
Advances in Immunomodulation and Immune Engineering Approaches to Improve Healing of Extremity Wounds

Abstract
1. Introduction
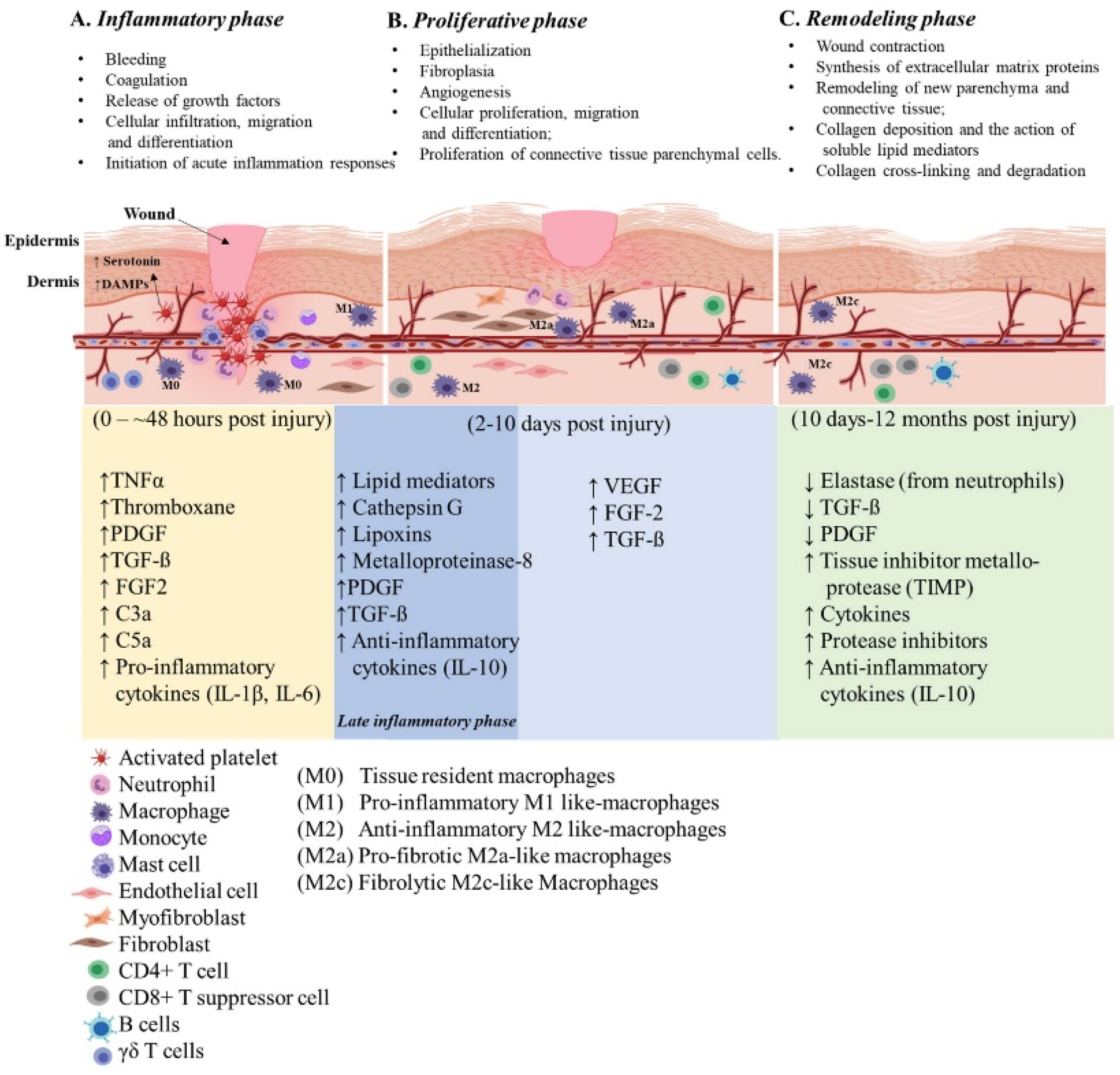
2. Cutaneous Wound Healing
2.1. Inflammatory Phase
2.2. Proliferative Phase
2.3. Remodeling Phase
3. Delayed Cutaneous Wound Healing
4. Deep Soft Tissue Injuries
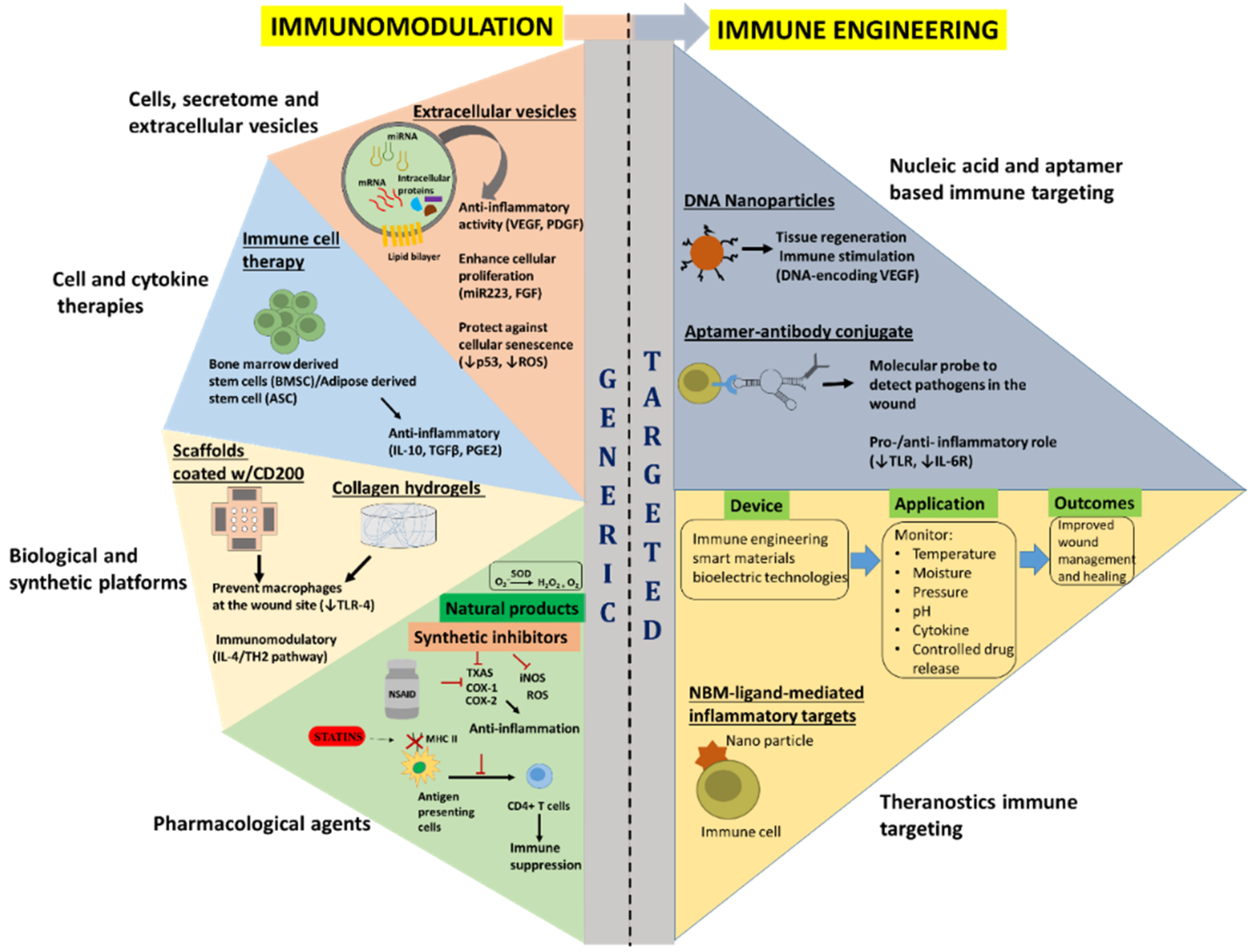
5. Current Immunomodulatory Approaches
5.1. Pharmacological Agents
5.2. Biological and Synthetic Platforms
5.3. Cell and Cytokine Therapies
5.4. Cell Secretome and Extracellular Vesicles
6. Immune Engineering Approaches to Modulate Inflammation
6.1. Nucleic Acid and Aptamers Based Immune Targeting
6.2. Theranostics Immune Targeting
7. FDA Position Statement
8. Conclusion and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
DOD Disclaimer
References
- Sun, B.K.; Siprashvili, Z.; Khavari, P.A. Advances in skin grafting and treatment of cutaneous wounds. Science 2014, 346, 941–945. [Google Scholar] [CrossRef]
- Champion, H.R.; Bellamy, R.F.; Roberts, C.P.; Leppaniemi, A. A profile of combat injury. J. Trauma Acute Care Surg. 2003, 54, S13–S19. [Google Scholar]
- Williams, D.T.; Harding, K. Healing responses of skin and muscle in critical illness. Crit. Care Med. 2003, 31, S547–S557. [Google Scholar] [CrossRef]
- Angele, M.K.; Knöferl, M.W.; Ayala, A.; Albina, J.E.; Cioffi, W.G.; Bland, K.I.; Chaudry, I.H. Trauma-hemorrhage delays wound healing potentially by increasing pro-inflammatory cytokines at the wound site. Surgery 1999, 126, 279–285. [Google Scholar] [CrossRef]
- Muire, P.J.; Schwacha, M.G.; Wenke, J.C. Systemic T Cell Exhaustion Dynamics Is Linked to Early High Mobility Group Box Protein 1 (HMGB1) Driven Hyper-Inflammation in a Polytrauma Rat Model. Cells 2021, 10, 1646. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.I.; Sørensen, A.M.; Perner, A.; Welling, K.L.; Wanscher, M.; Larsen, C.F.; Ostrowski, S.R. Disseminated intravascular coagulation or acute coagulopathy of trauma shock early after trauma? An observational study. Crit. Care 2011, 15, R272. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.V.; Bible, L.E.; Song, K.; Livingston, D.H.; Mohr, A.M.; Sifri, Z.C. Mesenchymal stem cells increase T-regulatory cells and improve healing following trauma and hemorrhagic shock (MSCs increase Tregs and improve healing After T/HS). J. Trauma Acute Care Surg. 2015, 79, 48. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Barbul, A. Understanding the role of immune regulation in wound healing. Am. J. Surg. 2004, 187, S11–S16. [Google Scholar] [CrossRef]
- Landén, N.X.; Li, D.; Ståhle, M. Transition from inflammation to proliferation: A critical step during wound healing. Cell. Mol. Life Sci. 2016, 73, 3861–3885. [Google Scholar] [CrossRef]
- Nagle, S.M.; Waheed, A.; Wilbraham, S.C. Wound Assessment; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Sinno, H.; Malhotra, M.; Lutfy, J.; Jardin, B.; Winocour, S.; Brimo, F.; Beckman, L.; Watters, K.; Philip, A.; Williams, B.; et al. Complements C3 and C5 Individually and in Combination Increase Early Wound Strength in a Rat Model of Experimental Wound Healing. Plast. Surg. Int. 2013, 2013, 243853. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Diegelmann, R.F.; Evans, M.C. Wound healing: An overview of acute, fibrotic and delayed healing. Front. Biosci. 2004, 9, 283–289. [Google Scholar] [CrossRef]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.L.; Flavell, R.A. Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef] [PubMed]
- Berton, A.; Levi-Schaffer, F.; Emonard, H.; Garbuzenko, E.; Gillery, P.; Maquart, F.X. Activation of fibroblasts in collagen lattices by mast cell extract: A model of fibrosis. Clin. Exp. Allergy 2000, 30, 485–492. [Google Scholar] [CrossRef]
- Gruber, B.L. Mast cells in the pathogenesis of fibrosis. Curr. Rheumatol. Rep. 2003, 5, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Oskeritzian, C.A. Mast Cells and Wound Healing. Adv. Wound Care 2012, 1, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Arase, H. Regulation of immune responses by neutrophils. Ann. N. Y. Acad. Sci. 2014, 1319, 66–81. [Google Scholar] [CrossRef]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A Myeloperoxidase-Containing Complex Regulates Neutrophil Elastase Release and Actin Dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Dalli, J. The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution. Semin. Immunol. 2015, 27, 200–215. [Google Scholar] [CrossRef]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2017, 18, 134–147. [Google Scholar] [CrossRef]
- van Kessel, K.P.; Bestebroer, J.; van Strijp, J.A. Neutrophil-Mediated Phagocytosis of Staphylococcus aureus. Front. Immunol. 2014, 5, 467. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 2019, 133, 2178–2185. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-C.; Wu, S.-Y.S.; Su, W.-Y.; Lin, Y.-C.; Lee, Y.-H.; Wu, W.-H.; Chen, C.-H.; Wen, Z.-H. Anti-inflammatory and burn injury wound healing properties of the shell of Haliotis diversicolor. BMC Complement. Altern. Med. 2016, 16, 487. [Google Scholar] [CrossRef]
- de Oliveira, S.; Rosowski, E.E.; Huttenlocher, A. Neutrophil migration in infection and wound repair: Going forward in reverse. Nat. Rev. Immunol. 2016, 16, 378–391. [Google Scholar] [CrossRef]
- Daley, J.M.; Brancato, S.K.; Thomay, A.A.; Reichner, J.; Albina, J.E. The phenotype of murine wound macrophages. J. Leukoc. Biol. 2009, 87, 59–67. [Google Scholar] [CrossRef]
- Das, A.; Sinha, M.; Datta, S.; Abas, M.; Chaffee, S.; Sen, C.K.; Roy, S. Monocyte and Macrophage Plasticity in Tissue Repair and Regeneration. Am. J. Pathol. 2015, 185, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Rappolee, D.A.; Mark, D.; Banda, M.J.; Werb, Z. Wound macrophages express TGF-alpha and other growth factors in vivo: Analysis by mRNA phenotyping. Science 1988, 241, 708–712. [Google Scholar] [CrossRef]
- Soehnlein, O.; Lindbom, L.; Weber, C. Mechanisms underlying neutrophil-mediated monocyte recruitment. Blood 2009, 114, 4613–4623. [Google Scholar] [CrossRef]
- Butterfield, T.A.; Best, T.M.; Merrick, M.A. The Dual Roles of Neutrophils and Macrophages in Inflammation: A Critical Balance Between Tissue Damage and Repair. J. Athl. Train. 2006, 41, 457–465. [Google Scholar]
- Gardai, S.J.; Bratton, N.L.; Ogden, C.A.; Henson, P.M. Recognition ligands on apoptotic cells: A perspective. J. Leukoc. Biol. 2006, 79, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, F.; Hsu, K.; Endoh, Y.; Geczy, C.L. FGF-2, IL-1beta and TGF-beta regulate fibroblast expression of S100A8. FEBS J. 2005, 272, 2811–2827. [Google Scholar] [CrossRef] [PubMed]
- Theilgaard-Mönch, K.; Knudsen, S.; Follin, P.; Borregaard, N. The Transcriptional Activation Program of Human Neutrophils in Skin Lesions Supports Their Important Role in Wound Healing. J. Immunol. 2004, 172, 7684–7693. [Google Scholar] [CrossRef] [PubMed]
- Boyce, D.E. The role of lymphocytes in human dermal wound healing. Br. J. Dermatol. 2000, 143, 59–65. [Google Scholar] [CrossRef]
- Fry, C.S.; Kirby, T.J.; Kosmac, K.; McCarthy, J.J.; Peterson, C.A. Myogenic Progenitor Cells Control Extracellular Matrix Production by Fibroblasts during Skeletal Muscle Hypertrophy. Cell Stem Cell 2017, 20, 56–69. [Google Scholar] [CrossRef]
- Tracy, L.E.; Minasian, R.A.; Caterson, E. Extracellular Matrix and Dermal Fibroblast Function in the Healing Wound. Adv. Wound Care 2016, 5, 119–136. [Google Scholar] [CrossRef]
