
Anti-Inflammatory Activities of an Anti-Histamine Drug, Loratadine, by Suppressing TAK1 in AP-1 Pathway

 , and
, and 
Abstract
:1. Introduction
2. Results
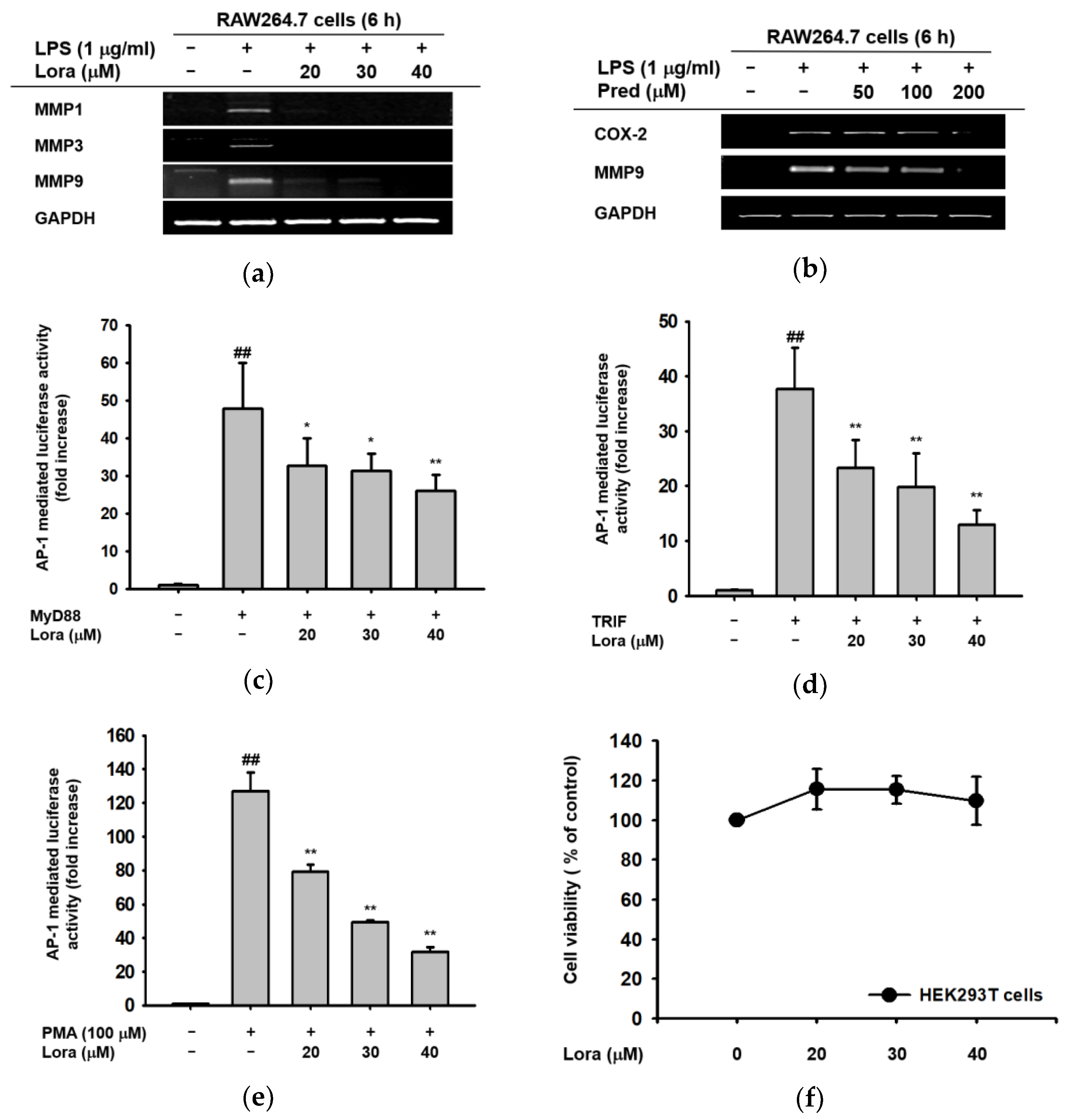
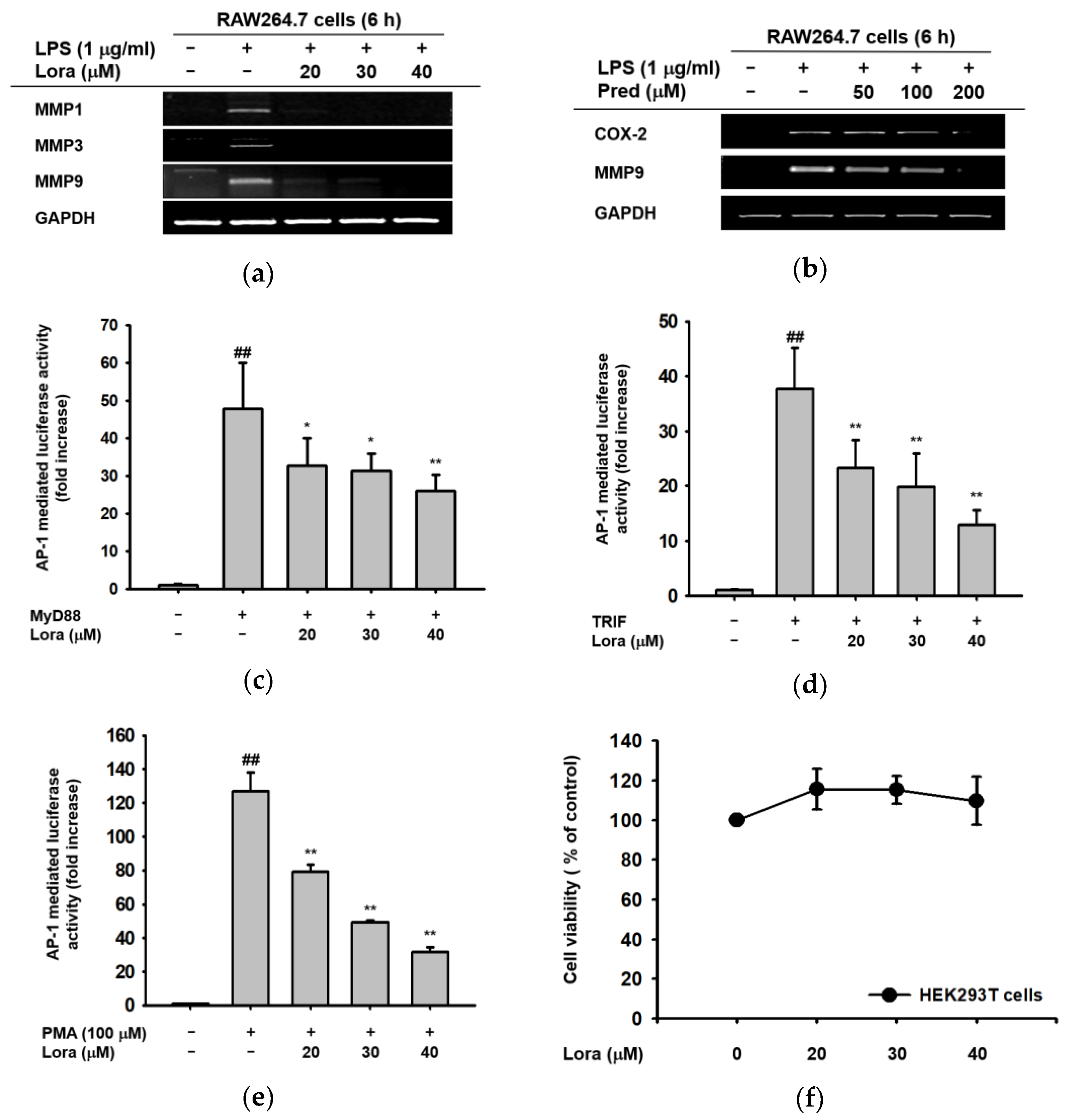
2.1. Anti-Inflammatory Effects of Loratadine Are Mediated by Its Transcriptional Regulation of Pro-Inflammatory Genes
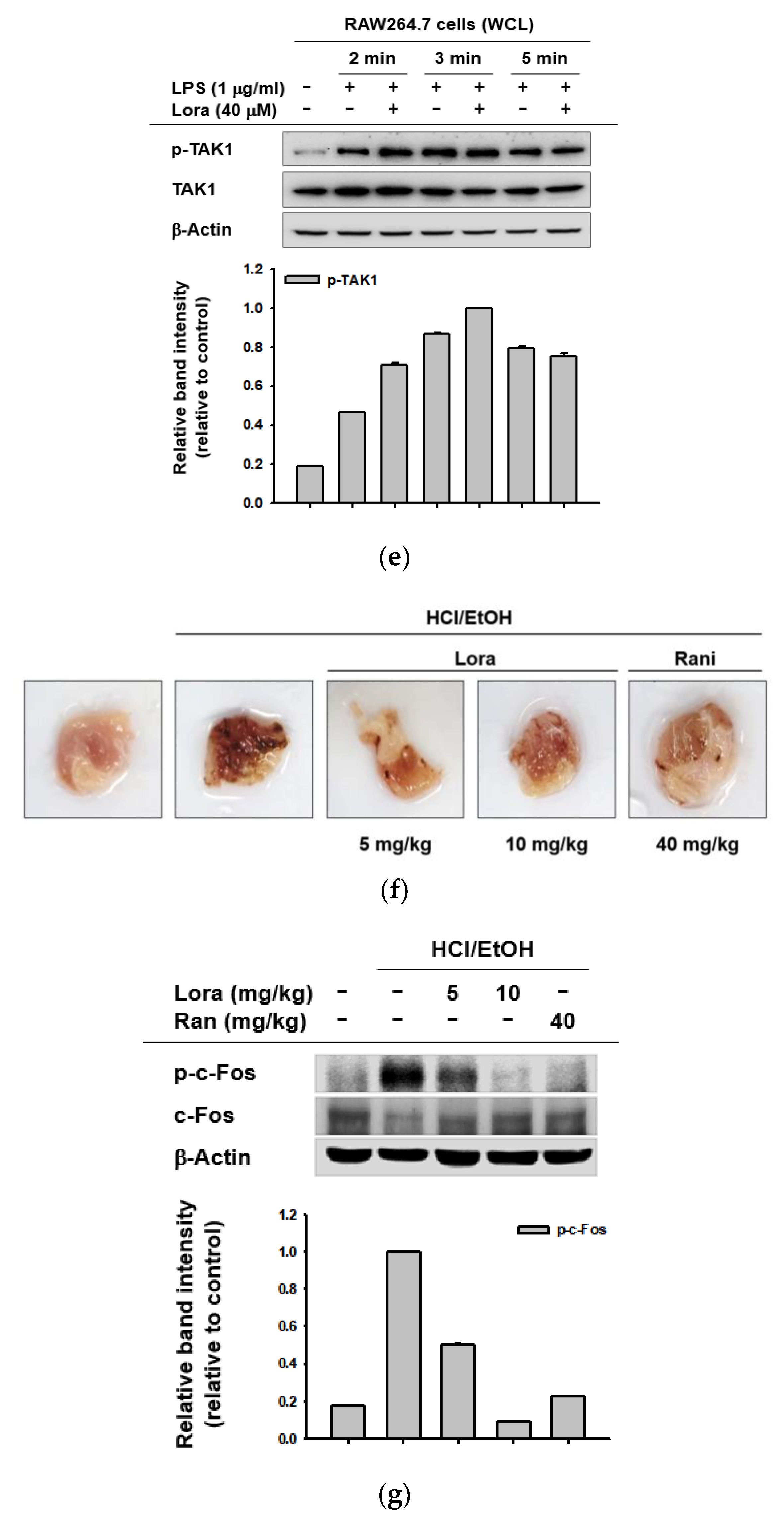
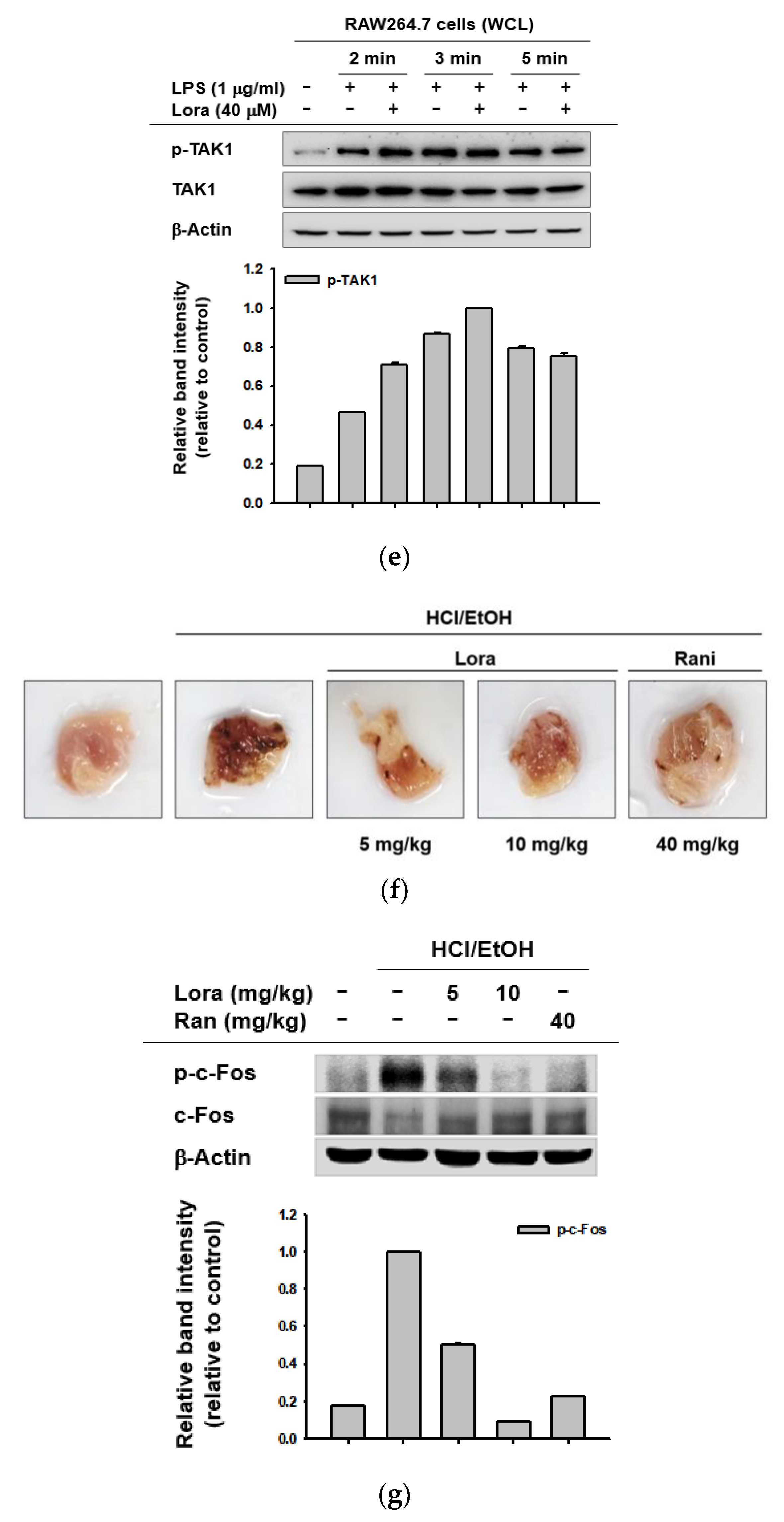
2.2. Loratadine Targets the AP-1 Signaling Pathway to Reduce Inflammatory Responses Both In Vitro and In Vivo
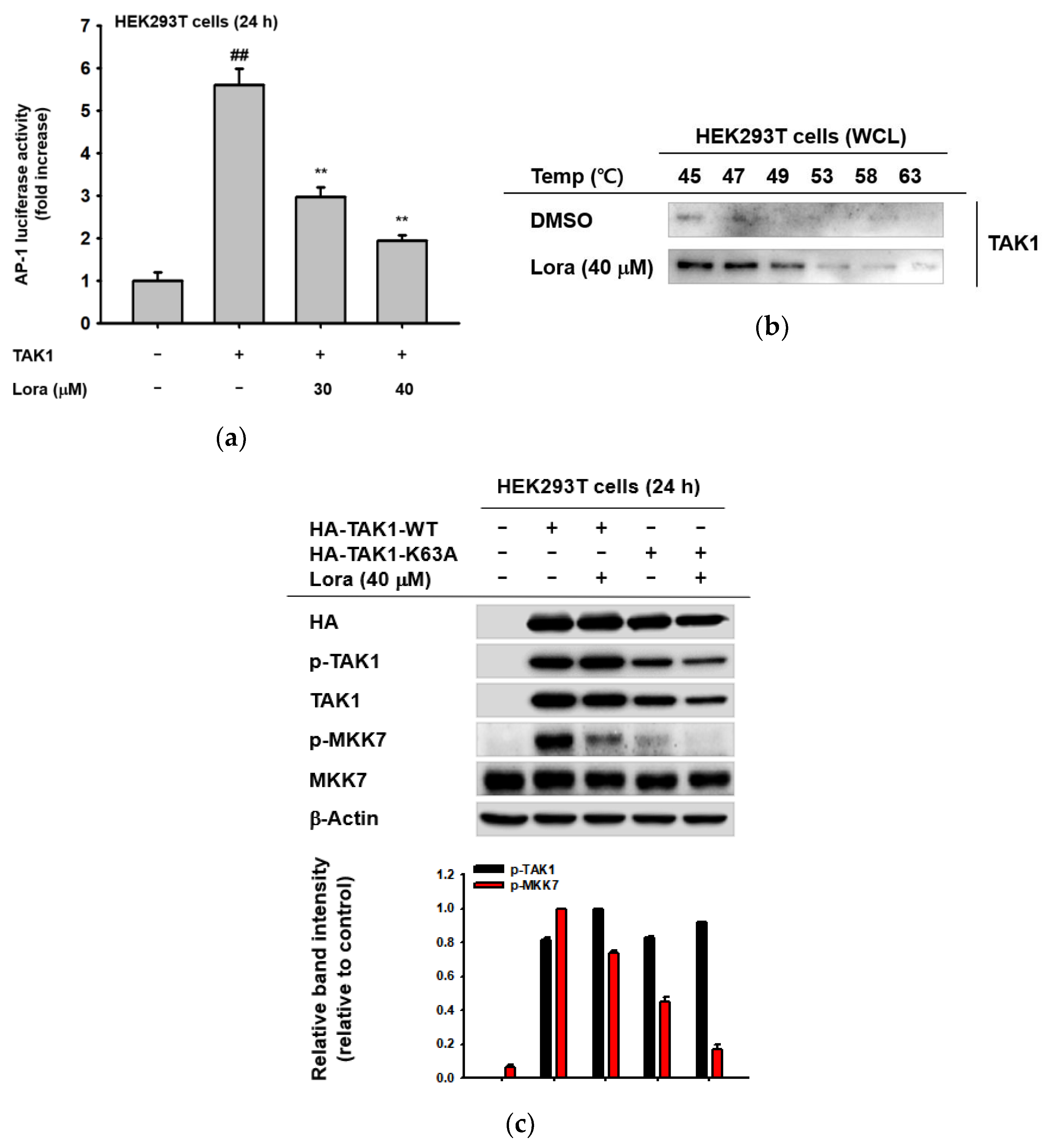
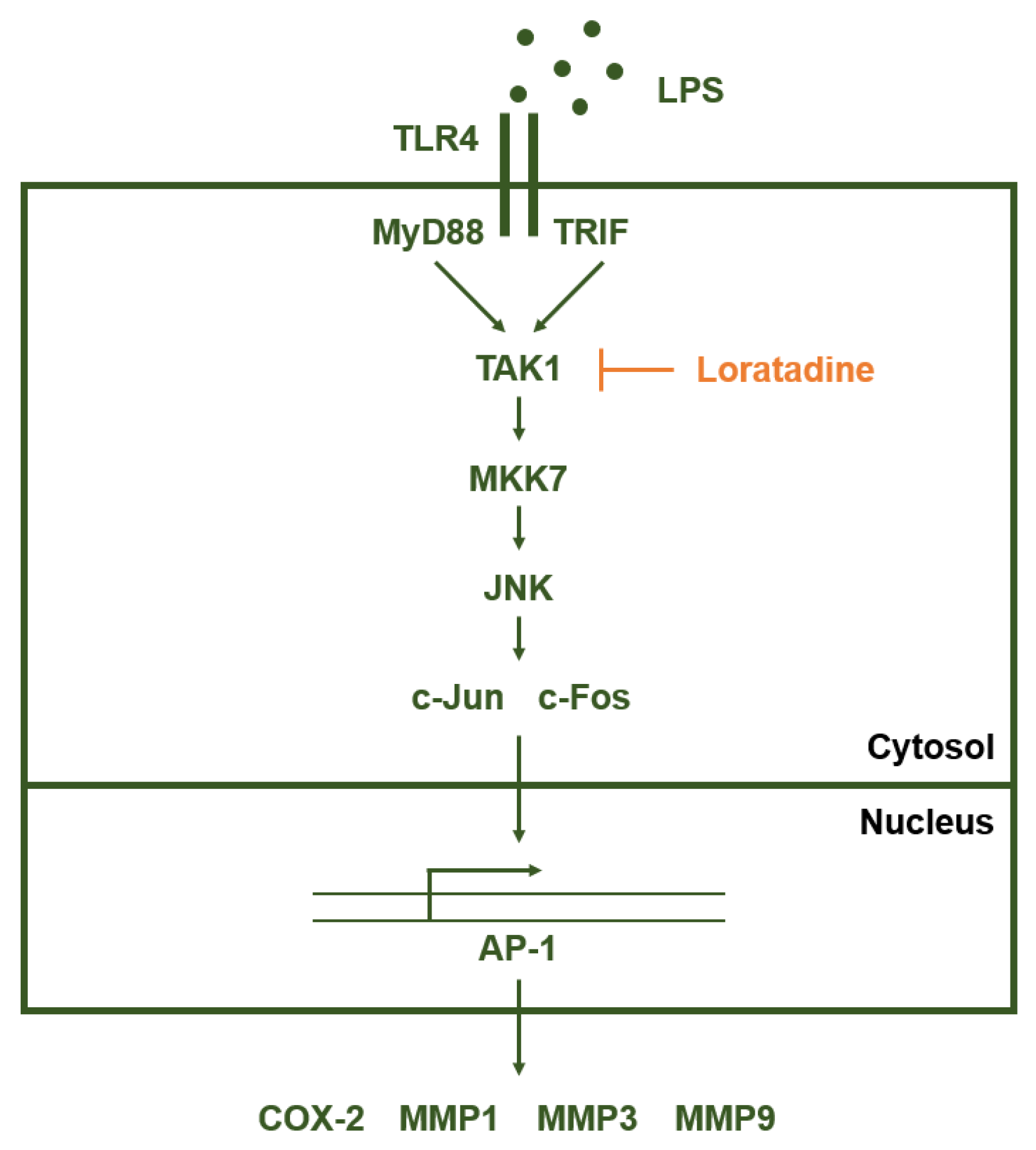
2.3. TAK1 Is the Prime Target Molecule of Loratadine
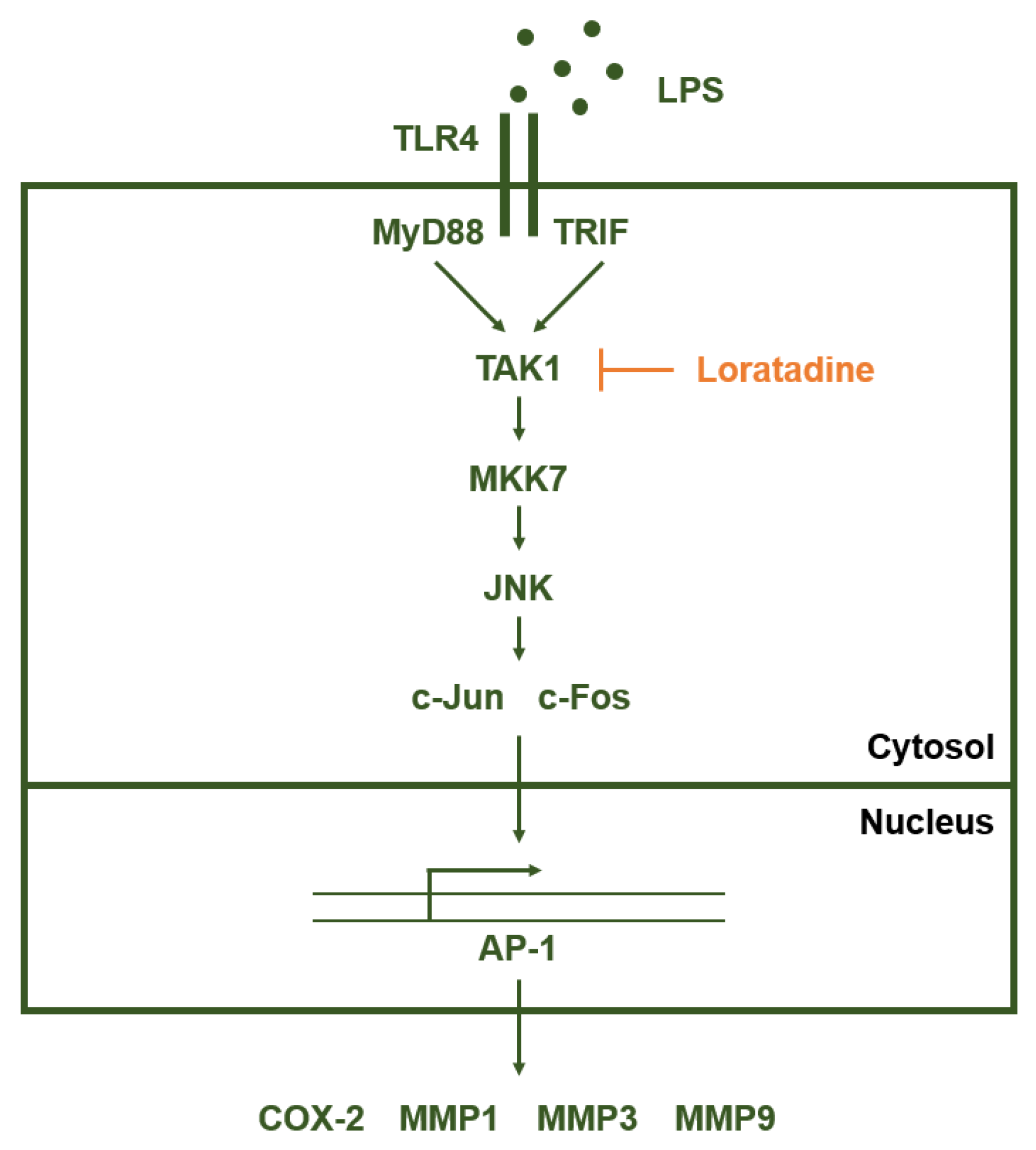
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Compound Preparation, Animals, and Cell Culture
4.3. Gastritis Mouse Model
4.4. mRNA Expression Analysis Using Reverse-Transcription Polymerase Chain Reaction
4.5. Luciferase Reporter Assay
4.6. Cell Viability Assay
4.7. Total Cell Lysate Preparation
4.8. Western Blotting
4.9. Gene Overexpression
4.10. Cellular Thermal Shift Assay
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hawiger, J. Innate immunity and inflammation: A transcriptional paradigm. Immunol. Res. 2001, 23, 99–110. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Larsen, G.L.; Henson, P.M. Mediators of inflammation. Annu. Rev. Immunol. 1983, 1, 335–359. [Google Scholar] [CrossRef]
- Gupta, S.C.; Kunnumakkara, A.B.; Aggarwal, S.; Aggarwal, B.B. Inflammation, a double-edge sword for cancer and other age-related diseases. Front. Immunol. 2018, 9, 2160. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [Green Version]
- Kotas, M.E.; Medzhitov, R. Homeostasis, inflammation, and disease susceptibility. Cell 2015, 160, 816–827. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.; Harmon, C.; O’Farrelly, C. Liver immunology and its role in inflammation and homeostasis. Cell. Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Ratan, Z.A.; Youn, S.H.; Kwak, Y.-S.; Han, C.-K.; Haidere, M.F.; Kim, J.K.; Min, H.; Jung, Y.-J.; Hosseinzadeh, H.; Hyun, S.H.; et al. Adaptogenic effects of Panax ginseng on modulation of immune functions. J. Ginseng Res. 2021, 45, 32–40. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signals that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef]
- Zindel, J.; Kubes, P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 493–518. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Yi, Y.-S.; Kim, M.-Y.; Cho, J.Y. Role of ginsenosides, the main active components of Panax ginseng, in inflammatory responses and diseases. J. Ginseng Res. 2017, 41, 435–443. [Google Scholar] [CrossRef] [Green Version]
- Ratan, Z.A.; Haidere, M.F.; Hong, Y.H.; Park, S.H.; Lee, J.-O.; Lee, J.; Cho, J.Y. Pharmacological potential of ginseng and its major component ginsenosides. J. Ginseng Res. 2021, 45, 199–210. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, H.G.; Kim, E.; Han, S.Y.; Aziz, N.; Yi, Y.-S.; Kim, S.; Lee, Y.; Yoo, B.C.; Han, J.-W.; et al. Isoprenylcysteine carboxyl methyltransferase and its substrate Ras are critical players regulating TLR-mediated inflammatory responses. Cells 2020, 9, 1216. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef]
