
Neuroprotection in Stroke—Focus on the Renin-Angiotensin System: A Systematic Review

Abstract
1. Introduction
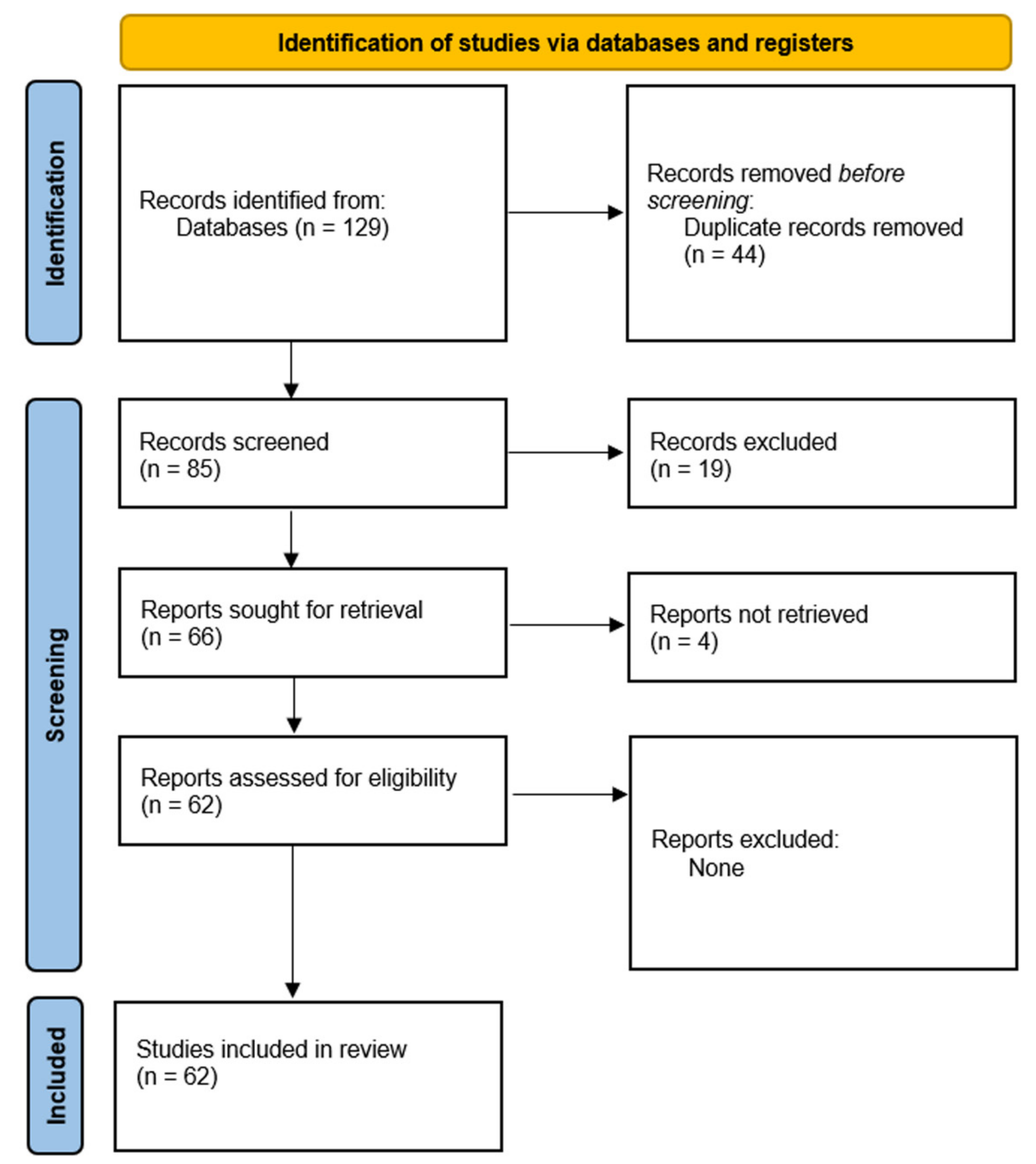
2. Methods
3. Results and Discussion
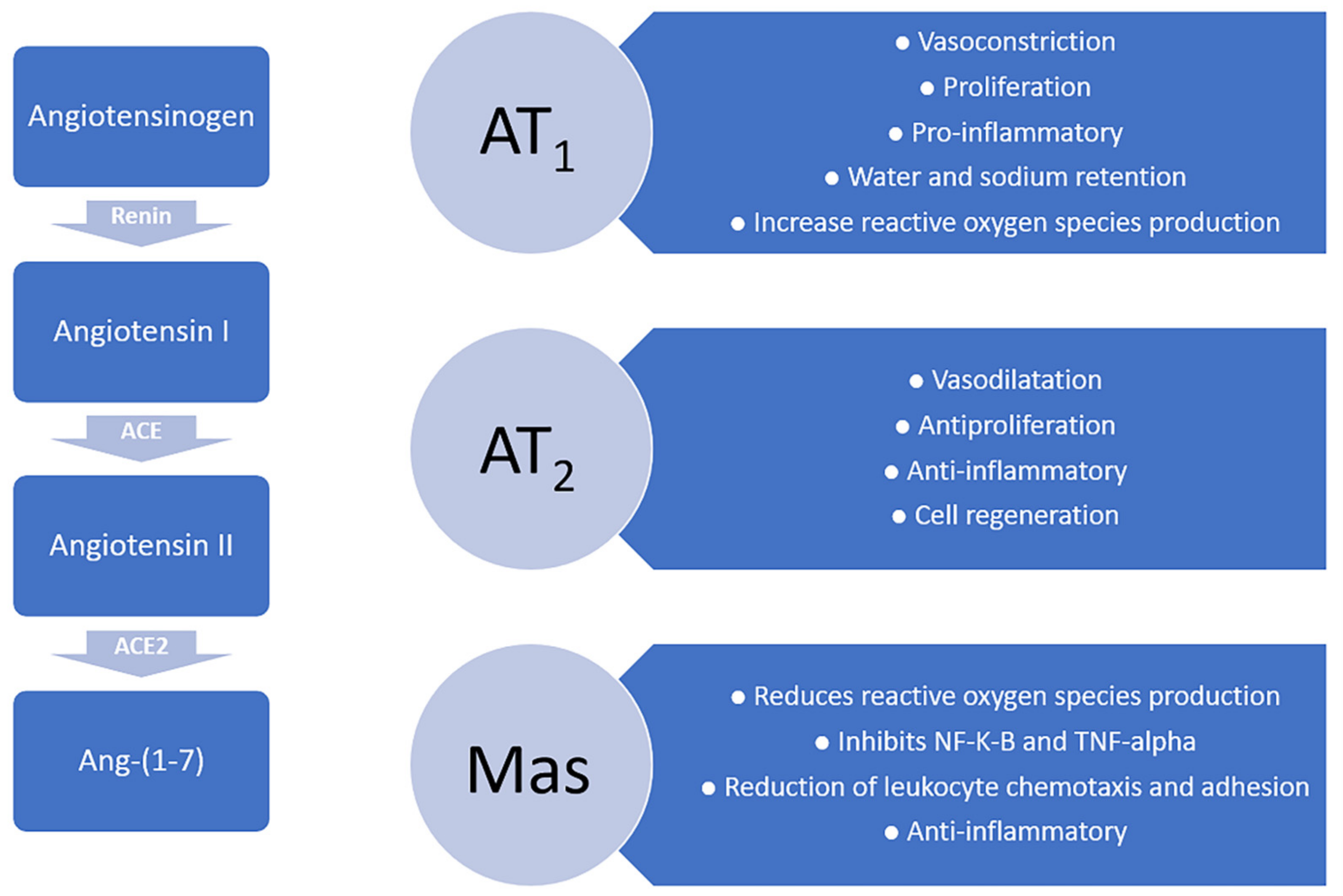
3.1. Angiotensin Receptors: AT1, AT2 and Mas
3.2. Angiotensin-Induced Neuroprotection Effects
3.3. Antihypertensive Drugs in Stroke: Focus on Angiotensin-Converting Enzyme Receptors and Angiotensin II Receptor Blockers
3.4. Clinical and Experimental Studies from the Prism of Neuroprotection
3.5. Angiotensin Receptor Antagonists—The Future of RAS-Neuroprotection Axis
4. Summary of Evidence
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Phipps, M.S.; Cronin, C.A. Management of Acute Ischemic Stroke. BMJ 2020, 368, l6983. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e153–e639. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Roth, G.A.; Naghavi, M.; Parmar, P.; Krishnamurthi, R.; Chugh, S.; Mensah, G.A.; Norrving, B.; Shiue, I.; Ng, M.; et al. Global Burden of Stroke and Risk Factors in 188 Countries, during 1990–2013: A Systematic Analysis for the Global Burden of Disease Study 2013. Lancet Neurol. 2016, 15, 913–924. [Google Scholar] [CrossRef]
- Schrader, J.; Kulschewski, A.; Dendorfer, A. Inhibition of the Renin-Angiotensin System and the Prevention of Stroke. Am. J. Cardiovasc. Drugs 2007, 7, 25–37. [Google Scholar] [CrossRef]
- Chalmers, J.; Chapman, N. Challenges for the Prevention of Primary and Secondary Stroke. The Importance of Lowering Blood Pressure and Total Cardiovascular Risk. Blood Press. 2001, 10, 344–351. [Google Scholar] [CrossRef]
- Mulvany, M.J. Small Artery Remodelling in Hypertension: Causes, Consequences and Therapeutic Implications. Med. Biol. Eng. Comput. 2008, 46, 461–467. [Google Scholar] [CrossRef]