- Martin, P.; Leibovich, S.J. Inflammatory cells during wound repair: The good, the bad and the ugly. Trends Cell Biol. 2005, 15, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Haller, H.L.; Sander, F.; Popp, D.; Rapp, M.; Hartmann, B.; Demircan, M.; Kamolz, L.P. Oxygen, pH, Lactate, and Metabolism-How Old Knowledge and New Insights Might Be Combined for New Wound Treatment. Medicina 2021, 57, 1190. [Google Scholar] [CrossRef]
- Dvorak, H.F. Vascular Permeability Factor/Vascular Endothelial Growth Factor: A Critical Cytokine in Tumor Angiogenesis and a Potential Target for Diagnosis and Therapy. J. Clin. Oncol. 2002, 20, 4368–4380. [Google Scholar] [CrossRef]
- Rusnati, M.; Presta, M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 159–178. [Google Scholar]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.-P.; Condorelli, F.; Park, J.; Ferrara, N. Differential Transcriptional Regulation of the Two Vascular Endothelial Growth Factor Receptor Genes: Flt-1, but not Flk-1/KDR, is up-regulated by hypoxia. J. Biol. Chem. 1997, 272, 23659–23667. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, H.Y.; Sylvester, J.; Mabrouk, M.E.; Zafarullah, M. TGF-beta-induced expression of tissue inhibitor of metalloproteinases-3 gene in chondrocytes is mediated by extracellular signal-regulated kinase pathway and Sp1 transcription factor. J. Cell Physiol. 2005, 203, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Maquart, F.X.; Monboisse, J.C. Extracellular matrix and wound healing. Pathol. Biol. 2014, 62, 91–95. [Google Scholar] [CrossRef]
- Cate, A.R.; Deporter, D.A. The degradative role of the fibroblast in the remodelling and turnover of collagen in soft connective tissue. Anat. Rec. 1975, 182, 1–13. [Google Scholar] [CrossRef]
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-wound Healing Phenotypes. Front. Physiol. 2018, 9, 419. [Google Scholar] [CrossRef]
- Kovtun, A.; Messerer, D.A.C.; Scharffetter-Kochanek, K.; Huber-Lang, M.; Ignatius, A. Neutrophils in Tissue Trauma of the Skin, Bone, and Lung: Two Sides of the Same Coin. J. Immunol. Res. 2018, 2018, 1–12. [Google Scholar] [CrossRef]
- Nguyen, H.X.; O’Barr, T.J.; Anderson, A.J. Polymorphonuclear leukocytes promote neurotoxicity through release of matrix metalloproteinases, reactive oxygen species, and TNF-α. J. Neurochem. 2007, 102, 900–912. [Google Scholar] [CrossRef]
- Behm, B.; Babilas, P.; Landthaler, M.; Schreml, S. Cytokines, chemokines and growth factors in wound healing. J. Eur. Acad. Dermatol. Venereol. 2011, 26, 812–820. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Moseley, R.; Stewart, J.E.; Stephens, P.; Waddington, R.J.; Thomas, D.W. Extracellular matrix metabolites as potential biomarkers of disease activity in wound fluid: Lessons learned from other inflammatory diseases? Br. J. Dermatol. 2004, 150, 401–413. [Google Scholar] [CrossRef]
- San Miguel, S.M.; Opperman, L.A.; Allen, E.P.; Zielinski, J.; Svoboda, K.K. Bioactive polyphenol antioxidants protect oral fibroblasts from ROS-inducing agents. Arch. Oral Biol. 2012, 57, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Strbo, N.; Yin, N.; Stojadinovic, O. Innate and Adaptive Immune Responses in Wound Epithelialization. Adv. Wound Care 2014, 3, 492–501. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, A.S.; Mansbridge, J.N. The Innate Immune System in Acute and Chronic Wounds. Adv. Wound Care 2016, 5, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Bullard, K.M.; Lund, L.; Mudgett, J.S.; Mellin, T.N.; Hunt, T.K.; Murphy, B.; Ronan, J.; Werb, Z.; Banda, M.J. Impaired Wound Contraction in Stromelysin-1–Deficient Mice. Ann. Surg. 1999, 230, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Pilcher, B.K.; Dumin, J.A.; Sudbeck, B.D.; Krane, S.M.; Welgus, H.G.; Parks, W.C. The Activity of Collagenase-1 Is Required for Keratinocyte Migration on a Type I Collagen Matrix. J. Cell Biol. 1997, 137, 1445–1457. [Google Scholar] [CrossRef]
- Reiss, M.J.; Han, Y.-P.; Garcia, E.; Goldberg, M.; Yu, H.; Garner, W.L. Matrix metalloproteinase-9 delays wound healing in a murine wound model. Surgery 2010, 147, 295–302. [Google Scholar] [CrossRef]
- Wang, Y.; Rosen, H.; Madtes, D.K.; Shao, B.; Martin, T.R.; Heinecke, J.W.; Fu, X. Myeloperoxidase inactivates TIMP-1 by oxidizing its N-terminal cysteine residue: An oxidative mechanism for regulating proteolysis during inflammation. J. Biol. Chem. 2007, 282, 31826–31834. [Google Scholar] [CrossRef]
- Gardner, J.; Ghorpade, A. Tissue inhibitor of metalloproteinase (TIMP)-1: The TIMPed balance of matrix metalloproteinases in the central nervous system. J. Neurosci. Res. 2003, 74, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Watari, M.; Watari, H.; DiSanto, M.E.; Chacko, S.; Shi, G.-P.; Strauss, J.F. Pro-Inflammatory Cytokines Induce Expression of Matrix-Metabolizing Enzymes in Human Cervical Smooth Muscle Cells. Am. J. Pathol. 1999, 154, 1755–1762. [Google Scholar] [CrossRef]
- Huggenberger, R.; Detmar, M. The Cutaneous Vascular System in Chronic Skin Inflammation. J. Investig. Dermatol. Symp. Proc. 2011, 15, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Stojadinovic, O.; Brem, H.; Vouthounis, C.; Lee, B.; Fallon, J.; Stallcup, M.; Tomic-Canic, M. Molecular pathogenesis of chronic wounds: The role of beta-catenin and c-myc in the inhibition of epithelialization and wound healing. Am. J. Pathol. 2005, 167, 59–69. [Google Scholar] [CrossRef]
- Wong, V.W.; Garg, R.K.; Sorkin, M.; Rustad, K.C.; Akaishi, S.; Levi, K.; Nelson, E.R.; Tran, M.; Rennert, R.; Liu, W.; et al. Loss of Keratinocyte Focal Adhesion Kinase Stimulates Dermal Proteolysis Through Upregulation of MMP9 in Wound Healing. Ann. Surg. 2014, 260, 1138–1146. [Google Scholar] [CrossRef]
- Barrientos, S.; Stojadinovic, O.; Golinko, M.S.; Brem, H.; Tomic-Canic, M. PERSPECTIVE ARTICLE: Growth factors and cytokines in wound healing. Wound Repair Regen. 2008, 16, 585–601. [Google Scholar] [CrossRef]
- Werner, S.; Grose, R. Regulation of Wound Healing by Growth Factors and Cytokines. Physiol. Rev. 2003, 83, 835–870. [Google Scholar] [CrossRef]
- Lopatina, T.; Bruno, S.; Tetta, C.; Kalinina, N.; Porta, M.; Camussi, G. Platelet-derived growth factor regulates the secretion of extracellular vesicles by adipose mesenchymal stem cells and enhances their angiogenic potential. Cell Commun. Signal. 2014, 12, 26. [Google Scholar] [CrossRef]
- Schultz, G.S.; Wysocki, A. Interactions between extracellular matrix and growth factors in wound healing. Wound Repair Regen. 2009, 17, 153–162. [Google Scholar] [CrossRef]
- Seppä, H.; Grotendorst, G.; Seppä, S.; Schiffmann, E.; Martin, G.R. Platelet-derived growth factor in chemotactic for fibroblasts. J. Cell Biol. 1982, 92, 584–588. [Google Scholar] [CrossRef]
- Weiner, O.D. Regulation of cell polarity during eukaryotic chemotaxis: The chemotactic compass. Curr. Opin. Cell Biol. 2002, 14, 196–202. [Google Scholar] [CrossRef]
