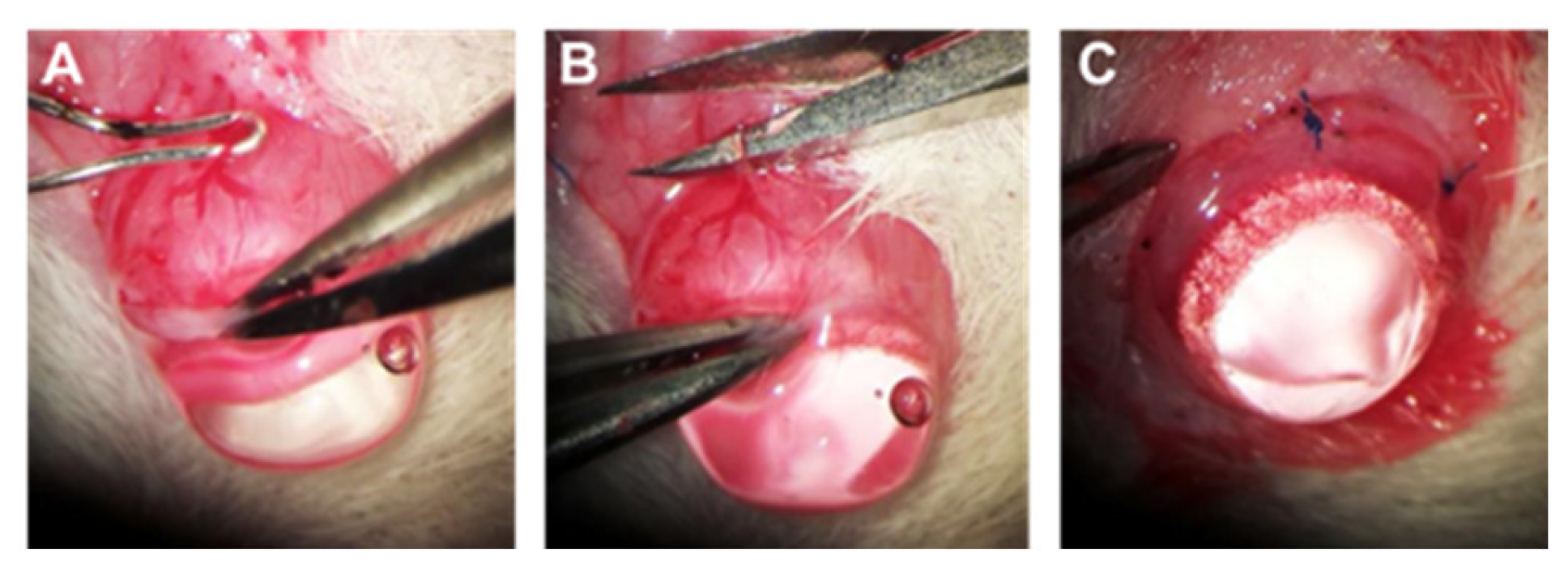
4.4. Chronic IOP Elevation by Episcleral Vein Occlusion (EVO)
4.5. Intravitreal Injection of Antibodies
4.6. RNFL Thickness Measurement by Optical Coherence Tomography (OCT)
4.7. Photopic Ganzfeld Electroretinogram (ERG)
4.8. Immunofluorescence Staining of Retinal Ganglion Cells of Retinal Flat Mounts
4.9. p-Phenylenediamine (PPD) Staining of Optic Nerve Cross-Sections
4.10. Discovery Proteomics
4.11. Analysis and Bioinformatics of MS Data
4.12. Antibody-Based Microarray
4.13. Microarray Data Processing
4.14. Statistical Analysis and Image Processing
4.15. Ethical Approval
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Manufacturer | Order Number |
|---|---|---|
| CCL2 | Sigma-Aldrich | HPA019163 |
| CD9 | Thermo Fisher Scientific | MA1-80307 |
| CTGF | Sigma-Aldrich | HPA031075 |
| CXCL8 | Sigma-Aldrich | HPA057179 |
| HMGB1 | Novus Biologicals | H00003146-M02 |
| NFkBp50 | Santa Cruz | sc-166588 |
| NFkBp52 | Santa Cruz | sc-7386 |
| NFkBp65 | Santa Cruz | sc-514451 |
| TGFβ2 | Sigma-Aldrich | SAB1409735 |
| TLR-2 | Abcam | ab13855 |
| TLR-4 | Abcam | ab13556 |
References
- Yu, S.; Tanabe, T.; Yoshimura, N. A rat model of glaucoma induced by episcleral vein ligation. Exp. Eye Res. 2006, 83, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, I.F. Normal tension glaucoma: Diagnostic features and comparisons with primary open angle glaucoma. Clin. Exp. Optom. 2000, 83, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.E.; Goldberg, J.L. Glaucoma 2.0: Neuroprotection, neuroregeneration, neuroenhancement. Ophthalmology 2012, 119, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, N.; Lamparter, J.; Gericke, A.; Grus, F.H.; Hoffmann, E.M.; Wahl, J. Neuroprotection of medical IOP-lowering therapy. Cell Tissue Res. 2013, 353, 245–251. [Google Scholar] [CrossRef]
- Lipton, S.A. The molecular basis of memantine action in Alzheimer’s disease and other neurologic disorders: Low-affinity, uncompetitive antagonism. Curr. Alzheimer Res. 2005, 2, 155–165. [Google Scholar] [CrossRef]
- Mursch-Edlmayr, A.S.; Bolz, M.; Strohmaier, C. Vascular Aspects in Glaucoma: From Pathogenesis to Therapeutic Approaches. Int. J. Mol. Sci. 2021, 22, 4662. [Google Scholar] [CrossRef]
- Tezel, G. Oxidative stress in glaucomatous neurodegeneration: Mechanisms and consequences. Prog. Retin. Eye Res. 2006, 25, 490–513. [Google Scholar] [CrossRef]
- Gramlich, O.W.; Beck, S.; von Thun Und Hohenstein-Blaul, N.; Boehm, N.; Ziegler, A.; Vetter, J.M.; Pfeiffer, N.; Grus, F.H. Enhanced insight into the autoimmune component of glaucoma: IgG autoantibody accumulation and pro-inflammatory conditions in human glaucomatous retina. PLoS ONE 2013, 8, e57557. [Google Scholar] [CrossRef]
- Bell, K.; Funke, S.; Pfeiffer, N.; Grus, F.H. Serum and antibodies of glaucoma patients lead to changes in the proteome, especially cell regulatory proteins, in retinal cells. PLoS ONE 2012, 7, e46910. [Google Scholar] [CrossRef]
- Boehm, N.; Wolters, D.; Thiel, U.; Lossbrand, U.; Wiegel, N.; Pfeiffer, N.; Grus, F.H. New insights into autoantibody profiles from immune privileged sites in the eye: A glaucoma study. Brain Behav. Immun. 2012, 26, 96–102. [Google Scholar] [CrossRef]
- Joachim, S.C.; Reichelt, J.; Berneiser, S.; Pfeiffer, N.; Grus, F.H. Sera of glaucoma patients show autoantibodies against myelin basic protein and complex autoantibody profiles against human optic nerve antigens. Graefe’s Arch. Clin. Exp. Ophthalmol. 2008, 246, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Joachim, S.C.; Bruns, K.; Lackner, K.J.; Pfeiffer, N.; Grus, F.H. Antibodies to alpha B-crystallin, vimentin, and heat shock protein 70 in aqueous humor of patients with normal tension glaucoma and IgG antibody patterns against retinal antigen in aqueous humor. Curr. Eye Res. 2007, 32, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Joachim, S.C.; Wax, M.B.; Boehm, N.; Dirk, D.R.; Pfeiffer, N.; Grus, F.H. Upregulation of antibody response to heat shock proteins and tissue antigens in an ocular ischemia model. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3468–3474. [Google Scholar] [CrossRef][Green Version]