- Agmon, Y.; Khandheria, B.K.; Meissner, I.; Schwartz, G.L.; Petterson, T.M.; O’Fallon, W.M.; Gentile, F.; Whisnant, J.P.; Wiebers, D.O.; Seward, J.B. Independent Association of High Blood Pressure and Aortic Atherosclerosis: A Population-Based Study. Circulation 2000, 102, 2087–2093. [Google Scholar] [CrossRef]
- Henskens, L.H.G.; Kroon, A.A.; van Oostenbrugge, R.J.; Gronenschild, E.H.B.M.; Fuss-Lejeune, M.M.J.J.; Hofman, P.A.M.; Lodder, J.; de Leeuw, P.W. Increased Aortic Pulse Wave Velocity Is Associated with Silent Cerebral Small-Vessel Disease in Hypertensive Patients. Hypertension 2008, 52, 1120–1126. [Google Scholar] [CrossRef]
- Arboix, A.; Jiménez, C.; Massons, J.; Parra, O.; Besses, C. Hematological Disorders: A Commonly Unrecognized Cause of Acute Stroke. Expert Rev. Hematol. 2016, 9, 891–901. [Google Scholar] [CrossRef]
- Dubow, J.; Fink, M.E. Impact of Hypertension on Stroke. Curr. Atheroscler. Rep. 2011, 13, 298–305. [Google Scholar] [CrossRef]
- Arroja, M.M.C.; Reid, E.; McCabe, C. Therapeutic Potential of the Renin Angiotensin System in Ischaemic Stroke. Exp. Trans. Stroke Med. 2016, 8, 8. [Google Scholar] [CrossRef]
- Brouns, R.; De Deyn, P.P. The Complexity of Neurobiological Processes in Acute Ischemic Stroke. Clin. Neurol. Neurosurg. 2009, 111, 483–495. [Google Scholar] [CrossRef]
- Chrysant, S.G. The Pathophysiologic Role of the Brain Renin-Angiotensin System in Stroke Protection: Clinical Implications. J. Clin. Hypertens. 2007, 9, 454–459. [Google Scholar] [CrossRef]
- Wang, K.; Gheblawi, M.; Oudit, G.Y. Angiotensin Converting Enzyme 2: A Double-Edged Sword. Circulation 2020, 142, 426–428. [Google Scholar] [CrossRef]
- Marcheselli, S.; Micieli, G. Renin-Angiotensin System and Stroke. Neurol. Sci. 2008, 29 (Suppl. 2), 277–278. [Google Scholar] [CrossRef]
- Phillips, M.I.; Sumners, C. Angiotensin II in Central Nervous System Physiology. Regul. Pept. 1998, 78, 1–11. [Google Scholar] [CrossRef]