- Dayer, C.; Stamenkovic, I. Recruitment of Matrix Metalloproteinase-9 (MMP-9) to the Fibroblast Cell Surface by Lysyl Hydroxylase 3 (LH3) Triggers Transforming Growth Factor-beta (TGF-beta) Activation and Fibroblast Differentiation. J. Biol. Chem. 2015, 290, 13763–13778. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Li, R.; Yan, N.; Chen, G.; Qian, W.; Jiang, H.L.; Bi, Z.G. Astragaloside IV controls collagen reduction in photoaging skin by improving transforming growth factor-beta/Smad signaling suppression and inhibiting matrix metalloproteinase-1. Mol. Med. Rep. 2015, 11, 3344–3348. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lim, S.L.; Du, H.; Zhang, M.; Kozak, I.; Hannum, G.; Zhang, K. High temperature requirement factor A1 (HTRA1) gene regulates angiogenesis through transforming growth factor-beta family member growth differentiation factor 6. J. Biol. Chem. 2012, 287, 1520–1526. [Google Scholar] [CrossRef]
- Johnson, K.E.; Wilgus, T.A. Vascular Endothelial Growth Factor and Angiogenesis in the Regulation of Cutaneous Wound Repair. Adv. Wound Care 2014, 3, 647–661. [Google Scholar] [CrossRef]
- Yang, L.; Kwon, J.; Popov, Y.V.; Gajdos, G.B.; Ordog, T.; Brekken, R.A.; Mukhopadhyay, D.; Schuppan, D.; Bi, Y.; Simonetto, D.; et al. Vascular Endothelial Growth Factor Promotes Fibrosis Resolution and Repair in Mice. Gastroenterology 2014, 146, 1339–1350.e1. [Google Scholar] [CrossRef] [PubMed]
- Sanes, J.R. The Basement Membrane/Basal Lamina of Skeletal Muscle. J. Biol. Chem. 2003, 278, 12601–12604. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, C.; Li, Y.; Miwa, T.; Liu, C.; Cui, W.; Song, W.-C.; Du, J. Complement C3a signaling facilitates skeletal muscle regeneration by regulating monocyte function and trafficking. Nat. Commun. 2017, 8, 2078. [Google Scholar] [CrossRef] [PubMed]
- Koffler, J.; Kaufman-Francis, K.; Shandalov, Y.; Egozi, D.; Amiad Pavlov, D.; Landesberg, A.; Levenberg, S. Improved vascular organization enhances functional integration of engineered skeletal muscle grafts. Proc. Natl. Acad. Sci. USA 2011, 108, 14789–14794. [Google Scholar] [CrossRef]
- Syverud, B.C.; VanDusen, K.; Larkin, L.M. Growth Factors for Skeletal Muscle Tissue Engineering. Cells Tissues Organs 2016, 202, 169–179. [Google Scholar] [CrossRef]
- Gillies, A.R.; Lieber, R.L. Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve 2011, 44, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Sass, F.A.; Sass, F.A.; Fuchs, M.; Pumberger, M.; Geissler, S.; Duda, G.N.; Perka, C.; Schmidt-Bleek, K. Immunology guides skeletal muscle regeneration. Int. J. Mol. Sci. 2018, 19, 835. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.J.; Badylak, S.F. Regeneration of skeletal muscle. Cell Tissue Res. 2012, 347, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Garg, K.; Corona, B.T.; Walters, T.J. Therapeutic strategies for preventing skeletal muscle fibrosis after injury. Front. Pharmacol. 2015, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Novak, M.L.; Weinheimer-Haus, E.M.; Koh, T.J. Macrophage activation and skeletal muscle healing following traumatic injury. J. Pathol. 2013, 232, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hu, P. Skeletal muscle regeneration is modulated by inflammation. J. Orthop. Transl. 2018, 13, 25–32. [Google Scholar] [CrossRef]
- Contreras, O.; Rebolledo, D.L.; Oyarzún, J.E.; Olguín, H.C.; Brandan, E. Connective tissue cells expressing fibro/adipogenic progenitor markers increase under chronic damage: Relevance in fibroblast-myofibroblast differentiation and skeletal muscle fibrosis. Cell Tissue Res. 2016, 364, 647–660. [Google Scholar] [CrossRef]
- Uezumi, A.; Fukada, S.; Yamamoto, N.; Ikemoto-Uezumi, M.; Nakatani, M.; Morita, M.; Tsuchida, K. Identification and characterization of PDGFR α+ mesenchymal progenitors in human skeletal muscle. Cell Death Dis. 2014, 5, e1186. [Google Scholar] [CrossRef]
- Nemcovsky Amar, D.; Epshtein, M.; Korin, N. Endothelial cell activation in an embolic ischemia-reperfusion injury microfluidic model. Micromachines 2019, 10, 857. [Google Scholar] [CrossRef]
- Kim, W.J.; Lee, S.-W.; Jung, K.H.; Jeong, C.H.; Seo, J.H.; Yoon, D.; Suh, J.-K.; Kim, K.-W. Inhibition of endothelial cell migration through the down-regulation of MMP-9 by A-kinase anchoring protein 12. Mol. Med. Rep. 2010, 4, 145–149. [Google Scholar] [CrossRef]
- Dvorak, H.F.; Brown, L.F.; Detmar, M.; Dvorak, A.M. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am. J. Pathol. 1995, 146, 1029–1039. [Google Scholar]
- Nagy, J.A.; Dvorak, A.M.; Dvorak, H.F. Vascular hyperpermeability, angiogenesis, and stroma generation. Cold Spring Harb. Perspect Med. 2012, 2, a006544. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-W.; West, X.Z.; Byzova, T.V. Inflammation and oxidative stress in angiogenesis and vascular disease. Klin. Wochenschr. 2013, 91, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Franz, S.; Rammelt, S.; Scharnweber, D.; Simon, J.C. Immune responses to implants–a review of the implications for the design of immunomodulatory biomaterials. Biomaterials 2011, 32, 6692–6709. [Google Scholar] [CrossRef]
- Saghazadeh, S.; Rinoldi, C.; Schot, M.; Kashaf, S.S.; Sharifi, F.; Jalilian, E.; Nuutila, K.; Giatsidis, G.; Mostafalu, P.; Derakhshandeh, H.; et al. Drug delivery systems and materials for wound healing applications. Adv. Drug Deliv. Rev. 2018, 127, 138–166. [Google Scholar] [CrossRef] [PubMed]
- Larouche, J.; Sheoran, S.; Maruyama, K.; Martino, M.M. Immune Regulation of Skin Wound Healing: Mechanisms and Novel Therapeutic Targets. Adv. Wound Care 2018, 7, 209–231. [Google Scholar] [CrossRef] [PubMed]
- Lisboa, F.A.; Bradley, M.J.; Hueman, M.T.; Schobel, S.A.; Gaucher, B.J.; Styrmisdottir, E.L.; Potter, B.K.; Forsberg, J.A.; Elster, E.A. Nonsteroidal anti-inflammatory drugs may affect cytokine response and benefit healing of combat-related extremity wounds. Surgery 2017, 161, 1164–1173. [Google Scholar] [CrossRef]
- Cámara-Lemarroy, C.R.; La Garza, F.J.G.-D.; Barrera-Oranday, E.A.; Cabello-García, A.J.; García-Tamez, A.; Fernández-Garza, N.E. Celecoxib accelerates functional recovery after sciatic nerve crush in the rat. J. Brachial Plex. Peripher. Nerve Inj. 2008, 3, e128–e131. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Qi, W.-N.; Liu, J.Q.; Urbaniak, J.R.; Folz, R.J.; Chen, L.-E. Inhibition of iNOS attenuates skeletal muscle reperfusion injury in extracellular superoxide dismutase knockout mice. Microsurgery 2005, 25, 606–613. [Google Scholar] [CrossRef]
- Bellot, G.L.; Dong, X.; Lahiri, A.; Sebastin, S.J.; Batinic-Haberle, I.; Pervaiz, S.; Puhaindran, M.E. MnSOD is implicated in accelerated wound healing upon Negative Pressure Wound Therapy (NPWT): A case in point for MnSOD mimetics as adjuvants for wound management. Redox Biol. 2018, 20, 307–320. [Google Scholar] [CrossRef]
- Mahmoud, R.; Safwat, N.; Fathy, M.; Mohamed, N.; El-Dek, S.; El-Banna, H.A.; Farghali, A.; El-Ela, F.I.A. Novel Anti-Inflammatory and Wound healing controlled released LDH-Curcumin nanocomposite via Intramuscular implantation, In-vivo Study. Arab. J. Chem. 2022, 15, 103646. [Google Scholar] [CrossRef]
- Abaricia, J.O.; Shah, A.H.; Musselman, R.M.; Olivares-Navarrete, R. Hydrophilic titanium surfaces reduce neutrophil inflammatory response and NETosis. Biomater. Sci. 2020, 8, 2289–2299. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Cheng, X.; Wang, S.; Zhao, Y.; Gao, Y.; Cai, H. Heparin-immobilized polymers as non-inflammatory and non-thrombogenic coating materials for arsenic trioxide eluting stents. Acta Biomater. 2010, 6, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.N.; Londono, R.; Tottey, S.; Zhang, L.; Kukla, K.A.; Wolf, M.T.; Badylak, S.F. Macrophage phenotype as a predictor of constructive remodeling following the implantation of biologically derived surgical mesh materials. Acta Biomater. 2012, 8, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.T.; Dearth, C.L.; Ranallo, C.A.; LoPresti, S.T.; Carey, L.E.; Daly, K.A.; Brown, B.N.; Badylak, S.F. Macrophage polarization in response to ECM coated polypropylene mesh. Biomaterials 2014, 35, 6838–6849. [Google Scholar] [CrossRef] [PubMed]
