
Dual Inhibition of H3K9me2 and H3K27me3 Promotes Tumor Cell Senescence without Triggering the Secretion of SASP

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
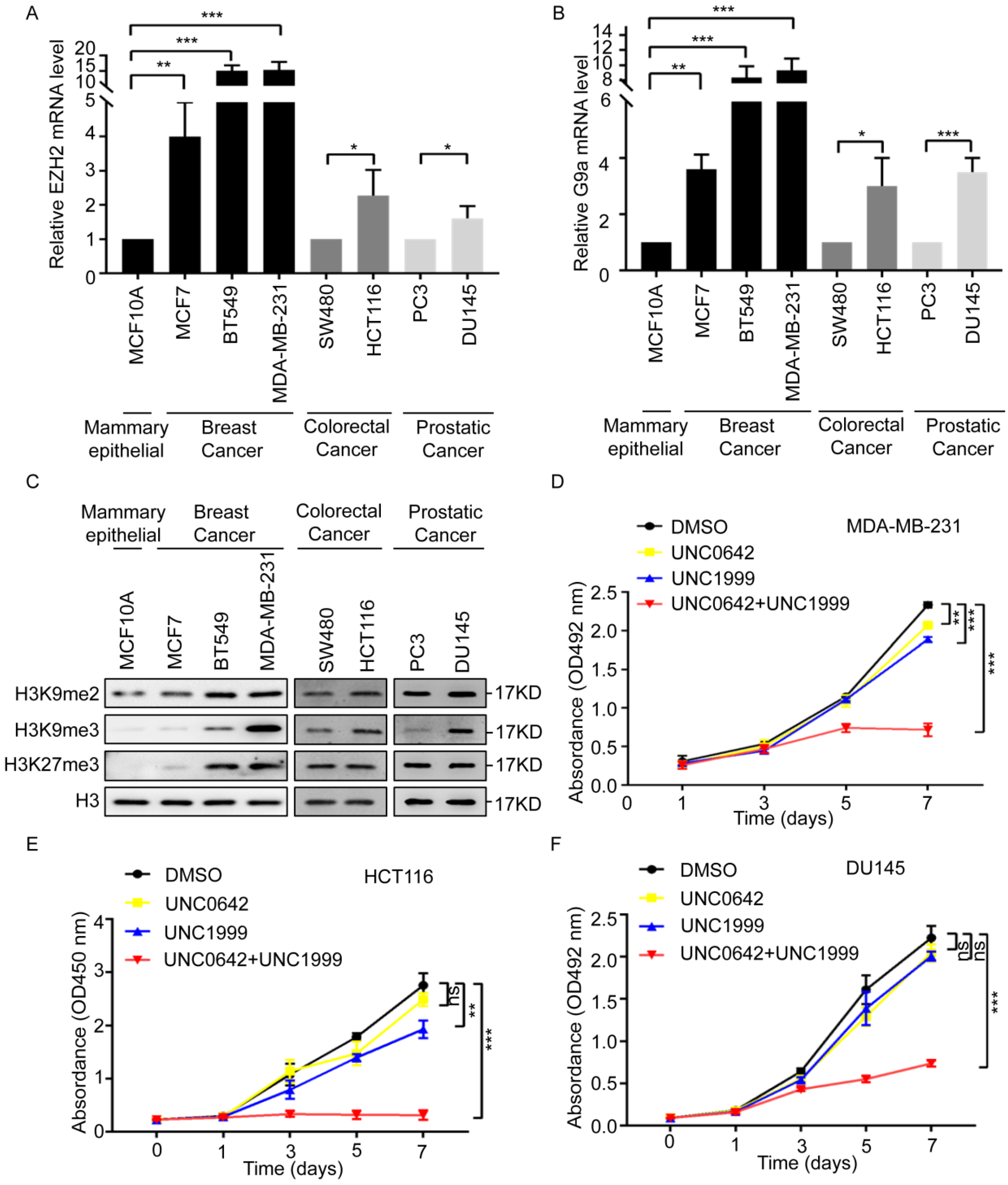
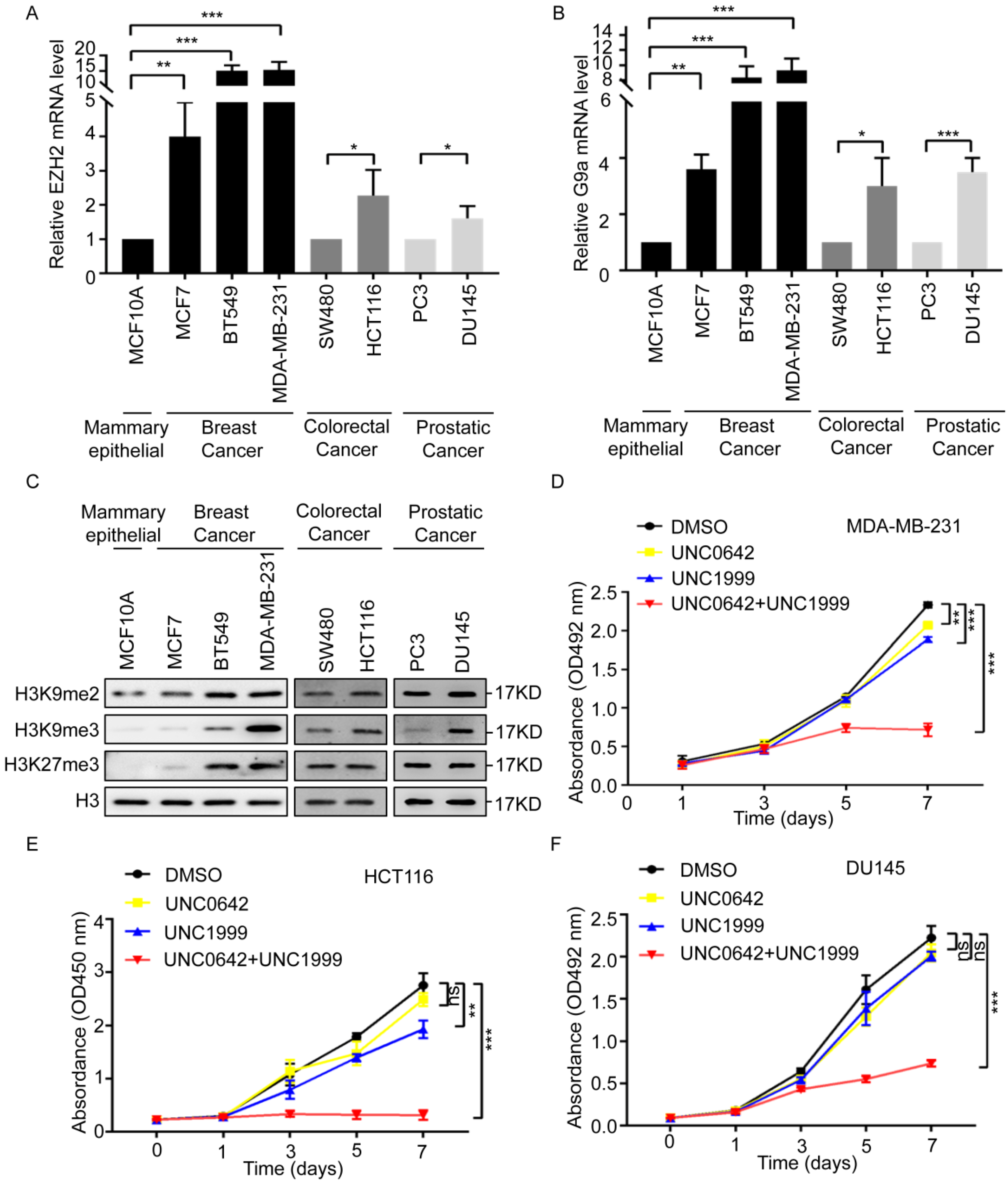
2.1. Dual Inhibition of H3K9me2 and H3K27me3 Inhibits the Proliferation and Migration of Highly Metastatic Tumor Cells
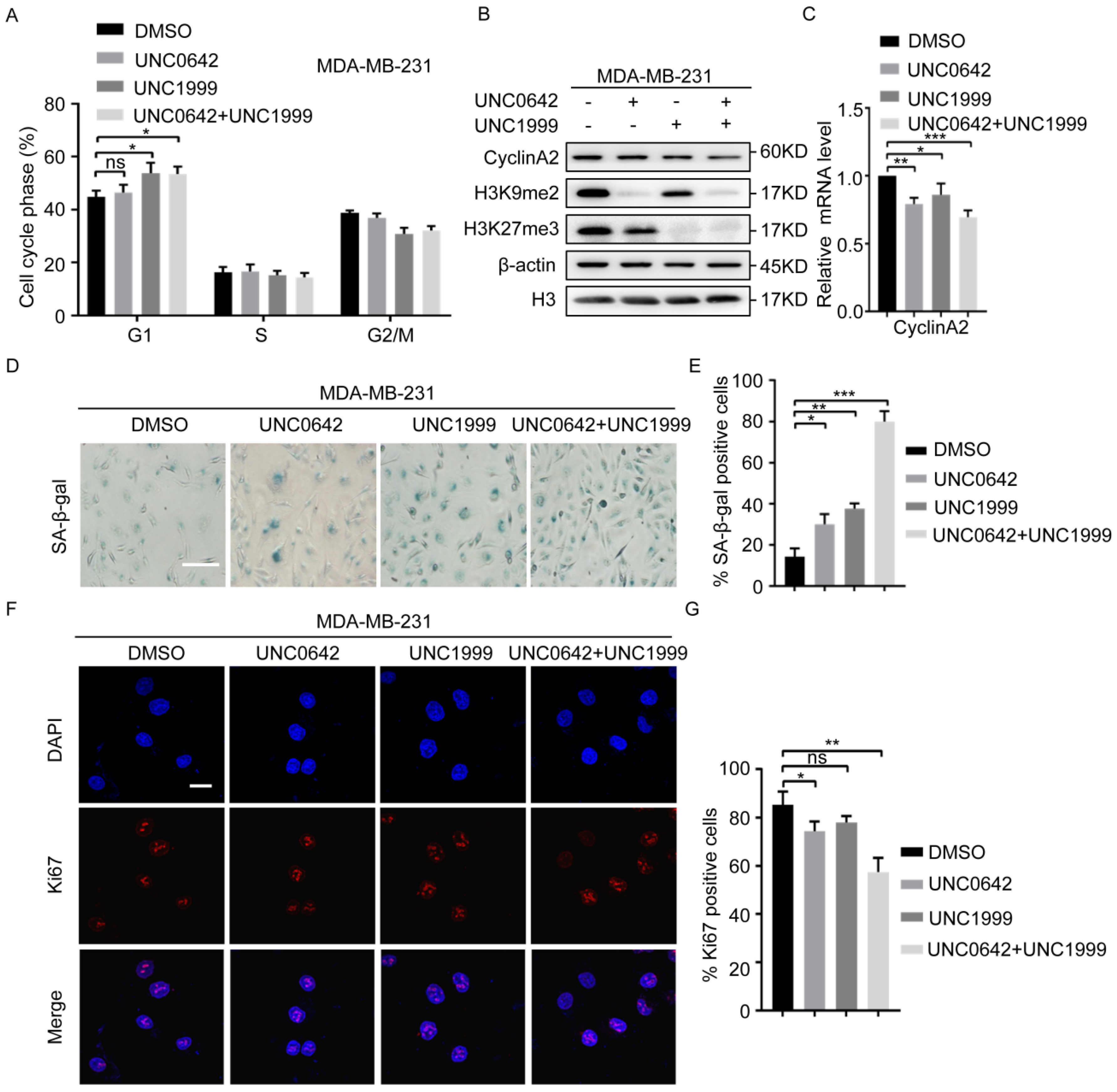
2.2. Dual Inhibition of H3K9me2 and H3K27me3 Has a Stronger Pro-Senescence Effect than Single Inhibition in Highly Metastatic Tumor Cells
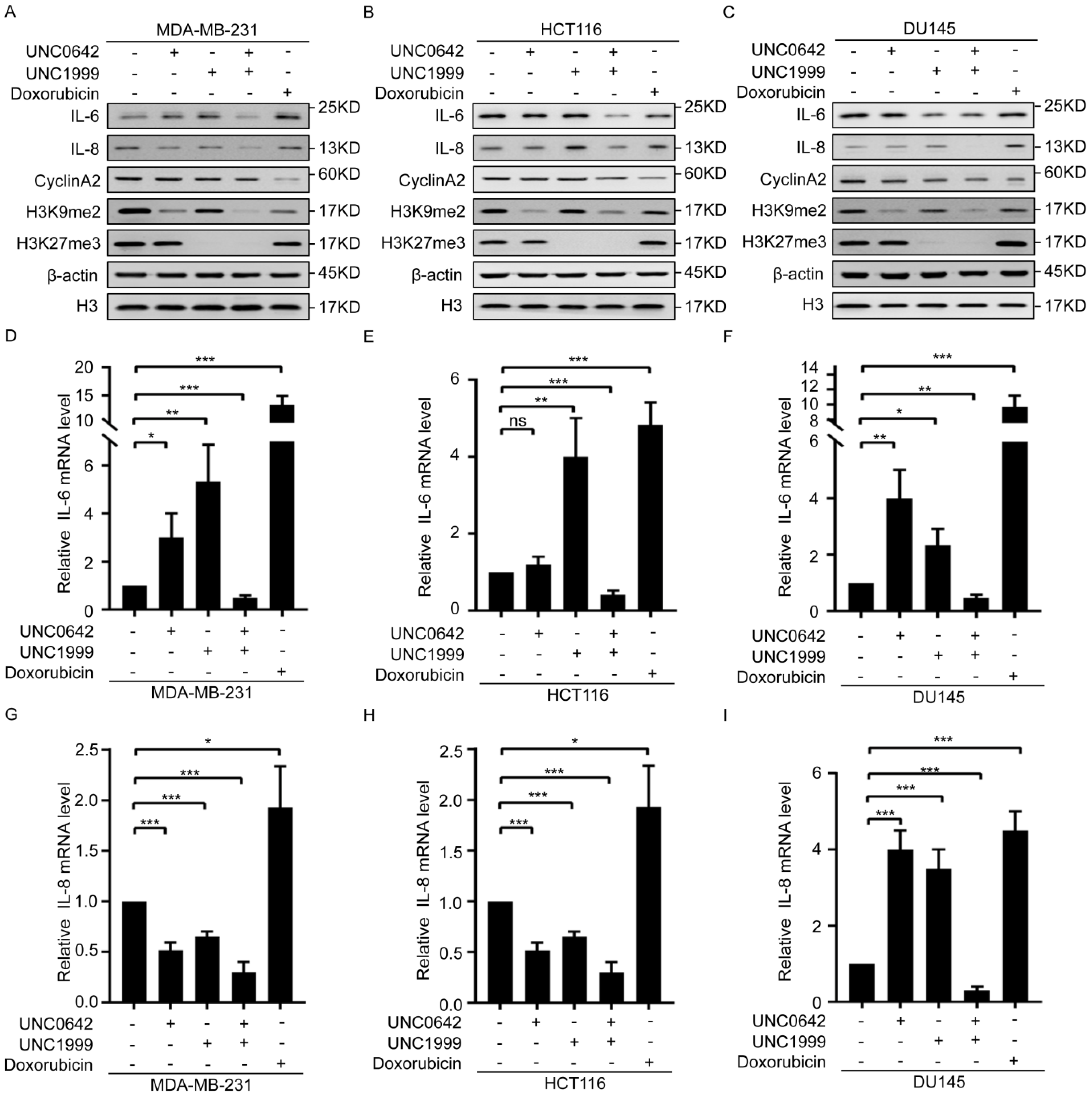
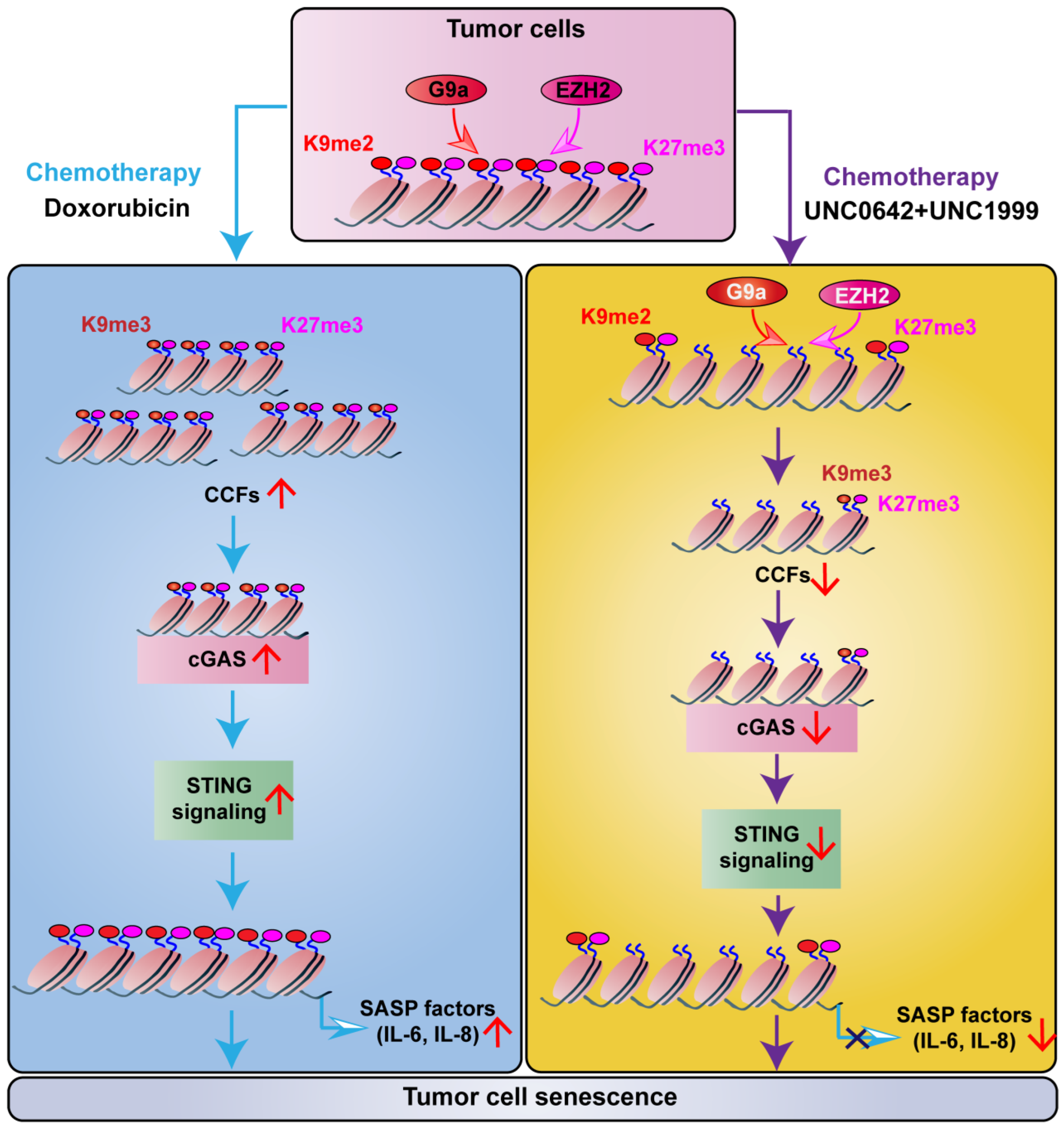
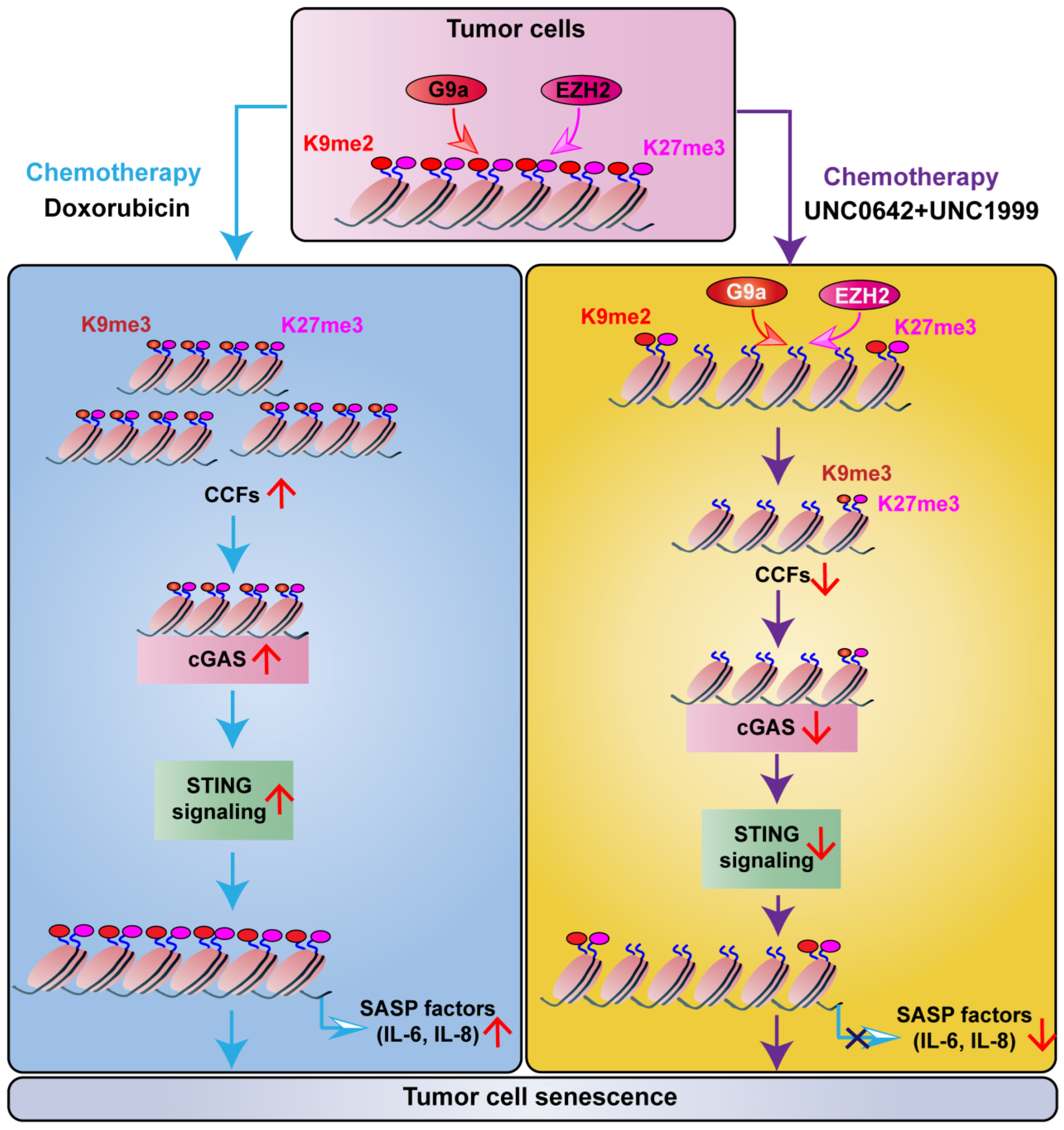
2.3. Dual Inhibition of H3K9me2 and H3K27me3 Induces Tumor Cell Senescence without Triggering SASP
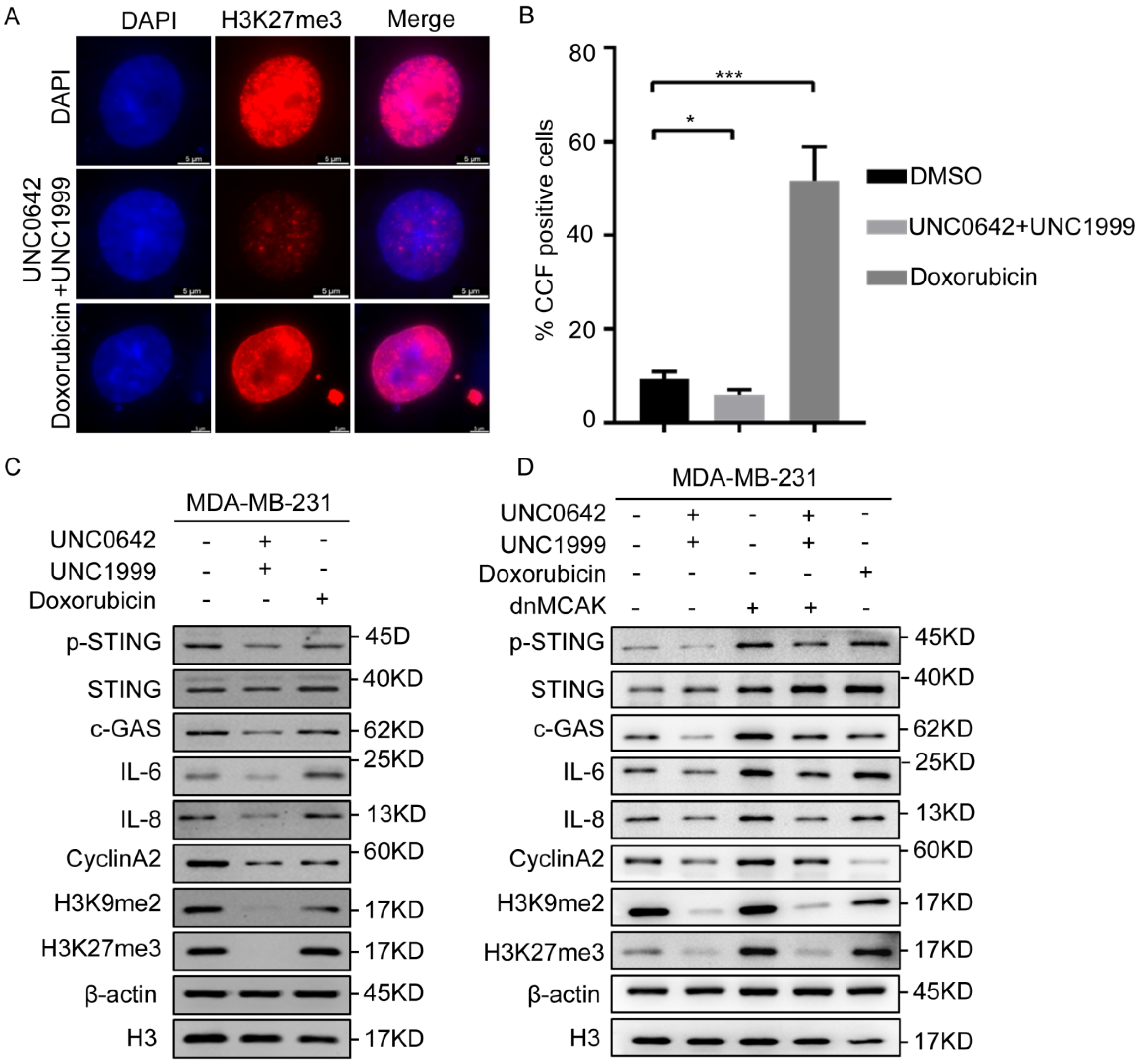
2.4. Dual Inhibition of H3K9me2 and H3K27me3 Suppresses cGAS-STING Signaling by Decreasing CCF
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Reagents and Drug Treatment
4.3. Plasmid and Retroviral Infection
4.4. Reverse Transcription, PCR and Real-Time PCR
4.5. Western Blot Analysis
4.6. Immunofluorescence
4.7. Wound Healing Assays
4.8. Flow Cytometric Analysis
4.9. SA-β-Gal Staining
4.10. Cell Viability Assays
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wynford-Thomas, D. Cellular senescence and cancer. J. Pathol. 2015, 187, 100–111. [Google Scholar] [CrossRef]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin. Radiat. Oncol. 