- Kim, H.G.; Kim, J.H.; Yang, W.S.; Kim, E.; Jeong, D.; Park, J.G.; Aziz, N.; Kim, S.; Parameswaran, N.; Choy, J.Y. Syk-MyD88 axis Is a critical determinant of inflammatory-response in activated macrophages. Front. Immunol. 2021, 12, 767366. [Google Scholar] [CrossRef]
- Fitzgerald, K.; Rowe, D.C.; Barnes, B.J.; Caffrey, D.R.; Visintin, A.; Latz, E.; Monks, B.; Pitha, P.M.; Golenbock, D.T. LPS-TLR4 signaling to IRF-3/7 and NF-κB involves the Toll adapters TRAM and TRIF. J. Exp. Med. 2003, 198, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Xue, Q.; He, N.; Wang, Z.; Fu, X.; Aung, L.H.H.; Liu, Y.; Li, M.; Cho, J.Y.; Yang, Y.; Yu, T. Functional roles and mechanisms of ginsenosides from Panax ginseng in atherosclerosis. J. Ginseng Res. 2021, 45, 22–31. [Google Scholar] [CrossRef]
- Ha, A.T.; Rahmawati, L.; You, L.; Hossain, M.A.; Kim, J.-H.; Cho, J.Y. Anti-inflammatory, antioxidant, moisturizing, and antimelanogenesis effects of quercetin 3-O-β-D-glucuronide in human keratinocytes and melanoma cells via activation of NF-κB and AP-1 pathways. Int. J. Mol. Sci. 2021, 23, 433. [Google Scholar] [CrossRef]
- Kim, E.; Ahuja, A.; Kim, M.-Y.; Cho, J.Y. DNA or protein methylation-dependent regulation of activator protein-1 function. Cells 2021, 10, 461. [Google Scholar] [CrossRef]
- Schonthaler, H.B.; Guinea-Viniegra, J.; Wagner, E.F. Targeting inflammation by modulating the Jun/AP-1 pathway. Ann. Rheum. 2011, 70, i109–i112. [Google Scholar] [CrossRef]
- Liu, X.; Yin, S.; Chen, Y.; Wu, Y.; Zheng, W.; Dong, H.; Bai, Y.; Qin, Y.; Li, J.; Feng, S.J.; et al. LPS-induced proinflammatory cytokine expression in human airway epithelial cells and macrophages via NF-κB, STAT3 or AP-1 activation. Mol. Med. Rep. 2018, 17, 5484–5491. [Google Scholar] [CrossRef] [Green Version]
- Whitmarsh, A.J.; Davis, R.J. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J. Mol. Med. 1996, 74, 589–607. [Google Scholar] [CrossRef]
- Silvers, A.L.; Bachelor, M.A.; Bowden, G.T. The Role of JNK and p38 MAPK Activities in UVA-induced signaling pathways leading to AP-1 activation and c-Fos expression. Neoplasia 2003, 5, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.; Robinson, M.; Smith, E.; Huntley, S.; Prime, S.; Paterson, I. Induction of an epithelial to mesenchymal transition in human immortal and malignant keratinocytes by TGF-β1 involves MAPK, Smad and AP-1 signalling pathways. J. Cell. Biochem. 2005, 95, 918–931. [Google Scholar] [CrossRef]
- Morse, D.; Pischke, S.E.; Zhou, Z.; Davis, R.J.; Flavell, R.A.; Loop, T.; Otterbein, S.L.; Otterbein, L.E.; Choi, A.M.K. Suppression of inflammatory cytokine production by carbon monoxide involves the JNK pathway and AP-1. J. Biol. Chem. 2003, 278, 36993–36998. [Google Scholar] [CrossRef] [Green Version]
- Kajanne, R.; Miettinen, P.; Mehlem, A.; Leivonen, S.-K.; Birrer, M.; Foschi, M.; Kähäri, V.-M.; Leppä, S. EGF-R regulates MMP function in fibroblasts through MAPK and AP-1 pathways. J. Cell. Physiol. 2007, 212, 489–497. [Google Scholar] [CrossRef]