- Fearing, B.; Van Dyke, M.E. In vitro response of macrophage polarization to a keratin biomaterial. Acta Biomater. 2014, 10, 3136–3144. [Google Scholar] [CrossRef]
- Bartneck, M.; Heffels, K.-H.; Pan, Y.; Bovi, M.; Zwadlo-Klarwasser, G.; Groll, J. Inducing healing-like human primary macrophage phenotypes by 3D hydrogel coated nanofibres. Biomaterials 2012, 33, 4136–4146. [Google Scholar] [CrossRef]
- Bouchlaka, M.N.; Moffitt, A.B.; Kim, J.; Kink, J.A.; Bloom, D.D.; Love, C.; Dave, S.; Hematti, P.; Capitini, C.M. Human Mesenchymal Stem Cell–Educated Macrophages Are a Distinct High IL-6–Producing Subset that Confer Protection in Graft-versus-Host-Disease and Radiation Injury Models. Biol. Blood Marrow Transplant. 2017, 23, 897–905. [Google Scholar] [CrossRef]
- Gronthos, S.; Franklin, D.M.; Leddy, H.A.; Robey, P.; Storms, R.W.; Gimble, J.M. Surface protein characterization of human adipose tissue-derived stromal cells. J. Cell. Physiol. 2001, 189, 54–63. [Google Scholar] [CrossRef]
- Lo Sicco, C.; Reverberi, D.; Balbi, C.; Ulivi, V.; Principi, E.; Pascucci, L.; Tasso, R. Mesenchymal Stem Cell-Derived Extracellular Vesicles as Mediators of Anti-Inflammatory Effects: Endorsement of Macrophage Polarization. Stem. Cells Transl. Med. 2017, 6, 1018–1028. [Google Scholar] [CrossRef]
- Mellows, B.; Mitchell, R.; Antonioli, M.; Kretz, O.; Chambers, D.; Zeuner, M.-T.; Denecke, B.; Musante, L.; Ramachandra, D.L.; Debacq-Chainiaux, F.; et al. Protein and Molecular Characterization of a Clinically Compliant Amniotic Fluid Stem Cell-Derived Extracellular Vesicle Fraction Capable of Accelerating Muscle Regeneration Through Enhancement of Angiogenesis. Stem Cells Dev. 2017, 26, 1316–1333. [Google Scholar] [CrossRef]
- Duggan, K.C.; Walters, M.J.; Musee, J.; Harp, J.M.; Kiefer, J.; Oates, J.A.; Marnett, L.J. Molecular Basis for Cyclooxygenase Inhibition by the Non-steroidal Anti-inflammatory Drug Naproxen. J. Biol. Chem. 2010, 285, 34950–34959. [Google Scholar] [CrossRef] [PubMed]
- Orlando, B.J.; Lucido, M.; Malkowski, M.G. The structure of ibuprofen bound to cyclooxygenase-2. J. Struct. Biol. 2014, 189, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Rouzer, C.A.; Marnett, L.J. Cyclooxygenases: Structural and functional insights. J. Lipid Res. 2009, 50, S29–S34. [Google Scholar] [CrossRef] [PubMed]
- Bombardier, C.; Laine, L.; Reicin, A.; Shapiro, D.; Burgos-Vargas, R.; Davis, B.; Day, R.; Ferraz, M.B.; Hawkey, C.J.; Hochberg, M.C.; et al. Comparison of Upper Gastrointestinal Toxicity of Rofecoxib and Naproxen in Patients with Rheumatoid Arthritis. N. Engl. J. Med. 2000, 343, 1520–1528. [Google Scholar] [CrossRef]
- Mukherjee, D.; Nissen, S.E.; Topol, E.J. Risk of Cardiovascular Events Associated with Selective COX-2 Inhibitors. JAMA 2001, 286, 954–959. [Google Scholar] [CrossRef]
- Radi, Z.A.; Khan, N.K. Effects of cyclooxygenase inhibition on the gastrointestinal tract. Exp. Toxicol. Pathol. 2006, 58, 163–173. [Google Scholar] [CrossRef]
- Chu, K.; Jeong, S.-W.; Jung, K.-H.; Han, S.-Y.; Lee, S.-T.; Kim, M.; Roh, J.-K. Celecoxib Induces Functional Recovery after Intracerebral Hemorrhage with Reduction of Brain Edema and Perihematomal Cell Death. J. Cereb. Blood Flow Metab. 2004, 24, 926–933. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Toth, B.; Kitchens, W.C.; Schwacha, M.G.; Bland, K.I.; Chaudry, I.H. Role of thromboxane in producing portal hypertension following trauma-hemorrhage. Am. J. Physiol. Liver Physiol. 2003, 285, G1293–G1299. [Google Scholar] [CrossRef]
- Chueh, T.-H.; Cheng, Y.-H.; Chen, K.-H.; Chien, C.-T. Thromboxane A2 Synthase and Thromboxane Receptor Deletion Reduces Ischaemia/Reperfusion-Evoked Inflammation, Apoptosis, Autophagy and Pyroptosis. Thromb. Haemost. 2019, 120, 329–343. [Google Scholar] [CrossRef]
- Patrono, C. Biosynthesis and pharmacological modulation of thromboxane in humans. Circulation 1990, 81, I12–I15. [Google Scholar]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, H.; Chang, K.; Shinozaki, S.; Yasukawa, T.; Ishimaru, K.; Yasuhara, S.; Kaneki, M. iNOS as a Driver of Inflammation and Apoptosis in Mouse Skeletal Muscle after Burn Injury: Possible Involvement of Sirt1 S-Nitrosylation-Mediated Acetylation of p65 NF-κB and p53. PLoS ONE 2017, 12, e0170391. [Google Scholar] [CrossRef]
- Zhang, L.; Looney, C.G.; Qi, W.-N.; Chen, L.-E.; Seaber, A.V.; Stamler, J.S.; Urbaniak, J.R. Reperfusion injury is reduced in skeletal muscle by inhibition of inducible nitric oxide synthase. J. Appl. Physiol. 2003, 94, 1473–1478. [Google Scholar] [CrossRef][Green Version]
- Kim, K. Interaction between HSP 70 and iNOS in skeletal muscle injury and repair. J. Exerc. Rehabilitation 2015, 11, 240–243. [Google Scholar] [CrossRef]
- Kitano, T.; Yamada, H.; Kida, M.; Okada, Y.; Saika, S.; Yoshida, M. Impaired Healing of a Cutaneous Wound in an Inducible Nitric Oxide Synthase-Knockout Mouse. Dermatol. Res. Pract. 2017, 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Whitely, M.E.; Collins, P.B.; Iwamoto, M.; Wenke, J.C. Administration of a selective retinoic acid receptor-γ agonist improves neuromuscular strength in a rodent model of volumetric muscle loss. J. Exp. Orthop. 2021, 8, 58. [Google Scholar] [CrossRef] [PubMed]
- Younus, H. Therapeutic potentials of superoxide dismutase. Int. J. Health Sci. 2018, 12, 88–93. [Google Scholar]
- Wittmann, C.; Chockley, P.; Singh, S.K.; Pase, L.; Lieschke, G.; Grabher, C. Hydrogen Peroxide in Inflammation: Messenger, Guide, and Assassin. Adv. Hematol. 2012, 2012, 1–6. [Google Scholar] [CrossRef]
- Kang, D.O.; Park, Y.; Seo, J.H.; Jeong, M.H.; Chae, S.C.; Ahn, T.H.; KAMIR-NIH Registry Investigators. Time-dependent prognostic effect of high sensitivity C-reactive protein with statin therapy in acute myocardial infarction. J. Cardiol. 2019, 74, 74–83. [Google Scholar] [CrossRef]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef]
- Pinal-Fernandez, I.; Casal-Dominguez, M.; Mammen, A.L. Statins: Pros and cons. Med. Clin. 2018, 150, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Akershoek, J.J.; Brouwer, K.M.; Vlig, M.; Boekema, B.K.H.L.; Beelen, R.H.J.; Middelkoop, E.; Ulrich, M.M.W. Differential effects of Losartan and Atorvastatin in partial and full thickness burn wounds. PLoS ONE 2017, 12, e0179350. [Google Scholar] [CrossRef] [PubMed]
- Van Ort, S.R.; Gerber, R.M. Topical application of insulin in the treatment of decubitus ulcers: A pilot study. Nurs. Res. 1976, 25, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Abdelkader, D.; Osman, M.A.; Elgizawy, S.A.; Faheem, A.M.; McCarron, P.A. The role of insulin in wound healing process: Mechanism of action and pharmaceutical applications. J. Anal. Pharm. Res. 2016, 2, 7. [Google Scholar]
- De Meyts, P. The Insulin Receptor and Its Signal Transduction Network. In Endotext; Feingold, K.R., Grunfeld, C., Anawalt, B., Boyce, A., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2016. Available online: https://www.ncbi.nlm.nih.gov/books/NBK378978/ (accessed on 15 February 2022).