2004, 14, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Laberge, R.M.; Awad, P.; Campisi, J. Epithelial-Mesenchymal Transition Induced by Senescent Fibroblasts. Cancer Microenviron. 2011, 5, 39–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; Doorn, R.V.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.D. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Peeper, D. Oncogene-induced cellular senescence relayed by a cell-autonomous interleukin-dependent network. Cancer Res. 2008, 68. [Google Scholar]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Alejandra, H.S.; Jamil, N.; Marco, D. Hallmarks of Cellular Senescence. Trends Cell. Biol. 2018, 28, 436–453. [Google Scholar]
- Freunda, A.; Laberge, R.-M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell. 2012, 23, 2066–2075. [Google Scholar] [CrossRef]
- Shah, P.P.; Donahue, G.; Otte, G.L.; Capell, B.C.; Nelson, D.M.; Cao, K.; Aggarwala, V.; Cruickshanks, H.A.; Rai, T.S.; McBryan, T.; et al. Lamin B1 Depletion in Senescent Cells Triggers Large-Scale Changes in Gene Expression and the Chromatin Landscape. Genes Dev. 2013, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.-W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP Is an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Science 2012, 339, 826–830. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [Green Version]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Han, X.J.; Chen, H.; Gong, H.; Tang, X.; Huang, N.; Xu, W.; Tai, H.; Zhang, G.; Zhao, T.; Gong, C.; et al. Autolysosomal degradation of cytosolic chromatin fragments antagonizes oxidative stress–induced senescence. J. Biol. Chem. 2020, 295, 4451–4463. [Google Scholar] [CrossRef] [Green Version]