- Tezel, G.; Wax, M.B. The mechanisms of hsp27 antibody-mediated apoptosis in retinal neuronal cells. J. Neurosci. 2000, 20, 3552–3562. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.; Wilding, C.; Funke, S.; Perumal, N.; Beck, S.; Wolters, D.; Holz-Muller, J.; Pfeiffer, N.; Grus, F.H. Neuroprotective effects of antibodies on retinal ganglion cells in an adolescent retina organ culture. J. Neurochem. 2016, 139, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.; Wilding, C.; Funke, S.; Pfeiffer, N.; Grus, F.H. Protective effect of 14-3-3 antibodies on stressed neuroretinal cells via the mitochondrial apoptosis pathway. BMC Ophthalmol. 2015, 15, 64. [Google Scholar] [CrossRef]
- Wilding, C.; Bell, K.; Funke, S.; Beck, S.; Pfeiffer, N.; Grus, F.H. GFAP antibodies show protective effect on oxidatively stressed neuroretinal cells via interaction with ERP57. J. Pharmacol. Sci. 2015, 127, 298–304. [Google Scholar] [CrossRef]
- Teister, J.; Anders, F.; Beck, S.; Funke, S.; von Pein, H.; Prokosch, V.; Pfeiffer, N.; Grus, F. Decelerated neurodegeneration after intravitreal injection of alpha-synuclein antibodies in a glaucoma animal model. Sci. Rep. 2017, 7, 6260. [Google Scholar] [CrossRef]
- Da Rocha Sobrinho, H.M.; Saar Gomes, R.; da Silva, D.J.; Quixabeira, V.B.L.; Joosten, L.A.B.; Ribeiro de Barros Cardoso, C.; Ribeiro-Dias, F. Toll-like receptor 10 controls TLR2-induced cytokine production in monocytes from patients with Parkinson’s disease. J. Neurosci. Res. 2021, 99, 2511–2524. [Google Scholar] [CrossRef]
- Rather, M.A.; Khan, A.; Alshahrani, S.; Rashid, H.; Qadri, M.; Rashid, S.; Alsaffar, R.M.; Kamal, M.A.; Rehman, M.U. Inflammation and Alzheimer’s Disease: Mechanisms and Therapeutic Implications by Natural Products. Mediat. Inflamm. 2021, 2021, 9982954. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Y.; Zhao, J.; Guo, X.; Hu, M.; Chen, Y. Possible inflammatory mechanisms and predictors of Parkinson’s disease patients with fatigue (Brief Review). Clin. Neurol. Neurosurg. 2021, 208, 106844. [Google Scholar] [CrossRef] [PubMed]
- Chi, M.; Ma, K.; Li, Y.; Quan, M.; Han, Z.; Ding, Z.; Liang, X.; Zhang, Q.; Song, L.; Liu, C. Immunological Involvement of MicroRNAs in the Key Events of Systemic Lupus Erythematosus. Front. Immunol. 2021, 12, 699684. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Uchida, K.; Nakagaki, K.; Kanazawa, H.; Trapnell, B.C.; Hoshino, Y.; Kagamu, H.; Yoshizawa, H.; Keicho, N.; Goto, H.; et al. Anti-cytokine autoantibodies are ubiquitous in healthy individuals. FEBS Lett. 2007, 581, 2017–2021. [Google Scholar] [CrossRef]
- Guan, Y.; Li, Y.; Zhao, G.; Li, Y. HMGB1 promotes the starvation-induced autophagic degradation of alpha-synuclein in SH-SY5Y cells Atg 5-dependently. Life Sci. 2018, 202, 1–10. [Google Scholar] [CrossRef]
- Kang, R.; Livesey, K.M.; Zeh, I.I.I.H.J.; Loze, M.T.; Tang, D. HMGB1: A novel Beclin 1-binding protein active in autophagy. Autophagy 2010, 6, 1209–1211. [Google Scholar] [CrossRef]
- Kang, R.; Chen, R.; Zhang, Q.; Hou, W.; Wu, S.; Cao, L.; Huang, J.; Yu, Y.; Fan, X.G.; Yan, Z.; et al. HMGB1 in health and disease. Mol. Asp. Med. 2014, 40, 1–116. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Svetkauskaite, D.; He, Q.; Kim, J.Y.; Strassheim, D.; Ishizaka, A.; Abraham, E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 2004, 279, 7370–7377. [Google Scholar] [CrossRef]
- Dupire, G.; Nicaise, C.; Gangji, V.; Soyfoo, M.S. Increased serum levels of high-mobility group box 1 (HMGB1) in primary Sjogren’s syndrome. Scand. J. Rheumatol. 2012, 41, 120–123. [Google Scholar] [CrossRef]
- Barnay-Verdier, S.; Fattoum, L.; Borde, C.; Kaveri, S.; Gibot, S.; Marechal, V. Emergence of autoantibodies to HMGB1 is associated with survival in patients with septic shock. Intensive Care Med. 2011, 37, 957–962. [Google Scholar] [CrossRef]