- Rezvani, O.; Shabbak, E.; Aslani, A.; Bidar, R.; Jafari, M.; Safarnezhad, S. A randomized, double-blind, placebo-controlled trial to determine the effects of topical insulin on wound healing. Ostomy/Wound Manag. 2009, 55, 22. [Google Scholar]
- Teplicki, E.; Ma, Q.; Castillo, D.E.; Zarei, M.; Hustad, A.P.; Chen, J.; Li, J. The Effects of Aloe vera on Wound Healing in Cell Proliferation, Migration, and Viability. Wounds Compend. Clin. Res. Pract. 2018, 30, 263–268. [Google Scholar]
- Hadagali, M.D.; Chua, L.S. The anti-inflammatory and wound healing properties of honey. Eur. Food Res. Technol. 2014, 239, 1003–1014. [Google Scholar] [CrossRef]
- Prasad, R.; Kumar, D.; Kant, V.; Tandan, S.K.; Kumar, D. Curcumin Enhanced Cutaneous Wound Healing by Modulating Cytokines and Transforming Growth Factor in Excision Wound Model in Rats. Int. J. Curr. Microbiol. Appl. Sci. 2017, 6, 2263–2273. [Google Scholar] [CrossRef]
- Wang, X. Overview on biocompatibilities of implantable biomaterials. In Advances in Biomaterials Science and Biomedical Applications in Biomedicine; Lazinica, R., Ed.; BoD–Books on Demand: Norderstedt, Germany, 2013; pp. 111–155. [Google Scholar]
- Taraballi, F.; Sushnitha, M.; Tsao, C.; Bauza, G.; Liverani, C.; Shi, A.; Tasciotti, E. Biomimetic tissue engineering: Tuning the immune and inflammatory response to implantable biomaterials. Adv. Healthc. Mater. 2018, 7, 1800490. [Google Scholar] [CrossRef]
- Mehrali, M.; Thakur, A.; Pennisi, C.P.; Talebian, S.; Arpanaei, A.; Nikkhah, M.; Dolatshahi-Pirouz, A. Nanoreinforced Hydrogels for Tissue Engineering: Biomaterials that are Compatible with Load-Bearing and Electroactive Tissues. Adv. Mater. 2016, 29, 1603612. [Google Scholar] [CrossRef]
- Smoak, M.M.; Mikos, A.G. Advances in biomaterials for skeletal muscle engineering and obstacles still to overcome. Mater. Today Bio. 2020, 7, 100069. [Google Scholar] [CrossRef] [PubMed]
- Chandorkar, Y.; Basu, B. The foreign body response demystified. ACS Biomater. Sci. Eng. 2018, 5, 19–44. [Google Scholar] [CrossRef] [PubMed]
- Morais, J.M.; Papadimitrakopoulos, F.; Burgess, D.J. Biomaterials/Tissue Interactions: Possible Solutions to Overcome Foreign Body Response. AAPS J. 2010, 12, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.; Maestas, D.R.; Housseau, F.; Elisseeff, J.H. Key players in the immune response to biomaterial scaffolds for regenerative medicine. Adv. Drug Deliv. Rev. 2017, 114, 184–192. [Google Scholar] [CrossRef]
- Cheung, H.-Y.; Lau, K.-T.; Lu, T.-P.; Hui, D. A critical review on polymer-based bio-engineered materials for scaffold development. Compos. Part B Eng. 2006, 38, 291–300. [Google Scholar] [CrossRef]
- Venkatraman, S.; Boey, F.; Lao, L.L. Implanted cardiovascular polymers: Natural, synthetic and bio-inspired. Prog. Polym. Sci. 2008, 33, 853–874. [Google Scholar] [CrossRef]
- Doloff, J.; Veiseh, O.; Vegas, A.J.; Tam, H.H.; Farah, S.; Ma, M.; Li, J.; Bader, A.; Chiu, A.; Sadraei, A.; et al. Colony stimulating factor-1 receptor is a central component of the foreign body response to biomaterial implants in rodents and non-human primates. Nat. Mater. 2017, 16, 671–680. [Google Scholar] [CrossRef]
- Boni, B.O.O.; Lamboni, L.; Souho, T.; Gauthier, M.; Yang, G. Immunomodulation and cellular response to biomaterials: The overriding role of neutrophils in healing. Mater. Horizons 2019, 6, 1122–1137. [Google Scholar] [CrossRef]
- Prame Kumar, K.; Nicholls, A.J.; Wong, C.H.Y. Partners in crime: Neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res. 2018, 371, 551–565. [Google Scholar] [CrossRef]
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal. 2017, 15, 23. [Google Scholar] [CrossRef]
- Finley, M.J.; Rauova, L.; Alferiev, I.S.; Weisel, J.W.; Levy, R.J.; Stachelek, S.J. Diminished adhesion and activation of platelets and neutrophils with CD47 functionalized blood contacting surfaces. Biomaterials 2012, 33, 5803–5811. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Popowich, Y.; Greco, R.S.; Haimovich, B. Neutrophil survival on biomaterials is determined by surface topography. J. Vasc. Surg. 2003, 37, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Que, R.; Wang, S.-W.; Liu, W.F. Modification of Biomaterials with a Self-Protein Inhibits the Macrophage Response. Adv. Health Mater. 2014, 3, 989–994. [Google Scholar] [CrossRef]
- Hayakawa, K.; Pham, L.D.D.; Seo, J.H.; Miyamoto, N.; Maki, T.; Terasaki, Y.; Lo, E.H. CD200 restrains macrophage attack on oligodendrocyte precursors via toll-like receptor 4 downregulation. J. Cereb. Blood Flow Metab. 2016, 36, 781–793. [Google Scholar] [CrossRef]
- Horsley, V.; Jansen, K.M.; Mills, S.T.; Pavlath, G.K. IL-4 Acts as a Myoblast Recruitment Factor during Mammalian Muscle Growth. Cell 2003, 113, 483–494. [Google Scholar] [CrossRef]
- Meng, J.; Zou, X.; Wu, R.; Zhong, R.; Zhu, D.; Zhang, Y. Accelerated regeneration of the skeletal muscle in RNF13-knockout mice is mediated by macrophage-secreted IL-4/IL-6. Protein Cell 2014, 5, 235–247. [Google Scholar] [CrossRef]
- Schiechl, G.; Hermann, F.J.; Gomez, M.R.; Kutzi, S.; Schmidbauer, K.; Talke, Y.; Neumayer, S.; Goebel, N.; Renner, K.; Brühl, H.; et al. Basophils Trigger Fibroblast Activation in Cardiac Allograft Fibrosis Development. Am. J. Transplant. 2016, 16, 2574–2588. [Google Scholar] [CrossRef]
- Zhang, S.; Marini, D.M.; Hwang, W.; Santoso, S. Design of nanostructured biological materials through self-assembly of peptides and proteins. Curr. Opin. Chem. Biol. 2002, 6, 865–871. [Google Scholar] [CrossRef]
- Aamodt, J.M.; Grainger, D.W. Extracellular matrix-based biomaterial scaffolds and the host response. Biomaterials 2016, 86, 68–82. [Google Scholar] [CrossRef]
- Eweida, A.; Marei, M. Naturally occurring extracellular matrix scaffolds for dermal regeneration: Do they really need cells? Bio. Med. Res. Int. 2015, 2015, 839694. [Google Scholar] [CrossRef]
- Cazzell, S.; Moyer, P.M.; Samsell, B.; Dorsch, K.; McLean, J.; Moore, M.A. A Prospective, Multicenter, Single-Arm Clinical Trial for Treatment of Complex Diabetic Foot Ulcers with Deep Exposure Using Acellular Dermal Matrix. Adv. Ski. Wound Care 2019, 32, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Hoshiba, T.; Kawazoe, N.; Koda, I.; Song, M.; Chen, G. Cultured cell-derived extracellular matrix scaffolds for tissue engineering. Biomaterials 2011, 32, 9658–9666. [Google Scholar] [CrossRef] [PubMed]
- Gentile, N.E.; Stearns, K.M.; Brown, E.H.; Rubin, J.P.; Boninger, M.; Dearth, C.L.; Ambrosio, F.; Badylak, S. Targeted Rehabilitation After Extracellular Matrix Scaffold Transplantation for the Treatment of Volumetric Muscle Loss. Am. J. Phys. Med. Rehabilitation 2014, 93, S79–S87. [Google Scholar] [CrossRef]
- Qiu, X.; Liu, S.; Zhang, H.; Zhu, B.; Su, Y.; Zheng, C.; Tian, R.; Wang, M.; Kuang, H.; Zhao, X.; et al. Mesenchymal stem cells and extracellular matrix scaffold promote muscle regeneration by synergistically regulating macrophage polarization toward the M2 phenotype. Stem Cell Res. Ther. 2018, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Sarrafian, T.L.; Bodine, S.C.; Murphy, B.; Grayson, J.K.; Stover, S.M. Extracellular matrix scaffolds for treatment of large volume muscle injuries: A review. Veter Surg. 2018, 47, 524–535. [Google Scholar] [CrossRef]
- Thompson, M.; Van Dyke, M. Natural Materials for Cell-Based Therapies. In Biomaterials for Cell Delivery; CRC Press: Boca Raton, FL, USA, 2018; pp. 1–24. [Google Scholar]
- Hill, P.; Brantley, H.; Van Dyke, M. Some properties of keratin biomaterials: Kerateines. Biomaterials 2010, 31, 585–593. [Google Scholar] [CrossRef]