- Bergman, M.R.; Cheng, S.; Honbo, N.; Piacentini, L.; Karliner, J.S.; Lovett, D.H. A functional activating protein 1 (AP-1) site regulates matrix metalloproteinase 2 (MMP-2) transcription by cardiac cells through interactions with JunB-Fra1 and JunB-FosB heterodimers. Biochem. J. 2003, 369, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Barchowsky, A.; Frleta, D.; Vincenti, M.P. Integration of the NF-κB and mitogen-activated protein kinase/AP-1 pathways at the collagenase-1 promoter: Divergence of IL-1 and TNF-dependent signal transduction in rabbit primary synovial fibroblasts. Cytokine 2000, 12, 1469–1479. [Google Scholar] [CrossRef]
- Subbaramaiah, K.; Norton, L.; Gerald, W.; Dannenberg, A.J. Cyclooxygenase-2 is overexpressed in HER-2/neu-positive breast cancer: Evidence for involvement of AP-1 and PEA3. J. Biol. Chem. 2002, 277, 18649–18657. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.-W.; Park, K.; Kweon, G.R.; Jang, B.-C.; Baek, W.-K.; Suh, M.-H.; Kim, C.-W.; Lee, K.-S.; Suh, S.-I. Curcumin inhibits the expression of COX-2 in UVB-irradiated human keratinocytes (HaCaT) by inhibiting activation of AP-1: p38 MAP kinase and JNK as potential upstream targets. Exp. Mol. Med. 2005, 37, 186–192. [Google Scholar] [CrossRef] [Green Version]
- Arrang, J.-M.; Garbarg, M.; Schwartz, J.-C. Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature 1983, 302, 832–837. [Google Scholar] [CrossRef]
- Lichtenstein, L.M.; Gillespie, E. Inhibition of histamine release by histamine controlled by H2 receptor. Nature 1973, 244, 278–288. [Google Scholar] [CrossRef]
- Thangam, E.B.; Jemima, E.A.; Singh, H.; Baig, M.S.; Khan, M.; Mathias, C.B.; Church, M.K.; Saluja, R. The role of histamine and histamine receptors in mast cell-mediated allergy and inflammation: The hunt for new therapeutic targets. Front. Immunol. 2018, 9, 1873. [Google Scholar] [CrossRef] [Green Version]
- Branco, A.C.C.C.; Yoshikawa, F.S.Y.; Pietrobon, A.J.; Sato, M.N. Role of histamine in modulating the immune response and inflammation. Mediat. Inflamm. 2018, 2018, 9524075. [Google Scholar] [CrossRef]
- Kreutner, W.; Chapman, R.W.; Gulbenkian, A.; Siegel, M.I. Antiallergic activity of loratadine, a non-sedating antihistamine. Allergy 1987, 42, 57–63. [Google Scholar] [CrossRef]
- Kay, G.; Harris, A.G. Loratadine: A non-sedating antihistamine. Review of its effects on cognition, psychomotor performance, mood and sedation. Clin. Exp. Allergy 1999, 29, 147–150. [Google Scholar] [CrossRef]
- Roumestan, C.; Henriquet, C.; Gougat, C.; Michel, A.; Bichon, F.; Portet, K.; Jaffuel, D.; Mathieu, M. Histamine H1-receptor antagonists inhibit nuclear factor-kappaB and activator protein-1 activities via H1-receptor-dependent and-independent mechanisms. Clin. Exp. Allergy 2008, 38, 947–956. [Google Scholar] [CrossRef]
- Juergens, U.R.; Gillissen, A.; Uen, S.; Racké, K.; Stöber, M.; Darlath, W.; Vetter, H. New evidence of H1-receptor independent COX-2 inhibition by fexofenadine HCl in vitro. Pharmacology 2006, 78, 129–135. [Google Scholar] [CrossRef]