- Issa, M.E.; Takhsha, F.S.; Chirumamilla, C.S.; Perez-Novo, C.; Berghe, W.V.; Cuendet, M. Epigenetic strategies to reverse drug resistance in heterogeneous multiple myeloma. Clin. Epigenetics 2017, 9, 17. [Google Scholar] [CrossRef] [Green Version]
- Mozzetta, C.; Boyarchuk, E.; Pontis, J.; Ait-Si-Ali, S. Sound of silence: The properties and functions of repressive Lys methyltransferases. Nat. Rev. Mol. Cell Biol. 2015, 16, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Hernando, H.; Gelato, K.A.; Lesche, R.; Beckmann, G.; Koehr, S.; Otto, S.; Steigemann, P.; Stresemann, C. EZH2 Inhibition Blocks Multiple Myeloma Cell Growth through Upregulation of Epithelial Tumor Suppressor Genes. Mol.Cancer Ther. 2016, 15, 287–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlyn, C.; Bright, M.D.; Buros, A.F.; Stein, C.K.; Walters, Z.; Aronson, L.I.; Mirabella, F.; Jones, J.R.; Kaiser, M.F.; Walker, B.A.; et al. Overexpression of EZH2 in multiple myeloma is associated with poor prognosis and dysregulation of cell cycle control. Blood Cancer J. 2017, 7, e549. [Google Scholar] [CrossRef] [PubMed]