- Thompson, M.; Giuffre, A.; McClenny, C.; Van Dyke, M. A keratin-based microparticle for cell delivery. J. Biomater. Appl. 2020, 35, 579–591. [Google Scholar] [CrossRef]
- Ledford, B.T.; Simmons, J.; Chen, M.; Fan, H.; Barron, C.; Liu, Z.; Van Dyke, M.; He, J.-Q. Keratose Hydrogels Promote Vascular Smooth Muscle Differentiation from C-kit-Positive Human Cardiac Stem Cells. Stem Cells Dev. 2017, 26, 888–900. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Raines, R.T. Collagen-based biomaterials for wound healing. Biopolymers 2014, 101, 821–833. [Google Scholar] [CrossRef]
- Janmey, P.A.; Winer, J.P.; Weisel, J.W. Fibrin gels and their clinical and bioengineering applications. J. R. Soc. Interface 2009, 6, 1–10. [Google Scholar] [CrossRef]
- Cañedo-Dorantes, L.; Cañedo-Ayala, M. Skin Acute Wound Healing: A Comprehensive Review. Int. J. Inflamm. 2019, 2019, 3706315. [Google Scholar] [CrossRef] [PubMed]
- Chargé, S.B.; Rudnicki, M. Cellular and Molecular Regulation of Muscle Regeneration. Physiol. Rev. 2004, 84, 209–238. [Google Scholar] [CrossRef] [PubMed]
- Howard, E.E.; Pasiakos, S.M.; Blesso, C.N.; Fussell, M.A.; Rodriguez, N.R. Divergent Roles of Inflammation in Skeletal Muscle Recovery from Injury. Front. Physiol. 2020, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- Singer, A. Healing Mechanisms in Cutaneous Wounds: Tipping the Balance. Tissue Eng. Part B Rev. 2021. [Google Scholar] [CrossRef]
- Kado, M.; Tanaka, R.; Arita, K.; Okada, K.; Ito-Hirano, R.; Fujimura, S.; Mizuno, H. Human peripheral blood mononuclear cells enriched in endothelial progenitor cells via quality and quantity controlled culture accelerate vascularization and wound healing in a porcine wound model. Cell Transplant. 2018, 27, 1068–1079. [Google Scholar] [CrossRef]
- Mizoguchi, T.; Ueno, K.; Takeuchi, Y.; Samura, M.; Suzuki, R.; Murata, T.; Hosoyama, T.; Morikage, N.; Hamano, K. Treatment of Cutaneous Ulcers with Multilayered Mixed Sheets of Autologous Fibroblasts and Peripheral Blood Mononuclear Cells. Cell. Physiol. Biochem. 2018, 47, 201–211. [Google Scholar] [CrossRef]
- Sîrbulescu, R.F.; Boehm, C.K.; Soon, E.; Wilks, M.Q.; Ilies, I.; Yuan, H.; Maxner, B.; Chronos, N.; Kaittanis, C.; Normandin, M.; et al. Mature B cells accelerate wound healing after acute and chronic diabetic skin lesions. Wound Repair Regen. 2017, 25, 774–791. [Google Scholar] [CrossRef]
- Vågesjö, E.; Öhnstedt, E.; Mortier, A.; Lofton, H.; Huss, F.; Proost, P.; Roos, S.; Phillipson, M. Accelerated wound healing in mice by on-site production and delivery of CXCL12 by transformed lactic acid bacteria. Proc. Natl. Acad. Sci. USA 2018, 115, 1895–1900. [Google Scholar] [CrossRef]
- Dong, Y.; Yang, Q.; Sun, X. Comprehensive Analysis of Cell Therapy on Chronic Skin Wound Healing: A Meta-Analysis. Hum. Gene Ther. 2021, 32, 787–795. [Google Scholar] [CrossRef]
- Corcione, A.; Benvenuto, F.; Ferretti, E.; Giunti, D.; Cappiello, V.; Cazzanti, F.; Risso, M.; Gualandi, F.; Mancardi, G.L.; Pistoia, V.; et al. Human mesenchymal stem cells modulate B-cell functions. Blood 2006, 107, 367–372. [Google Scholar] [CrossRef]
- Rea, S.; Giles, N.L.; Webb, S.; Adcroft, K.F.; Evill, L.M.; Strickland, D.H.; Wood, F.M.; Fear, M.W. Bone marrow-derived cells in the healing burn wound—More than just inflammation. Burns 2009, 35, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Kilroy, G.E.; Foster, S.J.; Wu, X.; Ruiz, J.; Sherwood, S.; Heifetz, A.; Ludlow, J.W.; Stricker, D.M.; Potiny, S.; Green, P.; et al. Cytokine profile of human adipose-derived stem cells: Expression of angiogenic, hematopoietic, and pro-inflammatory factors. J. Cell. Physiol. 2007, 212, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Choudhry, M.A.; Ahmed, Z.; Sayeed, M.M. PGE(2)-mediated inhibition of T cell p59(fyn) is independent of cAMP. Am. J. Physiol. 1999, 277, C302–C309. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.G.; Padilla, J.; Koumas, L.; Ray, D.; Phipps, R.P. Prostaglandins as modulators of immunity. Trends Immunol. 2002, 23, 144–150. [Google Scholar] [CrossRef]
- Ho, A.T.V.; Palla, A.R.; Blake, M.R.; Yucel, N.D.; Wang, Y.X.; Magnusson, K.E.G.; Holbrook, C.A.; Kraft, P.E.; Delp, S.L.; Blau, H.M. Prostaglandin E2 is essential for efficacious skeletal muscle stem-cell function, augmenting regeneration and strength. Proc. Natl. Acad. Sci. USA 2017, 114, 6675–6684. [Google Scholar] [CrossRef]
- Jin, D.K.; Shido, K.; Kopp, H.G.; Petit, I.; Shmelkov, S.V.; Young, L.M.; Rafii, S. Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat. Med. 2006, 12, 557–567. [Google Scholar] [CrossRef]
- Petit, I.; Jin, D.; Rafii, S. The SDF-1–CXCR4 signaling pathway: A molecular hub modulating neo-angiogenesis. Trends Immunol. 2007, 28, 299–307. [Google Scholar] [CrossRef]
- Aurora, A.; Wrice, N.; Walters, T.J.; Christy, R.J.; Natesan, S. A PEGylated platelet free plasma hydrogel based composite scaffold enables stable vascularization and targeted cell delivery for volumetric muscle loss. Acta Biomater. 2018, 65, 150–162. [Google Scholar] [CrossRef]
- Sato, Y.; Ohshima, T.; Kondo, T. Regulatory Role of Endogenous Interleukin-10 in Cutaneous Inflammatory Response of Murine Wound Healing. Biochem. Biophys. Res. Commun. 1999, 265, 194–199. [Google Scholar] [CrossRef]
- Gauglitz, G.G.; Finnerty, C.C.; Herndon, D.N.; Mlcak, R.P.; Jeschke, M.G. Are serum cytokines early predictors for the outcome of burn patients with inhalation injuries who do not survive? Crit. Care 2008, 12, R81. [Google Scholar] [CrossRef]
- Zahs, A.; Bird, M.D.; Ramirez, L.; Choudhry, M.A.; Kovacs, E.J. Anti-IL-6 antibody treatment but not IL-6 knockout improves intestinal barrier function and reduces inflammation after binge ethanol exposure and burn injury. Shock 2013, 39, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.T.; Friedrich, E.; Heuslein, J.L.; Pferdehirt, R.E.; Dangelo, N.M.; Natesan, S.; Christy, R.J.; Washburn, N.R. Reduction of burn progression with topical delivery of (antitumor necrosis factor-α)-hyaluronic acid conjugates. Wound Repair Regen. 2012, 20, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Simon, A.; Van Der Meer, J.W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [PubMed]
- Chen, Y.; Yang, W.; Zhang, X.; Yang, S.; Peng, G.; Wu, T.; Zhou, Y.; Huang, C.; Reinach, P.S.; Li, W.; et al. MK2 inhibitor reduces alkali burn-induced inflammation in rat cornea. Sci. Rep. 2016, 6, 28145. [Google Scholar] [CrossRef]
- Thuraisingam, T.; Xu, Y.Z.; Eadie, K.; Heravi, M.; Guiot, M.-C.; Greemberg, R.; Gaestel, M.; Radzioch, D. MAPKAPK-2 Signaling Is Critical for Cutaneous Wound Healing. J. Investig. Dermatol. 2010, 130, 278–286. [Google Scholar] [CrossRef]
- Zuloff-Shani, A.; Kachel, E.; Frenkel, O.; Orenstein, A.; Shinar, E.; Danon, D. Macrophage suspensions prepared from a blood unit for treatment of refractory human ulcers. Transfus. Apher. Sci. 2004, 30, 163–167. [Google Scholar] [CrossRef]
- Huang, G.; Sun, T.; Zhang, L.; Wu, Q.; Zhang, K.; Tian, Q.; Huo, R. Combined application of alginate dressing and human granulocyte-macrophage colony stimulating factor promotes healing in refractory chronic skin ulcers. Exp. Ther. Med. 2014, 7, 1772–1776. [Google Scholar] [CrossRef]
- Tanaka, R.; Masuda, H.; Kato, S.; Imagawa, K.; Kanabuchi, K.; Nakashioya, C.; Yoshiba, F.; Fukui, T.; Ito, R.; Kobori, M.; et al. Autologous G-CSF-Mobilized Peripheral Blood CD34+ Cell Therapy for Diabetic Patients with Chronic Nonhealing Ulcer. Cell Transplant. 2014, 23, 167–179. [Google Scholar] [CrossRef]
- NCT01785784. Available online: www.clincaltrial.gov (accessed on 15 February 2022).