- Mi, L.; Zhang, Y.; Xu, Y.; Zheng, X.; Zhang, X.; Wang, Z.; Xue, M.; Jin, X. HMGB1/RAGE pro-inflammatory axis promotes vascular endothelial cell apoptosis in limb ischemia/reperfusion injury. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 116, 109005. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Aamir, K.; Shaikh, M.F. Impact of HMGB1, RAGE, and TLR4 in Alzheimer’s Disease (AD): From Risk Factors to Therapeutic Targeting. Cells 2020, 9, 383. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Harris, H.E. The role of HMGB1 in the pathogenesis of rheumatic disease. Biochim. Biophys. Acta 2010, 1799, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Andersson, U. Targeting Inflammation Driven by HMGB1. Front. Immunol. 2020, 11, 484. [Google Scholar] [CrossRef]
- Yu, Y.; Tang, D.; Kang, R. Oxidative stress-mediated HMGB1 biology. Front. Physiol. 2015, 6, 93. [Google Scholar] [CrossRef]
- Chi, W.; Chen, H.; Li, F.; Zhu, Y.; Yin, W.; Zhuo, Y. HMGB1 promotes the activation of NLRP3 and caspase-8 inflammasomes via NF-kappaB pathway in acute glaucoma. J. Neuroinflamm. 2015, 12, 137. [Google Scholar] [CrossRef]
- Sakamoto, K.; Mizuta, A.; Fujimura, K.; Kurauchi, Y.; Mori, A.; Nakahara, T.; Ishii, K. High-mobility group Box-1 is involved in NMDA-induced retinal injury the in rat retina. Exp. Eye Res. 2015, 137, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Auler, N.; Tonner, H.; Pfeiffer, N.; Grus, F.H. Antibody and Protein Profiles in Glaucoma: Screening of Biomarkers and Identification of Signaling Pathways. Biology 2021, 10, 1296. [Google Scholar] [CrossRef]
- Entezari, M.; Javdan, M.; Antoine, D.J.; Morrow, D.M.; Sitapara, R.A.; Patel, V.; Wang, M.; Sharma, L.; Gorasiya, S.; Zur, M.; et al. Inhibition of extracellular HMGB1 attenuates hyperoxia-induced inflammatory acute lung injury. Redox Biol. 2014, 2, 314–322. [Google Scholar] [CrossRef]
- Nishibori, M.; Mori, S.; Takahashi, H.K. Anti-HMGB1 monoclonal antibody therapy for a wide range of CNS and PNS diseases. J. Pharmacol. Sci. 2019, 140, 94–101. [Google Scholar] [CrossRef]
- Funke, S.; Perumal, N.; Beck, S.; Gabel-Scheurich, S.; Schmelter, C.; Teister, J.; Gerbig, C.; Gramlich, O.W.; Pfeiffer, N.; Grus, F.H. Glaucoma related Proteomic Alterations in Human Retina Samples. Sci. Rep. 2016, 6, 29759. [Google Scholar] [CrossRef]
- Schallenberg, M.; Prokosch, V.; Thanos, S. Regulation of retinal proteome by topical antiglaucomatous eye drops in an inherited glaucoma rat model. PLoS ONE 2012, 7, e33593. [Google Scholar] [CrossRef] [PubMed]
- Shareef, S.R.; Garcia-Valenzuela, E.; Salierno, A.; Walsh, J.; Sharma, S.C. Chronic ocular hypertension following episcleral venous occlusion in rats. Exp. Eye Res. 1995, 61, 379–382. [Google Scholar] [CrossRef]
- Chrysostomou, V.; Crowston, J.G. The photopic negative response of the mouse electroretinogram: Reduction by acute elevation of intraocular pressure. Investig. Ophthalmol. Vis. Sci. 2013, 54, 4691–4697. [Google Scholar] [CrossRef] [PubMed]
- Anders, F.; Teister, J.; Liu, A.; Funke, S.; Grus, F.H.; Thanos, S.; von Pein, H.D.; Pfeiffer, N.; Prokosch, V. Intravitreal injection of beta-crystallin B2 improves retinal ganglion cell survival in an experimental animal model of glaucoma. PLoS ONE 2017, 12, e0175451. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y. Electrodiagnosis of Retinal Diseases; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Wilsey, L.; Gowrisankaran, S.; Cull, G.; Hardin, C.; Burgoyne, C.F.; Fortune, B. Comparing three different modes of electroretinography in experimental glaucoma: Diagnostic performance and correlation to structure. Doc. Ophthalmol. Adv. Ophthalmol. 2017, 134, 111–128. [Google Scholar] [CrossRef]
- Machida, S.; Tamada, K.; Oikawa, T.; Gotoh, Y.; Nishimura, T.; Kaneko, M.; Kurosaka, D. Comparison of photopic negative response of full-field and focal electroretinograms in detecting glaucomatous eyes. J. Ophthalmol. 2011, 2011, 564131. [Google Scholar] [CrossRef]
- Harazny, J.; Scholz, M.; Buder, T.; Lausen, B.; Kremers, J. Electrophysiological deficits in the retina of the DBA/2J mouse. Doc. Ophthalmol. Adv. Ophthalmol. 2009, 119, 181–197. [Google Scholar] [CrossRef]
- Husain, S.; Abdul, Y.; Crosson, C.E. Preservation of retina ganglion cell function by morphine in a chronic ocular-hypertensive rat model. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4289–4298. [Google Scholar] [CrossRef]