- Alzrigat, M.; Párraga, A.A.; Agarwal, P.; Zureigat, H.; Österborg, A.; Nahi, H.; Ma, A.; Jin, J.; Nilsson, K.; Öberg, F.; et al. EZH2 inhibition in multiple myeloma downregulates myeloma associated oncogenes and upregulates microRNAs with potential tumor suppressor functions. Oncotarget 2017, 8, 10213–10224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.P.; Sun, J.Y.; Li, M.Q.; Dong, Y.; Zhang, Y.H.; Yan, J.; Huang, R.M.; Yan, X. Inhibition of G9a by a small molecule inhibitor, UNC0642, induces apoptosis of human bladder cancer cells. Acta Pharmacol. Sin. 2019, 40, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Mozzetta, C.; Pontis, J.; Fritsch, L.; Robin, P.; Portoso, M.; Proux, C.; Margueron, R.; Ait-Si-Ali, S. The Histone H3 Lysine 9 Methyltransferases G9a and GLP Regulate Polycomb Repressive Complex 2-Mediated Gene Silencing. Mol. Cell. 2014, 53, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Coward, W.R.; Brand, O.J.; Pasini, A.; Jenkins, G.; Knox, A.; Pang, L. Interplay between EZH2 and G9a Regulates CXCL10 Gene Repression in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 449–460. [Google Scholar] [CrossRef] [Green Version]
- Ishiguro, K.; Kitajima, H.; Niinuma, T.; Maruyama, R.; Nishiyama, N.; Ohtani, H.; Sudo, G.; Toyota, M.; Sasaki, H.; Yamamoto, E.; et al. Dual EZH2 and G9a inhibition suppresses multiple myeloma cell proliferation by regulating the interferon signal and IRF4-MYC axis. Cell Death Discov. 2021, 7, 7. [Google Scholar] [CrossRef]