- Regenhardt, R.W.; Bennion, D.M.; Sumners, C. Cerebroprotective Action of Angiotensin Peptides in Stroke. Clin. Sci. 2014, 126, 195–205. [Google Scholar] [CrossRef]
- Kagiyama, T.; Kagiyama, S.; Phillips, M.I. Expression of Angiotensin Type 1 and 2 Receptors in Brain after Transient Middle Cerebral Artery Occlusion in Rats. Regul. Pept. 2003, 110, 241–247. [Google Scholar] [CrossRef]
- Steckelings, U.M.; Kaschina, E.; Unger, T. The AT2 Receptor—A Matter of Love and Hate. Peptides 2005, 26, 1401–1409. [Google Scholar] [CrossRef]
- Thöne-Reineke, C.; Steckelings, U.M.; Unger, T. Angiotensin Receptor Blockers and Cerebral Protection in Stroke. J. Hypertens. 2006, 24, S115–S121. [Google Scholar] [CrossRef]
- Stragier, B.; De Bundel, D.; Sarre, S.; Smolders, I.; Vauquelin, G.; Dupont, A.; Michotte, Y.; Vanderheyden, P. Involvement of Insulin-Regulated Aminopeptidase in the Effects of the Renin—Angiotensin Fragment Angiotensin IV: A Review. Heart Fail. Rev. 2008, 13, 321–337. [Google Scholar] [CrossRef]
- Chrysant, S.G. The Role of Angiotensin II Receptors in Stroke Protection. Curr. Hypertens. Rep. 2012, 14, 202–208. [Google Scholar] [CrossRef]
- Ginsberg, M.D. Adventures in the Pathophysiology of Brain Ischemia: Penumbra, Gene Expression, Neuroprotection: The 2002 Thomas Willis Lecture. Stroke 2003, 34, 214–223. [Google Scholar] [CrossRef]
- Wilms, H.; Rosenstiel, P.; Unger, T.; Deuschl, G.; Lucius, R. Neuroprotection with Angiotensin Receptor Antagonists. Am. J. Cardiovasc. Drugs 2005, 5, 245–253. [Google Scholar] [CrossRef]
- Tsukuda, K.; Mogi, M.; Iwanami, J.; Min, L.-J.; Sakata, A.; Jing, F.; Iwai, M.; Horiuchi, M. Cognitive Deficit in Amyloid-β–Injected Mice Was Improved by Pretreatment with a Low Dose of Telmisartan Partly Because of Peroxisome Proliferator-Activated Receptor-γ Activation. Hypertension 2009, 54, 782–787. [Google Scholar] [CrossRef]
- Horiuchi, M.; Mogi, M. Role of Angiotensin II Receptor Subtype Activation in Cognitive Function and Ischaemic Brain Damage. Br. J. Pharmacol. 2011, 163, 1122–1130. [Google Scholar] [CrossRef]