- Zhou, Y.; Gao, C.; Wang, H.; Liu, L.; Huang, Z.; Fa, X. Histamine H1 type receptor antagonist loratadine ameliorates oxidized LDL induced endothelial dysfunction. Biomed. Pharmacother. 2018, 106, 1448–1453. [Google Scholar] [CrossRef]
- Mitrea, D.-R.; Clichici, S.; Filip, A.; Olteanu, D.; Bâldea, I.; Moldovan, R.; Decea, N.; Hoteiuc, O.-A. Resveratrol and loratadine effects on oxidative stress induced by experimental inflammation. Studia UBB Chemia 2017, 62, 89–100. [Google Scholar] [CrossRef]
- Traidl-Hoffmann, C.; Münster, I.; Ring, J.; Behrendt, H. Impact of desloratadine and loratadine on the crosstalk between human keratinocytes and leukocytes: Implications for anti-inflammatory activity of antihistamines. Int. Arch. Allergy Immunol. 2006, 140, 315–320. [Google Scholar] [CrossRef]
- Hunto, S.T.; Kim, H.G.; Baek, K.-S.; Jeong, D.; Kim, E.; Kim, J.H.; Cho, J.Y. Loratadine, an antihistamine drug, exhibits anti-inflammatory activity through suppression of the NF-kB pathway. Biochem. Pharmacol. 2020, 177, 113949. [Google Scholar] [CrossRef]
- Benbow, U.; Brinckerhoff, C.E. The AP-1 site and MMP gene regulation: What is all the fuss about? Matrix Biol. 1997, 15, 519–526. [Google Scholar] [CrossRef]
- Liacini, A.; Sylvester, J.; Li, W.Q.; Zafarullah, M. Inhibition of interleukin-1-stimulated MAP kinases, activating protein-1 (AP-1) and nuclear factor kappa B (NF-κB) transcription factors down-regulates matrix metalloproteinase gene expression in articular chondrocytes. Matrix Biol. 2002, 21, 251–262. [Google Scholar] [CrossRef]
- Sakurai, H.; Shigemori, N.; Hisada, Y.; Ishizuka, T.; Kawashima, K.; Sugita, T. Suppression of NF-κB and AP-1 activation by glucocorticoids in experimental glomerulonephritis in rats: Molecular mechanisms of anti-nephritic action. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 1997, 1362, 252–262. [Google Scholar] [CrossRef] [Green Version]
- Angel, P.; Karin, M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta 1991, 1072, 129–157. [Google Scholar] [CrossRef]
- Boby, N.; Abbas, M.A.; Lee, E.-B.; Im, Z.-E.; Hsu, W.H.; Park, S.-C. Protective effect of Pyrus ussuriensis Maxim. extract against ethanol-induced gastritis in rats. Antioxidants 2021, 10, 439. [Google Scholar] [CrossRef]
- Hernández-Muñoz, R.; Montiel-Ruíz, F. Reversion by histamine H2-receptor antagonists of plasma membrane alterations in ethanol-induced gastritis. Am. J. Dig. Dis. Sci. 1996, 41, 2156–2165. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, S.-D.; Nam, W.; Nam, B.; Bae, C.H.; Kim, H.J.; Kim, J.; Lee, J.-L.; Sim, J.-H. Gastroprotective effects of Cudrania tricuspidata leaf extracts by suppressing gastric cAMP and increasing gastric mucins. Prev. Nutr. Food Sci. 2020, 25, 158–165. [Google Scholar] [CrossRef]
- Bradshaw, J.; Brittain, R.T.; Clitherow, J.W.; Daly, M.J.; Jack, D.; Price, B.J.; Stables, R. Ranitidine (AH 19065): A new potent, selective histamine H2-receptor antagonist [proceedings]. Br. J. Pharmacol. 1979, 66, 464P. [Google Scholar]