- Wozniak, R.J.; Klimecki, W.T.; Lau, S.S.; Feinstein, Y.; Futscher, B.W. 5-Aza-2′-deoxycytidine-mediated reductions in G9A histone methyltransferase and histone H3 K9 di-methylation levels are linked to tumor suppressorgene reactivation. Oncogene 2007, 26, 77. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Chiu, D.K.; Tsang, F.H.; Law, C.T.; Cheng, C.L.; Au, S.L.; Lee, J.M.; Wong, C.C.; Ngm, I.O.; Wong, C.M. Histone methyltransferase G9a promotes liver cancer development by epigenetic silencing of tumor suppressor gene RARRES3. J. Hepatol. 2017, 67, 758–769. [Google Scholar] [CrossRef]
- Ngollo, M.; Lebert, A.; Daures, M.; Judes, G.; Rifai, K.; Dubois, L.; Kemeny, J.L.; Penault-Llorca, F.; Bignon, Y.J.; Guy, L.; et al. Global analysis of H3K27me3 as an epigenetic marker in prostate cancer progression. BMC Cancer 2017, 17, 261. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, X.; Jiang, Y.; Liu, S.; Liu, H.; Sun, X.; Zhang, H.; Liu, Z.; Tao, Y.; Li, C.; et al. Elevating H3K27me3 level sensitizes colorectal cancer to oxaliplatin. J. Mol. Cell Biol. 2020, 12, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, S.W.; Rush, D.; Schimke, E. Defining Cellular senescence in IMR-90 Cells: A Flow Cytometric Analysis. Proc. Natl. Acad. Sci. USA 1988, 85, 9086–9090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, D.X.; Jackson, T.J.; Lin, A.W. Molecular signature of oncogenic ras-induced senescence. Oncogene 2004, 23, 9238–9246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, G.H.; Drullinger, L.F.; Robetorye, R.S.; Pereira-Smith, O.M.; Smith, J.R. Senescent cells fail to express cdc2, cycA, and cycB in response to mitogen stimulation. Proc. Natl. Acad. Sci. USA 1991, 88, 11012–11016. [Google Scholar] [CrossRef] [Green Version]