- Kong, Y.X.; Crowston, J.G.; Vingrys, A.J.; Trounce, I.A.; Bui, V.B. Functional changes in the retina during and after acute intraocular pressure elevation in mice. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5732–5740. [Google Scholar] [CrossRef]
- Li, B.; Barnes, G.E.; Holt, W.F. The decline of the photopic negative response (PhNR) in the rat after optic nerve transection. Doc. Ophthalmol. Adv. Ophthalmol. 2005, 111, 23–31. [Google Scholar] [CrossRef]
- Jiang, S.; Chen, X. HMGB1 siRNA can reduce damage to retinal cells induced by high glucose in vitro and in vivo. Drug Des. Dev. Ther. 2017, 11, 783–795. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Reinehr, S.; Gomes, S.C.; Gassel, C.J.; Asaad, M.A.; Stute, G.; Schargus, M.; Dick, H.B.; Joachim, S.C. Intravitreal Therapy Against the Complement Factor C5 Prevents Retinal Degeneration in an Experimental Autoimmune Glaucoma Model. Front Pharm. 2019, 10, 1381. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef] [PubMed]
- Chono, I.; Miyazaki, D.; Miyake, H.; Komatsu, N.; Ehara, F.; Nagase, D.; Kawamoto, Y.; Shimizu, Y.; Ideta, R.; Inoue, Y. High interleukin-8 level in aqueous humor is associated with poor prognosis in eyes with open angle glaucoma and neovascular glaucoma. Sci. Rep. 2018, 8, 14533. [Google Scholar] [CrossRef]
- Chua, J.; Vania, M.; Cheung, C.M.; Ang, M.; Chee, S.P.; Yang, H.; Li, J.; Wong, T.T. Expression profile of inflammatory cytokines in aqueous from glaucomatous eyes. Mol. Vis. 2012, 18, 431–438. [Google Scholar]
- De Oliveira, S.; Reyes-Aldasoro, C.C.; Candel, S.; Renshaw, S.A.; Mulero, V.; Calado, A. Cxcl8 (IL-8) mediates neutrophil recruitment and behavior in the zebrafish inflammatory response. J. Immunol. 2013, 190, 4349–4359. [Google Scholar] [CrossRef]
- Pantalon, A.; Obada, O.; Constantinescu, D.; Feraru, C.; Chiselita, D. Inflammatory model in patients with primary open angle glaucoma and diabetes. Int. J. Ophthalmol. 2019, 12, 795–801. [Google Scholar] [CrossRef]
- Takai, Y.; Tanito, M.; Ohira, A. Multiplex cytokine analysis of aqueous humor in eyes with primary open-angle glaucoma, exfoliation glaucoma, and cataract. Investig. Ophthalmol. Vis. Sci. 2012, 53, 241–247. [Google Scholar] [CrossRef]
- Khan, J.; Hassun, H.; Zusman, T.; Korczeniewska, O.; Eliav, E. Interleukin-8 levels in rat models of nerve damage and neuropathic pain. Neurosci. Lett. 2017, 657, 106–112. [Google Scholar] [CrossRef]
- Bose, S.; Cho, J. Role of chemokine CCL2 and its receptor CCR2 in neurodegenerative diseases. Arch. Pharmacal. Res. 2013, 36, 1039–1050. [Google Scholar] [CrossRef]
- Feng, C.; Wang, X.; Liu, T.; Zhang, M.; Xu, G.; Ni, Y. Expression of CCL2 and its receptor in activation and migration of microglia and monocytes induced by photoreceptor apoptosis. Mol. Vis. 2017, 23, 765–777. [Google Scholar] [PubMed]
- Semple, B.D.; Frugier, T.; Morganti-Kossmann, M.C. CCL2 modulates cytokine production in cultured mouse astrocytes. J. Neuroinflamm. 2010, 7, 67. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Liu, H.; Zhang, D.; Liu, Y.; Wang, C.; Liu, F.; Chen, J. HMGB1 Enhances the AGE-Induced Expression of CTGF and TGF-beta via RAGE-Dependent Signaling in Renal Tubular Epithelial Cells. Am. J. Nephrol. 2015, 41, 257–266. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Chen, Y.; Khankan, R.; Barron, E.; Burton, R.; Zhu, D.; Ryan, S.J.; Oliver, N.; Hinton, D.R. Connective tissue growth factor as a mediator of intraocular fibrosis. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4078–4088. [Google Scholar] [CrossRef]
- Junglas, B.; Kuespert, S.; Seleem, A.A.; Struller, T.; Ullmann, S.; Bosl, M.; Bosserhoff, A.; Kostler, J.; Wagner, R.; Tamm, E.R.; et al. Connective tissue growth factor causes glaucoma by modifying the actin cytoskeleton of the trabecular meshwork. Am. J. Pathol. 2012, 180, 2386–2403. [Google Scholar] [CrossRef]
- Van Setten, G.B.; Trost, A.; Schrodl, F.; Kaser-Eichberger, A.; Bogner, B.; van Setten, M.; Heindl, L.M.; Grabner, G.; Reitsamer, H.A. Immunohistochemical Detection of CTGF in the Human Eye. Curr. Eye Res. 2016, 41, 1571–1579. [Google Scholar] [CrossRef]