- Koch-Weser, J.; Zeldis, J.B.; Friedman, L.S.; Isselbacher, K.J. Ranitidine: A new H2-receptor antagonist. N. Engl. J. Med. 1983, 309, 1368–1373. [Google Scholar] [CrossRef]
- Adhikari, A.; Xu, M.; Chen, Z.J. Ubiquitin-mediated activation of TAK1 and IKK. Oncogene 2007, 26, 3214–3226. [Google Scholar] [CrossRef] [Green Version]
- Irie, T.; Muta, T.; Takeshige, K. TAK1 mediates an activation signal from toll-like receptor (s) to nuclear factor-κB in lipopolysaccharide-stimulated macrophages. FEBS Lett. 2000, 467, 160–164. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.; Vial, S.C.; Dedi, N.; Long, J.M.; Dunster, N.J.; Cheetham, G.M. Structural basis for the interaction of TAK1 kinase with its activating protein TAB1. J. Mol. Biol. 2005, 354, 1013–1020. [Google Scholar] [CrossRef]
- Sato, S.; Sanjo, H.; Takeda, K.; Ninomiya-Tsuji, J.; Yamamoto, M.; Kawai, T.; Matsumoto, K.; Takeuchi, O.; Akira, S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 2005, 6, 1087–1095. [Google Scholar] [CrossRef]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef]
- Vyse, T.J.; Todd, J.A. Genetic analysis of autoimmune disease. Cell 1996, 85, 311–318. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.; Hagen, P. Systemic inflammatory response syndrome. J. Br. Surg. 1997, 84, 920–935. [Google Scholar] [CrossRef]
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef]
- Shaulian, E.; Karin, M. AP-1 in cell proliferation and survival. Oncogene 2001, 20, 2390–2400. [Google Scholar] [CrossRef] [Green Version]
- Hess, J.; Angel, P.; Schorpp-Kistner, M. AP-1 subunits: Quarrel and harmony among siblings. J. Cell Sci. 2004, 117, 5965–5973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milde-Langosch, K.; Röder, H.; Andritzky, B.; Aslan, B.; Hemminger, G.; Brinkmann, A.; Bamberger, C.M.; Löning, T.; Bamberger, A.-M. The role of the AP-1 transcription factors c-Fos, FosB, Fra-1 and Fra-2 in the invasion process of mammary carcinomas. Breast Cancer Res. Treat. 2004, 86, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.K.; Tsugawa, K.; Tarnawski, A.S.; Baatar, D. Dual actions of nitric oxide on angiogenesis: Possible roles of PKC, ERK, and AP-1. Biochem. Biophys. Res. Commun. 2004, 318, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Chiu, R.; Boyle, W.J.; Meek, J.; Smeal, T.; Hunter, T.; Karin, M. The c-fos protein interacts with c-JunAP-1 to stimulate transcription of AP-1 responsive genes. Cell 1988, 54, 541–552. [Google Scholar] [CrossRef]
- Liebermann, D.A.; Gregory, B.; Hoffman, B. AP-1 (Fos/Jun) transcription factors in hematopoietic differentiation and apoptosis. Int. J. Oncol. 1998, 12, 685–1385. [Google Scholar] [CrossRef]
- Li, J.; Stickel, S.L.; Bouton-Verville, H.; Burgin, K.E.; Yu, X.; Wong, D.K.; Wagner, T.E.; Wei, Y. Fermented Noni exudate (fNE): A mediator between immune system and anti-tumor activity. Oncol. Rep. 2008, 20, 1505–1509. [Google Scholar] [CrossRef] [Green Version]
- Oltmanns, U.; Issa, R.; Sukkar, M.B.; John, M.; Chung, K.F. Role of c-jun N-terminal kinase in the induced release of GM-CSF, RANTES and IL-8 from human airway smooth muscle cells. Br. J. Pharmacol. 2003, 139, 1228–1234. [Google Scholar] [CrossRef] [Green Version]