- Yan, D.; Liu, S.; Zhao, X.; Bian, H.; Yao, X.; Xing, J.; Sun, W.; Chen, X. Recombinant human granulocyte macrophage colony stimulating factor in deep second-degree burn wound healing. Medicine 2017, 96, e6881. [Google Scholar] [CrossRef]
- Yan, H.; Chen, J.; Peng, X. Recombinant human granulocyte-macrophage colony-stimulating factor hydrogel promotes healing of deep partial thickness burn wounds. Burns 2012, 38, 877–881. [Google Scholar] [CrossRef]
- Camões, S.P.; Bulut, O.; Yazar, V.; Gaspar, M.M.; Simões, S.; Ferreira, R.; Vitorino, R.; Santos, J.M.; Gursel, I.; Miranda, J.P. 3D-MSCs A151 ODN-loaded exosomes are immunomodulatory and reveal a proteomic cargo that sustains wound resolution. J. Adv. Res. 2022; in press. [Google Scholar] [CrossRef]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [PubMed]
- Yamashita, T.; Takahashi, Y.; Takakura, Y. Possibility of Exosome-Based Therapeutics and Challenges in Production of Exosomes Eligible for Therapeutic Application. Biol. Pharm. Bull. 2018, 41, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Miyaki, S.; Ishitobi, H.; Matsuyama, S.; Nakasa, T.; Kamei, N.; Akimoto, T.; Higashi, Y.; Ochi, M. Mesenchymal-stem-cell-derived exosomes accelerate skeletal muscle regeneration. FEBS Lett. 2015, 589, 1257–1265. [Google Scholar] [CrossRef]
- Nakasa, T.; Ishikawa, M.; Shi, M.; Shibuya, H.; Adachi, N.; Ochi, M. Acceleration of muscle regeneration by local injection of muscle-specific microRNAs in rat skeletal muscle injury model. J. Cell. Mol. Med. 2009, 14, 2495–2505. [Google Scholar] [CrossRef]
- Sinha, A.; Principe, S.; Alfaro, J.; Ignatchenko, A.; Ignatchenko, V.; Kislinger, T. Proteomic Profiling of Secreted Proteins, Exosomes, and Microvesicles in Cell Culture Conditioned Media. Methods Mol. Biol. 2017, 1722, 91–102. [Google Scholar]
- Jiang, N. Immune engineering: From systems immunology to engineering immunity. Curr. Opin. Biomed. Eng. 2017, 1, 54–62. [Google Scholar] [CrossRef]
- Kim, S.; Shah, S.B.; Graney, P.L.; Singh, A. Multiscale engineering of immune cells and lymphoid organs. Nat. Rev. Mater. 2019, 4, 355–378. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, T.; Sun, X. Vascular Endothelial Growth Factor Gene Delivery by Magnetic DNA Nanospheres Ameliorates Limb Ischemia in Rabbits1. J. Surg. Res. 2005, 126, 48–54. [Google Scholar] [CrossRef]
- Gui, L.; Chen, Y.; Diao, Y.; Chen, Z.; Duan, J.; Liang, X.; Li, Y. ROS-responsive nanoparticle-mediated delivery of CYP2J2 gene for therapeutic angiogenesis in severe hindlimb ischemia. Mater Today Bio. 2022, 13, 100192. [Google Scholar] [CrossRef]
- Wang, P.; Huang, S.; Hu, Z.; Yang, W.; Lan, Y.; Zhu, J.; Hancharou, A.; Guo, R.; Tang, B. In situ formed anti-inflammatory hydrogel loading plasmid DNA encoding VEGF for burn wound healing. Acta Biomater. 2019, 100, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Pang, Q.; Lou, D.; Li, S.; Wang, G.; Qiao, B.; Dong, S.; Ma, L.; Gao, C.; Wu, Z. Smart Flexible Electronics-Integrated Wound Dressing for Real-Time Monitoring and On-Demand Treatment of Infected Wounds. Adv. Sci. 2020, 7, 1902673. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Zhan, Y.; Mao, C.; Xie, X.; Lin, Y. The biological applications of DNA nanomaterials: Current challenges and future directions. Signal Transduct. Target. Ther. 2021, 6, 351. [Google Scholar]
- Lou, D.; Luo, Y.; Pang, Q.; Tan, W.-Q.; Ma, L. Gene-activated dermal equivalents to accelerate healing of diabetic chronic wounds by regulating inflammation and promoting angiogenesis. Bioact. Mater. 2020, 5, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Bi, S.; Xiu, B.; Ye, J.; Dong, Y. Target-Catalyzed DNA Four-Way Junctions for CRET Imaging of MicroRNA, Concatenated Logic Operations, and Self-Assembly of DNA Nanohydrogels for Targeted Drug Delivery. ACS Appl. Mater. Interfaces 2015, 7, 23310–23319. [Google Scholar] [CrossRef] [PubMed]
- Douglas, S.M.; Bachelet, I.; Church, G.M. A Logic-Gated Nanorobot for Targeted Transport of Molecular Payloads. Science 2012, 335, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, M.; Gao, X.; Yu, Z.; Pan, W.; Wang, H.; Tang, B. A DNA Tetrahedron Nanoprobe with Controlled Distance of Dyes for Multiple Detection in Living Cells and in Vivo. Anal. Chem. 2017, 89, 6670–6677. [Google Scholar] [CrossRef]
- Lv, Y.; Hu, R.; Zhu, G.; Zhang, X.; Mei, L.; Liu, Q.; Qiu, L.; Wu, C.; Tan, W. Preparation and biomedical applications of programmable and multifunctional DNA nanoflowers. Nat. Protoc. 2015, 10, 1508–1524. [Google Scholar] [CrossRef]
- Wu, C.; Han, D.; Chen, T.; Peng, L.; Zhu, G.; You, M.; Qiu, L.; Sefah, K.; Zhang, X.; Tan, W. Building a Multifunctional Aptamer-Based DNA Nanoassembly for Targeted Cancer Therapy. J. Am. Chem. Soc. 2013, 135, 18644–18650. [Google Scholar] [CrossRef]
- Bock, L.C.; Griffin, L.C.; Latham, J.A.; Vermaas, E.H.; Toole, J.J. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature 1992, 355, 564–566. [Google Scholar] [CrossRef]
- Wang, K.Y.; McCurdy, S.; Shea, R.G.; Swaminathan, S.; Bolton, P.H. A DNA aptamer which binds to and inhibits thrombin exhibits a new structural motif for DNA. Biochemistry 1993, 32, 1899–1904. [Google Scholar] [CrossRef] [PubMed]
- Stoltenburg, R.; Reinemann, C.; Strehlitz, B. SELEX—A (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng. 2007, 24, 381–403. [Google Scholar] [PubMed]
- Tuerk, C.; Gold, L. Systematic Evolution of Ligands by Exponential Enrichment: RNA Ligands to Bacteriophage T4 DNA Polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- McGuire, V.A.; Arthur, J.S. Subverting Toll-Like Receptor Signaling by Bacterial Pathogens. Front. Immunol. 2015, 6, 607. [Google Scholar] [CrossRef]
- Berezhnoy, A.; Stewart, C.A.; Ii, J.O.M.; Thiel, W.; Giangrande, P.; Trinchieri, G.; Gilboa, E. Isolation and Optimization of Murine IL-10 Receptor Blocking Oligonucleotide Aptamers Using High-throughput Sequencing. Mol. Ther. 2012, 20, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Hirota, M.; Murakami, I.; Ishikawa, Y.; Suzuki, T.; Sumida, S.-I.; Ibaragi, S.; Kasai, H.; Horai, N.; Drolet, D.W.; Gupta, S.; et al. Chemically Modified Interleukin-6 Aptamer Inhibits Development of Collagen-Induced Arthritis in Cynomolgus Monkeys. Nucleic Acid Ther. 2016, 26, 10–19. [Google Scholar] [CrossRef]
- Engelen, S.E.; Robinson, A.J.; Zurke, Y.X.; Monaco, C. Therapeutic strategies targeting inflammation and immunity in atherosclerosis: How to proceed? Nat. Rev. Cardiol. 2022, 1–21. [Google Scholar]
- Ford, M.L.; Adams, A.; Pearson, T.C. Targeting co-stimulatory pathways: Transplantation and autoimmunity. Nat. Rev. Nephrol. 2014, 10, 14–24. [Google Scholar] [CrossRef]
- Hirsh, V.; Paz-Ares, L.; Boyer, M.; Rosell, R.; Middleton, G.; Eberhardt, W.E.; Szczesna, A.; Reiterer, P.; Saleh, M.; Arrieta, O.; et al. Randomized Phase III Trial of Paclitaxel/Carboplatin with or Without PF-3512676 (Toll-Like Receptor 9 Agonist) As First-Line Treatment for Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2011, 29, 2667–2674. [Google Scholar] [CrossRef]
- Hwang, B.; Han, K.; Lee, S.-W. Prevention of passively transferred experimental autoimmune myasthenia gravis by an in vitro selected RNA aptamer. FEBS Lett. 2003, 548, 85–89. [Google Scholar] [CrossRef]
- Cao, X.; Li, S.; Chen, L.; Ding, H.; Xu, H.; Huang, Y.; Li, J.; Liu, N.; Cao, W.; Zhu, Y.; et al. Combining use of a panel of ssDNA aptamers in the detection of Staphylococcus aureus. Nucleic Acids Res. 2009, 37, 4621–4628. [Google Scholar] [CrossRef] [PubMed]
- Ramlal, S.; Mondal, B.; Lavu, P.S.; Bhavanashri, N.; Kingston, J. Capture and detection of Staphylococcus aureus with dual labeled aptamers to cell surface components. Int. J. Food Microbiol. 2018, 265, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Nguyen, D.T.; Yeo, T.; Bin Lim, S.; Tan, W.X.; Madden, L.E.; Jin, L.; Long, J.Y.K.; Aloweni, F.A.B.; Liew, Y.J.A.; et al. A flexible multiplexed immunosensor for point-of-care in situ wound monitoring. Sci. Adv. 2021, 7, eabg9614. [Google Scholar] [CrossRef] [PubMed]
- National Academy Press. Frontiers of Engineering: Reports on Leading-Edge Engineering from the 2017 Symposium; National Academy of Engineering; National Academy Press: Washington, DC, USA, 2018. [Google Scholar] [CrossRef]