- Joyner, D.E.; Bastar, J.D.; Randall, R.L. Doxorubicin induces cell senescence preferentially over apoptosis in the FU-SY-1 synovial sarcoma cell line. J. Orthop. Res. 2010, 24, 1163–1169. [Google Scholar] [CrossRef]
- Jackson, J.G.; Pereira-Smith, O.M. Primary and Compensatory Roles for RB Family Members at Cell Cycle Gene Promoters That Are Deacetylated and Downregulated in Doxorubicin-Induced Senescence of Breast Cancer Cells. Mol. Cell Biol. 2006, 26, 2501–2510. [Google Scholar] [CrossRef] [Green Version]
- Bakhoum, S.F.; Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [Green Version]
- Fioravanti, R.; Stazi, G.; Zwergel, C.; Valente, S.; Mai, A. Six Years (2012–2018) of Researches on Catalytic EZH2 Inhibitors: The Boom of the 2-Pyridone Compounds. Chem. Rec. 2018, 18, 1818–1832. [Google Scholar] [CrossRef]
- Wang, Y.F.; Zhang, J.; Su, Y.; Shen, Y.Y.; Jiang, D.X.; Hou, Y.Y.; Geng, M.Y.; Ding, J.; Chen, Y. G9a regulates breast cancer growth by modulating iron homeostasis through the repression of ferroxidase hephaestin. Nat.Commun. 2017, 8, 274. [Google Scholar] [CrossRef] [Green Version]
- Dang, N.N.; Jiao, J.; Meng, X.; An, Y.; Han, C.; Huang, S. Abnormal overexpression of G9a in melanoma cells promotes cancer progression via upregulation of the Notch1 signaling pathway. Aging 2020, 12, 2393–2407. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Du, Y.; Liu, X.; Chen, H.; Weng, X.; Guo, J.; Wang, M.; Wang, X.; Wang, L. EZH2 inhibition suppresses bladder cancer cell growth and metastasis via the JAK2/STAT3 signaling pathway. Oncol. Lett. 2019, 18, 907–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biol, N. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar]
- Brenner, E.; Schörg, B.; Ahmetlić, F.; Wieder, T.; Hilke, F.J.; Simon, N.; Schroeder, C.; Demidov, G.; Riedel, T.; Fehrenbacher, B.; et al. Cancer immune control needs senescence induction by interferon-dependent cell cycle regulator pathways in tumours. Nat. Commun. 2020, 11, 1335. [Google Scholar] [CrossRef] [Green Version]
- Bracken, A.P.; Kleine-Kohlbrecher, D.; Dietrich, N.; Pasini, D.; Gargiulo, G.; Beekman, C.; Theilgaard-Mönch, K.; Minucci, S.; Porse, B.T.; Marine, J.C.; et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007, 21, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Stewart, C.A.; Gay, C.M.; Xi, Y.; Sivajothi, S.; Sivakamasundari, V.; Fujimoto, J.; Bolisetty, M.; Hartsfield, P.M.; Balasubramaniyan, V.; Chalishazar, M.D.; et al. Single-cell analyses reveal increased intratumoral heterogeneity after the onset of therapy resistance in small-cell lung cancer. Nat. Cancer 2020, 1, 423–436. [Google Scholar] [CrossRef]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, D.; Shang, M.; Han, Y.; Liu, J.; Liu, J.; Kong, S.H.; Hou, J.; Huang, B.; Lu, J.; Zhang, Y. EZH2-CCF-cGAS Axis Promotes Breast Cancer Metastasis. Int. J. Mol. Sci. 2022, 23, 1788. [Google Scholar] [CrossRef]