- Chaqour, B.; Karrasch, C. Eyeing the Extracellular Matrix in Vascular Development and Microvascular Diseases and Bridging the Divide between Vascular Mechanics and Function. Int. J. Mol. Sci. 2020, 21, 3487. [Google Scholar] [CrossRef]
- Kuiper, E.J.; Van Nieuwenhoven, F.A.; de Smet, M.D.; van Meurs, J.C.; Tanck, M.W.; Oliver, N.; Klaassen, I.; Van Noorden, C.J.; Goldschmeding, R.; Schlingemann, R.O. The angio-fibrotic switch of VEGF and CTGF in proliferative diabetic retinopathy. PLoS ONE 2008, 3, e2675. [Google Scholar] [CrossRef]
- Chintala, H.; Liu, H.; Parmar, R.; Kamalska, M.; Kim, Y.J.; Lovett, D.; Grant, M.B.; Chaqour, B. Connective tissue growth factor regulates retinal neovascularization through p53 protein-dependent transactivation of the matrix metalloproteinase (MMP)-2 gene. J. Biol. Chem. 2012, 287, 40570–40585. [Google Scholar] [CrossRef]
- He, S.; Jin, M.L.; Worpel, V.; Hinton, D.R. A role for connective tissue growth factor in the pathogenesis of choroidal neovascularization. Arch. Ophthalmol. 2003, 121, 1283–1288. [Google Scholar] [CrossRef]
- Kuiper, E.J.; Witmer, A.N.; Klaassen, I.; Oliver, N.; Goldschmeding, R.; Schlingemann, R.O. Differential expression of connective tissue growth factor in microglia and pericytes in the human diabetic retina. Br. J. Ophthalmol. 2004, 88, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Deissler, H.; Kuhn, E.M.; Lang, G.E.; Deissler, H. Tetraspanin CD9 is involved in the migration of retinal microvascular endothelial cells. Int. J. Mol. Med. 2007, 20, 643–652. [Google Scholar] [PubMed]
- Sanchez-Lopez, E.; Rayego, S.; Rodrigues-Diez, R.; Rodriguez, J.S.; Rodrigues-Diez, R.; Rodriguez-Vita, J.; Carvajal, G.; Aroeira, L.S.; Selgas, R.; Mezzano, S.A.; et al. CTGF promotes inflammatory cell infiltration of the renal interstitium by activating NF-kappaB. J. Am. Soc. Nephrol. 2009, 20, 1513–1526. [Google Scholar] [CrossRef] [PubMed]
- Dreyfuss, G.; Matunis, M.J.; Pinol-Roma, S.; Burd, C.G. hnRNP proteins and the biogenesis of mRNA. Annu. Rev. Biochem. 1993, 62, 289–321. [Google Scholar] [CrossRef] [PubMed]
- Nakielny, S.; Dreyfuss, G. Transport of proteins and RNAs in and out of the nucleus. Cell 1999, 99, 677–690. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, H.; Deaton, S.K.; Szaro, B.G. Heterogeneous nuclear ribonucleoprotein K, an RNA-binding protein, is required for optic axon regeneration in Xenopus laevis. J. Neurosci. 2012, 32, 3563–3574. [Google Scholar] [CrossRef]
- Gomes, C.; Merianda, T.T.; Lee, S.J.; Yoo, S.; Twiss, J.L. Molecular determinants of the axonal mRNA transcriptome. Dev. Neurobiol. 2014, 74, 218–232. [Google Scholar] [CrossRef]
- Jung, H.; Gkogkas, C.G.; Sonenberg, N.; Holt, C.E. Remote control of gene function by local translation. Cell 2014, 157, 26–40. [Google Scholar] [CrossRef]
- Jung, H.; Yoon, B.C.; Holt, C.E. Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat. Rev. Neurosci. 2012, 13, 308–324. [Google Scholar] [CrossRef]
- Gautrey, H.; Jackson, C.; Dittrich, A.L.; Browell, D.; Lennard, T.; Tyson-Capper, A. SRSF3 and hnRNP H1 regulate a splicing hotspot of HER2 in breast cancer cells. RNA Biol. 2015, 12, 1139–1151. [Google Scholar] [CrossRef]
- Bi, H.S.; Yang, X.Y.; Yuan, J.H.; Yang, F.; Xu, D.; Guo, Y.J.; Zhang, L.; Zhou, C.C.; Wang, F.; Sun, S.H. H19 inhibits RNA polymerase II-mediated transcription by disrupting the hnRNP U-actin complex. Biochim. Biophys. Acta 2013, 1830, 4899–4906. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Nikodem, V.M. hnRNP U inhibits carboxy-terminal domain phosphorylation by TFIIH and represses RNA polymerase II elongation. Mol. Cell. Biol. 1999, 19, 6833–6844. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Oses-Prieto, J.A.; Kawaguchi, R.; Sahoo, P.K.; Kar, A.N.; Rozenbaum, M.; Oliver, D.; Chand, S.; Ji, H.; Shtutman, M.; et al. hnRNPs Interacting with mRNA Localization Motifs Define Axonal RNA Regulons. Mol. Cell. Proteom. 2018, 17, 2091–2106. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, H.J.; Rauvala, H. Amphoterin as an extracellular regulator of cell motility: From discovery to disease. J. Intern. Med. 2004, 255, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Hock, R.; Furusawa, T.; Ueda, T.; Bustin, M. HMG chromosomal proteins in development and disease. Trends Cell Biol. 2007, 17, 72–79. [Google Scholar] [CrossRef]