- Wincewicz, D.; Braszko, J.J. Angiotensin II AT1 Receptor Blockade by Telmisartan Reduces Impairment of Spatial Maze Performance Induced by Both Acute and Chronic Stress. J. Renin-Angiotensin-Aldosterone Syst. 2015, 16, 495–505. [Google Scholar] [CrossRef]
- Li, J.; Culman, J.; Hörtnagl, H.; Zhao, Y.; Gerova, N.; Timm, M.; Blume, A.; Zimmermann, M.; Seidel, K.; Dirnagl, U.; et al. Angiotensin AT2 Receptor Protects against Cerebral Ischemia-induced Neuronal Injury. FASEB J. 2005, 19, 1–25. [Google Scholar] [CrossRef]
- Arboix, A.; Cabeza, N.; García-Eroles, L.; Massons, J.; Oliveres, M.; Targa, C.; Balcells, M. Relevance of Transient Ischemic Attack to Early Neurological Recovery after Nonlacunar Ischemic Stroke. Cerebrovasc. Dis. 2004, 18, 304–311. [Google Scholar] [CrossRef]
- Magy, L.; Vincent, F.; Faure, S.; Messerli, F.; Wang, J.; Achard, J.; Fournier, A. The Renin-Angiotensin Systems: Evolving Pharmacological Perspectives for Cerebroprotection. Curr. Pharm. Des. 2005, 11, 3275–3291. [Google Scholar] [CrossRef]
- Iadecola, C.; Zhang, F.; Xu, S.; Casey, R.; Ross, M.E. Inducible Nitric Oxide Synthase Gene Expression in Brain Following Cerebral Ischemia. J. Cereb. Blood Flow Metab. 1995, 15, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Ren, J.; Chan, K.; Chen, H. Angiotensin-(1–7) Regulates Angiotensin II-Induced VCAM-1 Expression on Vascular Endothelial Cells. Biochem. Biophys. Res. Commun. 2013, 430, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Da Silveira, K.D.; Coelho, F.M.; Vieira, A.T.; Sachs, D.; Barroso, L.C.; Costa, V.V.; Bretas, T.L.B.; Bader, M.; de Sousa, L.P.; da Silva, T.A.; et al. Anti-Inflammatory Effects of the Activation of the Angiotensin-(1–7) Receptor, Mas, in Experimental Models of Arthritis. J. Immunol. 2010, 185, 5569–5576. [Google Scholar] [CrossRef] [PubMed]