- Schwabe, R.F.; Bradham, C.A.; Uehara, T.; Hatano, E.; Bennett, B.L.; Schoonhoven, R.; Brenner, D.A. c-Jun-N-terminal kinase drives cyclin D1 expression and proliferation during liver regeneration. Hepatology 2003, 37, 824–832. [Google Scholar] [CrossRef]
- Wulf, G.M.; Ryo, A.; Wulf, G.G.; Lee, S.W.; Niu, T.; Petkova, V.; Lu, K.P. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J. 2001, 20, 3459–3472. [Google Scholar] [CrossRef] [Green Version]
- Eferl, R.; Ricci, R.; Kenner, L.; Zenz, R.; David, J.-P.; Rath, M.; Wagner, E.F. Liver tumor development: C-Jun antagonizes the proapoptotic activity of p53. Cell 2003, 112, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Stepniak, E.; Ricci, R.; Eferl, R.; Sumara, G.; Sumara, I.; Rath, M.; Hui, L.; Wagner, E.F. c-Jun/AP-1 controls liver regeneration by repressing p53/p21 and p38 MAPK activity. Genes Dev. 2006, 20, 2306–2314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Shen, J.; Wang, Y.; Chen, X.; Yu, S.; Shi, H.; Huo, K. Up-regulation of c-Fos associated with neuronal apoptosis following intracerebral hemorrhage. Cell. Mol. Neurobiol. 2015, 35, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-H.; Grégori, G.; Hullinger, R.L.; Andrisani, O.M. Sustained activation of p38 mitogen-activated protein kinase and c-Jun N-terminal kinase pathways by hepatitis B virus X protein mediates apoptosis via induction of Fas/FasL and tumor necrosis factor (TNF) receptor 1/TNF-α expression. Mol. Cell. Biol. 2004, 24, 10352–10365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litherland, G.J.; Elias, M.S.; Hui, W.; Macdonald, C.D.; Catterall, J.B.; Barter, M.J.; Farren, M.J.; Jefferson, M.; Rowan, A.D. Protein kinase C isoforms ζ and ι mediate collagenase expression and cartilage destruction via STAT3- and ERK-dependent c-fos induction. J. Biol. Chem. 2010, 285, 22414–22425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makino, H.; Seki, S.; Yahara, Y.; Shiozawa, S.; Aikawa, Y.; Motomura, H.; Nogami, M.; Watanabe, K.; Sainoh, T.; Ito, H.; et al. A selective inhibition of c-Fos/activator protein-1 as a potential therapeutic target for intervertebral disc degeneration and associated pain. Sci. Rep. 2017, 7, 16983. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Ye, D.-X.; Zhang, W.-B.; Pan, H.-Y.; Zhang, Z.-Y.; Zhang, L. Overexpression of c-fos promotes cell invasion and migration via CD44 pathway in oral squamous cell carcinoma. J. Oral Pathol. Med. 2015, 44, 353–360. [Google Scholar] [CrossRef]
- Lamb, R.F.; Hennigan, R.F.; Turnbull, K.; Katsanakis, K.D.; MacKenzie, E.D.; Birnie, G.D.; Ozanne, B.W. AP-1-mediated invasion requires increased expression of the hyaluronan receptor CD44. Mol. Cell. Biol. 1997, 17, 963–976. [Google Scholar] [CrossRef] [Green Version]
- Motomura, H.; Seki, S.; Shiozawa, S.; Aikawa, Y.; Nogami, M.; Kimura, T. A selective c-Fos/AP-1 inhibitor prevents cartilage destruction and subsequent osteophyte formation. Biochem. Biophys. Res. Commun. 2018, 497, 756–761. [Google Scholar] [CrossRef]