- Saarela, J.; Kallio, S.P.; Chen, D.; Montpetit, A.; Jokiaho, A.; Choi, E.; Asselta, R.; Bronnikov, D.; Lincoln, M.R.; Sadovnick, A.D.; et al. PRKCA and multiple sclerosis: Association in two independent populations. PLoS Genet. 2006, 2, e42. [Google Scholar] [CrossRef]
- Abbadi, D.; Yang, M.; Chenette, D.M.; Andrews, J.J.; Schneider, R.J. Muscle development and regeneration controlled by AUF1-mediated stage-specific degradation of fate-determining checkpoint mRNAs. Proc. Natl. Acad. Sci. USA 2019, 116, 11285–11290. [Google Scholar] [CrossRef]
- Chenette, D.M.; Cadwallader, A.B.; Antwine, T.L.; Larkin, L.C.; Wang, J.; Olwin, B.B.; Schneider, R.J. Targeted mRNA Decay by RNA Binding Protein AUF1 Regulates Adult Muscle Stem Cell Fate, Promoting Skeletal Muscle Integrity. Cell Rep. 2016, 16, 1379–1390. [Google Scholar] [CrossRef]
- Eftekharzadeh, B.; Daigle, J.G.; Kapinos, L.E.; Coyne, A.; Schiantarelli, J.; Carlomagno, Y.; Cook, C.; Miller, S.J.; Dujardin, S.; Amaral, A.S.; et al. Tau Protein Disrupts Nucleocytoplasmic Transport in Alzheimer’s Disease. Neuron 2018, 99, 925–940.e7. [Google Scholar] [CrossRef]
- Maziuk, B.F.; Apicco, D.J.; Cruz, A.L.; Jiang, L.; Ash, P.E.A.; da Rocha, E.L.; Zhang, C.; Yu, W.H.; Leszyk, J.; Abisambra, J.F.; et al. RNA binding proteins co-localize with small tau inclusions in tauopathy. Acta Neuropathol. Commun. 2018, 6, 71. [Google Scholar] [CrossRef]
- Beusch, I.; Barraud, P.; Moursy, A.; Clery, A.; Allain, F.H. Tandem hnRNP A1 RNA recognition motifs act in concert to repress the splicing of survival motor neuron exon 7. eLife 2017, 6, e25736. [Google Scholar] [CrossRef] [PubMed]
- Gilpin, K.M.; Chang, L.; Monteiro, M.J. ALS-linked mutations in ubiquilin-2 or hnRNPA1 reduce interaction between ubiquilin-2 and hnRNPA1. Hum. Mol. Genet. 2015, 24, 2565–2577. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, Y.; Gervasi, C.; Szaro, B.G. A crucial role for hnRNP K in axon development in Xenopus laevis. Development 2008, 135, 3125–3135. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Carrier, F. The UV-inducible RNA-binding protein A18 (A18 hnRNP) plays a protective role in the genotoxic stress response. J. Biol. Chem. 2001, 276, 47277–47284. [Google Scholar] [CrossRef]
- Cooper, J.A.; Sept, D. New insights into mechanism and regulation of actin capping protein. Int. Rev. Cell Mol. Biol. 2008, 267, 183–206. [Google Scholar] [CrossRef]
- Rauvala, H.; Rouhiainen, A. Physiological and pathophysiological outcomes of the interactions of HMGB1 with cell surface receptors. Biochim. Biophys. Acta 2010, 1799, 164–170. [Google Scholar] [CrossRef]
- Saenz, R.; Futalan, D.; Leutenez, L.; Eekhout, F.; Fecteau, J.F.; Sundelius, S.; Sundqvist, S.; Larsson, M.; Hayashi, T.; Minev, B.; et al. TLR4-dependent activation of dendritic cells by an HMGB1-derived peptide adjuvant. J. Transl. Med. 2014, 12, 211. [Google Scholar] [CrossRef]
- Reinhard, J.; Roll, L.; Faissner, A. Tenascins in Retinal and Optic Nerve Neurodegeneration. Front. Integr. Neurosci. 2017, 11, 30. [Google Scholar] [CrossRef]
- Liao, H.; Bu, W.Y.; Wang, T.H.; Ahmed, S.; Xiao, Z.C. Tenascin-R plays a role in neuroprotection via its distinct domains that coordinate to modulate the microglia function. J. Biol. Chem. 2005, 280, 8316–8323. [Google Scholar] [CrossRef]
- Miller, C.G.; Budoff, G.; Prenner, J.L.; Schwarzbauer, J.E. Minireview: Fibronectin in retinal disease. Exp. Biol. Med. 2017, 242, 1–7. [Google Scholar] [CrossRef]
- Zuliani-Alvarez, L.; Marzeda, A.M.; Deligne, C.; Schwenzer, A.; McCann, F.E.; Marsden, B.D.; Piccinini, A.M.; Midwood, K.S. Mapping tenascin-C interaction with toll-like receptor 4 reveals a new subset of endogenous inflammatory triggers. Nat. Commun. 2017, 8, 1595. [Google Scholar] [CrossRef] [PubMed]
- Becker, T.; Anliker, B.; Becker, C.G.; Taylor, J.; Schachner, M.; Meyer, R.L.; Bartsch, U. Tenascin-R inhibits regrowth of optic fibers in vitro and persists in the optic nerve of mice after injury. Glia 2000, 29, 330–346. [Google Scholar] [CrossRef]