- Dellacherie, M.O.; Seo, B.R.; Mooney, D.J. Macroscale biomaterials strategies for local immunomodulation. Nat. Rev. Mater. 2019, 4, 379–397. [Google Scholar] [CrossRef]
- Scott, E.A.; Karabin, N.B.; Augsornworawat, P. Overcoming Immune Dysregulation with Immunoengineered Nanobiomaterials. Annu. Rev. Biomed. Eng. 2017, 19, 57–84. [Google Scholar] [CrossRef]
- Xie, J.; Lee, S.; Chen, X. Nanoparticle-based theranostic agents. Adv. Drug Deliv. Rev. 2010, 62, 1064–1079. [Google Scholar] [CrossRef]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef]
- Truskewycz, A.; Truong, V.K.; Ball, A.S.; Houshyar, S.; Nassar, N.; Yin, H.; Murdoch, B.J.; Cole, I. Fluorescent Magnesium Hydroxide Nanosheet Bandages with Tailored Properties for Biocompatible Antimicrobial Wound Dressings and pH Monitoring. ACS Appl. Mater. Interfaces 2021, 13, 27904–27919. [Google Scholar] [CrossRef]
- Lenzer, J. FDA advisers warn: COX 2 inhibitors increase risk of heart attack and stroke. BMJ 2005, 330, 440. [Google Scholar] [CrossRef]
- Stone Ii, R.; Natesan, S.; Kowalczewski, C.J.; Mangum, L.H.; Clay, N.E.; Clohessy, R.M.; Carlsson, A.H.; Tassin, D.H.; Chan, R.K.; Rizzo, J.A.; et al. Advancements in Regenerative Strategies Through the Continuum of Burn Care. Front. Pharmacol. 2018, 9, 672. [Google Scholar]
- Watson, D.C.; Yung, B.C.; Bergamaschi, C.; Chowdhury, B.; Bear, J.; Stellas, D.; Pavlakis, G.N. Scalable, cGMP-compatible purification of extracellular vesicles carrying bioactive human heterodimeric IL-15/lactadherin complexes. J. Extracell. Vesicles 2018, 7, 1442088. [Google Scholar] [CrossRef] [PubMed]
- Yoo, K.W.; Li, N.; Makani, V.; Singh, R.; Atala, A.; Lu, B. Large-Scale Preparation of Extracellular Vesicles Enriched with Specific microRNA. Tissue Eng. Part C Methods 2018, 24, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Rohde, E.; Pachler, K.; Gimona, M. Manufacturing and characterization of extracellular vesicles from umbilical cord–derived mesenchymal stromal cells for clinical testing. Cytotherapy 2019, 21, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Pachler, K.; Lener, T.; Streif, D.; Dunai, Z.A.; Desgeorges, A.; Feichtner, M.; Öller, M.; Schallmoser, K.; Rohde, E.; Gimona, M. A Good Manufacturing Practice–grade standard protocol for exclusively human mesenchymal stromal cell–derived extracellular vesicles. Cytotherapy 2017, 19, 458–472. [Google Scholar] [CrossRef] [PubMed]
- Andriolo, G.; Provasi, E.; Cicero, V.L.; Brambilla, A.; Soncin, S.; Torre, T.; Milano, G.; Biemmi, V.; Vassalli, G.; Turchetto, L.; et al. Exosomes from Human Cardiac Progenitor Cells for Therapeutic Applications: Development of a GMP-Grade Manufacturing Method. Front. Physiol. 2018, 9, 1169. [Google Scholar] [CrossRef]
- Silva, A.K.A.; Morille, M.; Piffoux, M.; Arumugam, S.; Mauduit, P.; Larghero, J.; Banzet, S. Development of extracellular vesicle-based medicinal products: A position paper of the group “Extracellular Vesicle translatiOn to clinicaL perspectiVEs-EVOLVE France”. Adv. Drug Deliv. Rev. 2021, 179, 114001. [Google Scholar] [CrossRef]
- Zipkin, M. Exosome redux. Nat. Biotechnol. 2019, 37, 1395–1400. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Growth Factors | Cell Source | Functions | Ref. |
|---|---|---|---|
| Fibroblast growth factor 2 (FGF2) |
| Chemotactic for fibroblasts; Mitogenic for fibroblasts and keratinocytes; Stimulates keratinocyte migration, angiogenesis wound contraction and matrix production. | [65,66] |
| Epidermal growth factor (EGF) |
| Mitogenic for keratinocytes and fibroblasts; Stimulates re-epithelialization and granulation tissue formation. | [65] |
| Platelet derived growth factor (PDGF) |
| Chemotactic for neutrophils macrophages, fibroblasts and smooth muscle cells; Stimulates production of matrix metalloproteinases, fibronectin and hyaluronic acid; Stimulates angiogenesis. | [65,67,68,69,70] |
| Transforming growth factor β (TGFβ) |
| Most important factor in wound healing; Maintains monocyte chemotaxis, fibroblast migration and differentiation; Angiogenesis and fibronectin synthesis; Regulates increased synthesis of collagen and extracellular matrix and decreased degradation by matrix metalloproteinase. | [65,71,72,73,74] |
| Vascular Endothelial growth factor (VEGF) |
| Increases vascular permeability; Mitogenic for endothelial cells. | [65,75,76] |
| Approach | Injury Type | Outcomes | Limitations | Ref. |
|---|---|---|---|---|
| Pharmacological agents | ||||
| NSAIDs | Debrided combat-related extremity wounds |
|
| [97] |
| COX-2 Inhibitor (Celecoxib) | Sciatic Nerve Crush |
|
| [98] |
| Skeletal muscle ischemia/reperfusion (I/R) injury | Inducible nitric oxide synthase (iNOS) inhibitor (1400W) |
| [99] | |
| Manganese superoxide dismutase (MnSOD) mimetic molecule, MnE | Dermal full-thickness excision injury |
| [100] | |
| Injectable curcumin-loaded Zn-Al layer double hydroxide nanocomposites | Intramuscular implantation |
| [101] | |
| Biological and synthetic platforms | ||||
| Modification to surface topography and hydrophilicity | In vitro, neutrophil activation and macrophage polarization |
|
| [102] |
| Heparin-immobilized copolymers of L--lactide (LA) and 5-methyl-5-benzyloxycar-bonate-1,3-dioxan-2-one (MBC) on metal stents | Porcine coronary artery injury model |
|
| [103] |
| Biologically derived surgical mesh materials | In situ polarization of macrophages responding to implanted mesh materials |
|
| [104] |
| Dermal ECM (D-ECM) or Urinary bladder matrix ECM (UBM-ECM) coating polypropylene mesh | In vivo macrophage polarization following mesh implantation in a rodent model |
|
| [105] |
| Keratin and Collagen coatings (films) | In vitro macrophage polarization |
| [106] | |
| Cell and cytokine therapies | ||||
| Macrophage polarization | In vitro model—monocytes embedded in modified hydrogel |
|
| [107] |
| Mesenchymal stem cells (MSCs) | Mouse lethal radiation injury |
| [108] | |
| Human bone maow stromal cells (BM-SC) | Specialized in vitro culture for modulating cell phenotype |
| [109] | |
| Cell secretome and extracellular vesicles | ||||
| Mesenchymal Stem Cells (MSC) Extracellular vesicles (EVs) | Bone marrow-derived macrophage polarization, Cardiotoxin-induced skeletal muscle injury |
| [110] | |
| Amniotic fluid stem cell-derived extracellular vesicle | Cardiotoxin induced tibialis anterior mouse muscle injury |
| [111] | |
| Approach | Injury Type | Outcomes | Ref. |
|---|---|---|---|
| Nucleic acid and aptamers based immune targeting | |||
| Intra-arterial VEGF gene delivery by magnetic DNA nano spheres | Rabbit limb ischemia model |
| [214] |
| Nanoparticle-based pcDNA3.1-CYP2J2 plasmid DNA (pDNA) delivery system (nanoparticle/pDNA complex) | Mouse limb ischemia model |
| [215] |
| Hydrogel loading plasmid DNA encoding VEGF | Mouse burn wound model |
| [216] |
| Theranostics immune targeting | |||
| Smart flexible electronics-integrated wound dressing | Pig full thickness wound model |
| [217] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muire, P.J.; Thompson, M.A.; Christy, R.J.; Natesan, S. Advances in Immunomodulation and Immune Engineering Approaches to Improve Healing of Extremity Wounds. Int. J. Mol. Sci. 2022, 23, 4074. https://doi.org/10.3390/ijms23084074
Muire PJ, Thompson MA, Christy RJ, Natesan S. Advances in Immunomodulation and Immune Engineering Approaches to Improve Healing of Extremity Wounds. International Journal of Molecular Sciences. 2022; 23(8):4074. https://doi.org/10.3390/ijms23084074
Chicago/Turabian StyleMuire, Preeti J., Marc A. Thompson, Robert J. Christy, and Shanmugasundaram Natesan. 2022. "Advances in Immunomodulation and Immune Engineering Approaches to Improve Healing of Extremity Wounds" International Journal of Molecular Sciences 23, no. 8: 4074. https://doi.org/10.3390/ijms23084074
APA StyleMuire, P. J., Thompson, M. A., Christy, R. J., & Natesan, S. (2022). Advances in Immunomodulation and Immune Engineering Approaches to Improve Healing of Extremity Wounds. International Journal of Molecular Sciences, 23(8), 4074. https://doi.org/10.3390/ijms23084074