- Aikawa, Y.; Morimoto, K.; Yamamoto, T.; Chaki, H.; Hashiramoto, A.; Narita, H.; Hirono, S.; Shiozawa, S. Treatment of arthritis with a selective inhibitor of c-Fos/activator protein-1. Nat. Biotechnol. 2008, 26, 817–823. [Google Scholar] [CrossRef]
- Totzke, J.; Gurbani, D.; Raphemot, R.; Hughes, P.F.; Bodoor, K.; Carlson, D.A.; Loiselle, D.R.; Bera, A.K.; Eibschutz, L.S.; Perkins, M.M. Takinib, a selective TAK1 inhibitor, broadens the therapeutic efficacy of TNF-α inhibition for cancer and autoimmune disease. Cell Chem. Biol. 2017, 24, 1029–1039.e1027. [Google Scholar] [CrossRef] [Green Version]
- Scarneo, S.A.; Eibschutz, L.S.; Bendele, P.J.; Yang, K.W.; Totzke, J.; Hughes, P.; Fox, D.A.; Haystead, T.A.J. Pharmacological inhibition of TAK1, with the selective inhibitor takinib, alleviates clinical manifestation of arthritis in CIA mice. Arthritis Res. Ther. 2019, 21, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, L.; Chunxia, Y.; Wei, L. Inhibition of ovarian cancer cell growth by a novel TAK1 inhibitor LYTAK1. Cancer Chemother. Pharmacol. 2015, 76, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.; Lu, J.; Zhao, Y.; Woodfield, S.E.; Zhang, H.; Xu, X.; Yu, Y.; Zhao, J.; Bieerkehazhi, S.; Liang, H.; et al. TAK1 inhibitor 5Z-7-oxozeaenol sensitizes cervical cancer to doxorubicin-induced apoptosis. Oncotarget 2017, 8, 33666–33675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Powell, F.; Larsen, N.A.; Lai, Z.; Byth, K.F.; Read, J.; Gu, R.-F.; Roth, M.; Toader, D.; Saeh, J.C.; et al. Mechanism and in vitro pharmacology of TAK1 inhibition by (5Z)-7-oxozeaenol. ACS Chem. Biol. 2013, 8, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Cheng, J.; Vasudevan, S.A.; Patel, R.H.; Liang, L.; Xu, X.; Zhao, Y.; Jia, W.; Lu, F.; Zhang, H.; et al. TAK1 inhibitor 5Z-7-oxozeaenol sensitizes neuroblastoma to chemotherapy. Apoptosis 2013, 18, 1224–1234. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-O.; Hwang, S.-H.; Shen, T.; Kim, J.H.; You, L.; Hu, W.; Cho, J.Y. Enhancement of skin barrier and hydration-related molecules by protopanaxatriol in human keratinocytes. J. Ginseng Res. 2021, 45, 354–360. [Google Scholar] [CrossRef]
- Jang, J.; Lee, J.S.; Jang, Y.-J.; Choung, E.S.; Li, W.Y.; Lee, S.W.; Kim, E.; Kim, J.-H.; Cho, J.Y. Sorbaria kirilowii ethanol extract exerts anti-inflammatory effects in vitro and in vivo by targeting Src/nuclear factor (NF)-κB. Biomolecules 2020, 10, 741. [Google Scholar] [CrossRef]
- Kim, S.H.; Park, J.G.; Sung, G.-H.; Yang, S.; Yang, W.S.; Kim, E.; Kim, J.H.; Ha, V.T.; Kim, H.G.; Yi, Y.-S.; et al. Kaempferol, a dietary flavonoid, ameliorates acute inflammatory and nociceptive symptoms in gastritis, pancreatitis, and abdominal pain. Mol. Nutr. Food Res. 2015, 59, 1400–1405. [Google Scholar] [CrossRef]
- Lee, J.-O.; Kim, J.H.; Kim, S.; Kim, M.-Y.; Hong, Y.H.; Kim, H.G.; Cho, J.Y. Gastroprotective effects of the nonsaponin fraction of Korean Red Ginseng through cyclooxygenase-1 upregulation. J. Ginseng Res. 2020, 44, 655–663. [Google Scholar] [CrossRef]