- Siddiqui, S.; Horvat-Brocker, A.; Faissner, A. The glia-derived extracellular matrix glycoprotein tenascin-C promotes embryonic and postnatal retina axon outgrowth via the alternatively spliced fibronectin type III domain TNfnD. Neuron Glia Biol. 2008, 4, 271–283. [Google Scholar] [CrossRef]
- Apostolova, I.; Irintchev, A.; Schachner, M. Tenascin-R restricts posttraumatic remodeling of motoneuron innervation and functional recovery after spinal cord injury in adult mice. J. Neurosci. 2006, 26, 7849–7859. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.G.; Becker, T.; Meyer, R.L.; Schachner, M. Tenascin-R inhibits the growth of optic fibers in vitro but is rapidly eliminated during nerve regeneration in the salamander Pleurodeles waltl. J. Neurosci. 1999, 19, 813–827. [Google Scholar] [CrossRef]
- Becker, C.G.; Schweitzer, J.; Feldner, J.; Becker, T.; Schachner, M. Tenascin-R as a repellent guidance molecule for developing optic axons in zebrafish. J. Neurosci. 2003, 23, 6232–6237. [Google Scholar] [CrossRef][Green Version]
- Becker, T.; Becker, C.G. Axonal regeneration in zebrafish. Curr. Opin. Neurobiol. 2014, 27, 186–191. [Google Scholar] [CrossRef]
- Pena, J.D.; Varela, H.J.; Ricard, C.S.; Hernandez, M.R. Enhanced tenascin expression associated with reactive astrocytes in human optic nerve heads with primary open angle glaucoma. Exp. Eye Res. 1999, 68, 29–40. [Google Scholar] [CrossRef]
- Reinhard, J.; Renner, M.; Wiemann, S.; Shakoor, D.A.; Stute, G.; Dick, H.B.; Faissner, A.; Joachim, S.C. Ischemic injury leads to extracellular matrix alterations in retina and optic nerve. Sci. Rep. 2017, 7, 43470. [Google Scholar] [CrossRef]
- Keller, K.E.; Vranka, J.A.; Haddadin, R.I.; Kang, M.H.; Oh, D.J.; Rhee, D.J.; Yang, Y.F.; Sun, Y.Y.; Kelley, M.J.; Acott, T.S. The effects of tenascin C knockdown on trabecular meshwork outflow resistance. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5613–5623. [Google Scholar] [CrossRef]
- Wiemann, S.; Reinhard, J.; Reinehr, S.; Cibir, Z.; Joachim, S.C.; Faissner, A. Loss of the Extracellular Matrix Molecule Tenascin-C Leads to Absence of Reactive Gliosis and Promotes Anti-inflammatory Cytokine Expression in an Autoimmune Glaucoma Mouse Model. Front. Immunol. 2020, 11, 566279. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, B.A.; Lukaszew, R.A.; Mullins, C.M.; Penn, J.S. Impaired hyaloidal circulation function and uncoordinated ocular growth patterns in experimental retinopathy of prematurity. Investig. Ophthalmol. Vis. Sci. 1998, 39, 391–396. [Google Scholar]
- Hu, B.; Zhang, Y.; Zeng, Q.; Han, Q.; Zhang, L.; Liu, M.; Li, X. Intravitreal injection of ranibizumab and CTGF shRNA improves retinal gene expression and microvessel ultrastructure in a rodent model of diabetes. Int. J. Mol. Sci. 2014, 15, 1606–1624. [Google Scholar] [CrossRef] [PubMed]
- Anders, F.; Liu, A.; Mann, C.; Teister, J.; Lauzi, J.; Thanos, S.; Grus, F.H.; Pfeiffer, N.; Prokosch, V. The Small Heat Shock Protein alpha-Crystallin B Shows Neuroprotective Properties in a Glaucoma Animal Model. Int. J. Mol. Sci. 2017, 18, 2418. [Google Scholar] [CrossRef]
- Nadal-Nicolas, F.M.; Jimenez-Lopez, M.; Sobrado-Calvo, P.; Nieto-Lopez, L.; Canovas-Martinez, I.; Salinas-Navarro, M.; Vidal-Sanz, M.; Agudo, M. Brn3a as a marker of retinal ganglion cells: Qualitative and quantitative time course studies in naive and optic nerve-injured retinas. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3860–3868. [Google Scholar] [CrossRef]
- Maddineni, P.; Kasetti, R.B.; Patel, P.D.; Millar, J.C.; Kiehlbauch, C.; Clark, A.F.; Zode, G.S. CNS axonal degeneration and transport deficits at the optic nerve head precede structural and functional loss of retinal ganglion cells in a mouse model of glaucoma. Mol. Neurodegener. 2020, 15, 48. [Google Scholar] [CrossRef]
- Schmelter, C.; Fomo, K.N.; Perumal, N.; Manicam, C.; Bell, K.; Pfeiffer, N.; Grus, F.H. Synthetic Polyclonal-Derived CDR Peptides as an Innovative Strategy in Glaucoma Therapy. J. Clin. Med. 2019, 8, 1222. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef]