- Zhang, N.; Ji, J.; Zhou, D.; Liu, X.; Zhang, X.; Liu, Y.; Xiang, W.; Wang, M.; Zhang, L.; Wang, G.; et al. The Interaction of the Senescent and Adjacent Breast Cancer Cells Promotes the Metastasis of Heterogeneous Breast Cancer Cells through Notch Signaling. Int. J. Mol. Sci. 2021, 22, 849. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, N.; Shang, M.; Li, H.; Wu, L.; Dong, M.; Huang, B.; Lu, J.; Zhang, Y. Dual Inhibition of H3K9me2 and H3K27me3 Promotes Tumor Cell Senescence without Triggering the Secretion of SASP. Int. J. Mol. Sci. 2022, 23, 3911. https://doi.org/10.3390/ijms23073911
Zhang N, Shang M, Li H, Wu L, Dong M, Huang B, Lu J, Zhang Y. Dual Inhibition of H3K9me2 and H3K27me3 Promotes Tumor Cell Senescence without Triggering the Secretion of SASP. International Journal of Molecular Sciences. 2022; 23(7):3911. https://doi.org/10.3390/ijms23073911
Chicago/Turabian StyleZhang, Na, Mengjie Shang, Hongxin Li, Lan Wu, Meichen Dong, Baiqu Huang, Jun Lu, and Yu Zhang. 2022. "Dual Inhibition of H3K9me2 and H3K27me3 Promotes Tumor Cell Senescence without Triggering the Secretion of SASP" International Journal of Molecular Sciences 23, no. 7: 3911. https://doi.org/10.3390/ijms23073911
APA StyleZhang, N., Shang, M., Li, H., Wu, L., Dong, M., Huang, B., Lu, J., & Zhang, Y. (2022). Dual Inhibition of H3K9me2 and H3K27me3 Promotes Tumor Cell Senescence without Triggering the Secretion of SASP. International Journal of Molecular Sciences, 23(7), 3911. https://doi.org/10.3390/ijms23073911