- Yu, T.; Wang, Z.; Jie, W.; Fu, X.; Li, B.; Xu, H.; Liu, Y.; Li, M.; Kim, E.; Yang, Y.; et al. The kinase inhibitor BX795 suppresses the inflammatory response via multiple kinases. Biochem. Pharmacol. 2020, 174, 113797. [Google Scholar] [CrossRef]
- Song, C.; Lorz, L.R.; Lee, J.; Cho, J.Y. In vitro photoprotective, anti-inflammatory, moisturizing, and antimelanogenic effects of a methanolic extract of Chrysophyllum lucentifolium Cronquist. Plants 2021, 11, 94. [Google Scholar] [CrossRef]
- Ahuja, A.; Kim, M.-Y.; Cho, J.Y. Protium javanicum Burm. methanol extract attenuates LPS-induced inflammatory activities in macrophage-like RAW264.7 cells. Evidence-Based Complement. Altern. Med. 2019, 2019, 2910278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, S.-H.; Lorz, L.R.; Yi, D.-K.; Noh, J.K.; Yi, Y.-S.; Cho, J.Y. Viburnum pichinchense methanol extract exerts anti-inflammatory effects via targeting the NF-κB and caspase-11 non-canonical inflammasome pathways in macrophages. J. Ethnopharmacol. 2019, 245, 112161. [Google Scholar] [CrossRef] [PubMed]
- Aziz, N.; Hong, Y.H.; Jo, M.K.; Kim, J.K.; Kim, K.-H.; Ashktorab, H.; Smoot, D.T.; Hur, H.; Yoo, B.C.; Cho, J.Y. Molecular signatures of JMJD10/MINA53 in gastric cancer. Cancers 2020, 12, 1141. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Choi, E.; Hong, Y.H.; Kim, H.; Jang, Y.-J.; Lee, J.S.; Choung, E.S.; Woo, B.Y.; Hong, Y.D.; Lee, S. Syk/NF-κB-targeted anti-inflammatory activity of Melicope accedens (Blume) TG Hartley methanol extract. J. Ethnopharmacol. 2021, 271, 113887. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Sequence (5′ to 3′) | |
|---|---|---|
| MMP-1 | F | GCCTGCGTCCATCAACACT |
| R | CCCTCCTCGTCCACCTCAA | |
| MMP-3 | F | ACTCCCTGGGACTCTACCAC |
| R | TTCTTCACGGTTGCAGGGAG | |
| MMP-9 | F | TCTTCCCCAAAGACCTGAAA |
| R | TGATGTTATGATGGTCCCAC | |
| COX-2 | F | GGGAGTCTGGAACATTGTGAA |
| R | GCACATTGTAAGTAGGTGGACTGT | |
| GAPDH | F | CAATGAATACGGCRACAGCA |
| R | AGGGAGATGCTGGTTGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, J.; Hunto, S.T.; Kim, J.W.; Lee, H.P.; Kim, H.G.; Cho, J.Y. Anti-Inflammatory Activities of an Anti-Histamine Drug, Loratadine, by Suppressing TAK1 in AP-1 Pathway. Int. J. Mol. Sci. 2022, 23, 3986. https://doi.org/10.3390/ijms23073986
Jang J, Hunto ST, Kim JW, Lee HP, Kim HG, Cho JY. Anti-Inflammatory Activities of an Anti-Histamine Drug, Loratadine, by Suppressing TAK1 in AP-1 Pathway. International Journal of Molecular Sciences. 2022; 23(7):3986. https://doi.org/10.3390/ijms23073986
Chicago/Turabian StyleJang, Jiwon, Stephanie Triseptya Hunto, Ji Won Kim, Hwa Pyoung Lee, Han Gyung Kim, and Jae Youl Cho. 2022. "Anti-Inflammatory Activities of an Anti-Histamine Drug, Loratadine, by Suppressing TAK1 in AP-1 Pathway" International Journal of Molecular Sciences 23, no. 7: 3986. https://doi.org/10.3390/ijms23073986
APA StyleJang, J., Hunto, S. T., Kim, J. W., Lee, H. P., Kim, H. G., & Cho, J. Y. (2022). Anti-Inflammatory Activities of an Anti-Histamine Drug, Loratadine, by Suppressing TAK1 in AP-1 Pathway. International Journal of Molecular Sciences, 23(7), 3986. https://doi.org/10.3390/ijms23073986