- Beutgen, V.M.; Pfeiffer, N.; Grus, F.H. Serological Levels of Anti-clathrin Antibodies Are Decreased in Patients With Pseudoexfoliation Glaucoma. Front. Immunol. 2021, 12, 616421. [Google Scholar] [CrossRef]









| Experimental Group | RNFLT for the Indicated Regions (%) | ||||||
|---|---|---|---|---|---|---|---|
| AV | NS | N | NI | TS | T | TI | |
| HMGB1 OS | 94.8 | 91.9 | 97.6 | 92.1 | 90.3 | 97.1 | 99.4 |
| IgG OS | 77.3 | 68.9 | 69.8 | 72.4 | 72.6 | 81.6 | 87.1 |
| HMGB1 OS–IgG OS | 17.5 | 22.5 | 27.8 | 19.7 | 17.7 | 15.5 | 12.3 |
| GO Terms | 5623 | 5737 | 5634 | 5739 | 5886 | 30529 | 5576 |
|---|---|---|---|---|---|---|---|
| GO description | Cell | Cytoplasm | Nucleus | Mitochondrion | Plasma membrane | Ribonucleo-protein complex | Extracellular region |
| Frequency | 24/25 96.0% | 20/25 80.0% | 9/25 36.0% | 7/25 28.0% | 6/25 24.0% | 5/25 20.0% | 3/25 12.0% |
| Genes | MTPN, FDPS, GPR37, NONO, IDH2, HNRNPU, ABAT PSMA7, CTGF, NAP1L4, CALB2, ATP5B, SLC25A18, ILF3, IMPA1, C1QBP, RPL14, HNRNPD, TNR, CD9, HNRNPH2, RBBP7, PPID | MTPN, FDPS, GPR37, IDH2, HNRNPU, ABAT, PSMA7, CTGF, NAP1L4, ATP5B, SLC25A18, ILF3, IMPA1, C1QBP, RPL14, HNRNPD, CD9, HNRNPH2, PPID | ILF3, C1QBP, NONO, HNRNPD, HNRNPU, HNRNPH2, RBBP7, PSMA7, NAP1L4 | ATP5B, FDPS, SLC25A18, ILF3, C1QBP, IDH2, ABAT | CALB2, ATP5B, GPR37, C1QBP, CD9, CTGF | ILF3, RPL14, HNRNPD, HNRNPU, HNRNPH2 | CXCL8, TNR, CTGF |
| GO Terms | 5515 | 166 | 3676 | 3723 |
|---|---|---|---|---|
| GO description | Protein binding | Nucleotide binding | Nucleic acid binding | RNA binding |
| frequency | 21/27 77.7% | 7/27 25.9% | 6/27 22.2% | 6/27 22.2% |
| Genes | MTPN, CXCL8, NONO, PDXP, HNRNPU, ABAT, PSMA7, CTGF, NAP1L4, ATP5B, ILF3, IMPA1, C1QBP, RPL14, HNRNPD, TNR CD9, HNRNPH2, RBBP7, PPID | ATP5B, NONO, IDH2, HNRNPD, HNRNPU, HNRNPH2, GTPBP1 | ILF3, NONO, RPL14, HNRNPD, HNRNPU, HNRNPH2 | ILF3, NONO, RPL14, HNRNPD, HNRNPU, HNRNPH2 |
| GO Terms | 65007 | 8152 | 50896 | 23052 | 10468 | 6950 | 23060 | 16070 |
|---|---|---|---|---|---|---|---|---|
| GO description | Biological regulation | Metabolic process | Response to stimulus | Signaling | Regulation of gene expression | Response to stress | Signal transmission | RNA metabolic process |
| Frequency | 16/26 61.5% | 15/26 57.6% | 11/26 42.3% | 9/26 34.6% | 7/26 26.9% | 7/26 26.9% | 7/26 26.9% | 5/26 19.2% |
| Genes | MTPN, CXCL8, NONO, HNRNPU, ABAT, GTPBP1, PSMA7, CTGF, ATP5B, ILF3, IMPA1, HNRNPD, TNR, CD9, RBBP7 | MTPN, FDPS, NONO, IDH2, HNRNPU, ABAT, PSMA7, CTGF, ATP5B, IMPA1, RPL14, HNRNPD, HNRNPH2, RBBP7, PPID | MTPN, CXCL8, IMPA1, C1QBP, NONO, PDXP, CD9, ABAT, RBBP7, GTPBP1, CTGF | CXCL8, GPR37, IMPA1, TNR, CD9, ABAT, GTPBP1, CTGF | MTPN, ILF3, NONO, HNRNPD, HNRNPU, RBBP7, CTGF | MTPN, CXCL8, NONO, CD9, ABAT, RBBP7, CTGF | CXCL8, IMPA1, TNR, CD9, ABAT, GTPBP1 | NONO, RPL14, HNRNPD, HNRNPU, HNRNPH2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tonner, H.; Hunn, S.; Auler, N.; Schmelter, C.; Beutgen, V.M.; von Pein, H.D.; Pfeiffer, N.; Grus, F.H. A Monoclonal Anti-HMGB1 Antibody Attenuates Neurodegeneration in an Experimental Animal Model of Glaucoma. Int. J. Mol. Sci. 2022, 23, 4107. https://doi.org/10.3390/ijms23084107
Tonner H, Hunn S, Auler N, Schmelter C, Beutgen VM, von Pein HD, Pfeiffer N, Grus FH. A Monoclonal Anti-HMGB1 Antibody Attenuates Neurodegeneration in an Experimental Animal Model of Glaucoma. International Journal of Molecular Sciences. 2022; 23(8):4107. https://doi.org/10.3390/ijms23084107
Chicago/Turabian StyleTonner, Henrik, Selina Hunn, Nadine Auler, Carsten Schmelter, Vanessa M. Beutgen, Harald D. von Pein, Norbert Pfeiffer, and Franz H. Grus. 2022. "A Monoclonal Anti-HMGB1 Antibody Attenuates Neurodegeneration in an Experimental Animal Model of Glaucoma" International Journal of Molecular Sciences 23, no. 8: 4107. https://doi.org/10.3390/ijms23084107
APA StyleTonner, H., Hunn, S., Auler, N., Schmelter, C., Beutgen, V. M., von Pein, H. D., Pfeiffer, N., & Grus, F. H. (2022). A Monoclonal Anti-HMGB1 Antibody Attenuates Neurodegeneration in an Experimental Animal Model of Glaucoma. International Journal of Molecular Sciences, 23(8), 4107. https://doi.org/10.3390/ijms23084107