- Mecca, A.P.; Regenhardt, R.W.; O’Connor, T.E.; Joseph, J.P.; Raizada, M.K.; Katovich, M.J.; Sumners, C. Cerebroprotection by Angiotensin-(1-7) in Endothelin-1-Induced Ischaemic Stroke: Angiotensin-(1-7) Cerebroprotection during Stroke. Exp. Physiol. 2011, 96, 1084–1096. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, J.M. Brain and Pituitary Angiotensin. Endocr. Rev. 1992, 13, 329–380. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Pavel, J.; Macova, M.; Yu, Z.-X.; Imboden, H.; Ge, L.; Nishioku, T.; Dou, J.; Delgiacco, E.; Saavedra, J.M. AT1 Receptor Blockade Regulates the Local Angiotensin II System in Cerebral Microvessels from Spontaneously Hypertensive Rats. Stroke 2006, 37, 1271–1276. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.; Olah, L.; Harms, C.; Maul, B.; Bader, M.; Hörtnagl, H.; Schultheiss, H.-P.; Mies, G. Ischemic Injury in Experimental Stroke Depends on Angiotensin II. FASEB J. 2002, 16, 169–176. [Google Scholar] [CrossRef]
- Iwai, M.; Liu, H.-W.; Chen, R.; Ide, A.; Okamoto, S.; Hata, R.; Sakanaka, M.; Shiuchi, T.; Horiuchi, M. Possible Inhibition of Focal Cerebral Ischemia by Angiotensin II Type 2 Receptor Stimulation. Circulation 2004, 110, 843–848. [Google Scholar] [CrossRef]
- Farmer, J.A.; Torre-Amione, G. The Renin Angiotensin System as a Risk Factor for Coronary Artery Disease. Curr. Atheroscler. Rep. 2001, 3, 117–124. [Google Scholar] [CrossRef]
- Gorelick, P.B. Stroke Prevention Therapy Beyond Antithrombotics: Unifying Mechanisms in Ischemic Stroke Pathogenesis and Implications for Therapy: An Invited Review. Stroke 2002, 33, 862–875. [Google Scholar] [CrossRef]
- Markus, H.S.; Barley, J.; Lunt, R.; Bland, J.M.; Jeffery, S.; Carter, N.D.; Brown, M.M. Angiotensin-Converting Enzyme Gene Deletion Polymorphism: A New Risk Factor for Lacunar Stroke but Not Carotid Atheroma. Stroke 1995, 26, 1329–1333. [Google Scholar] [CrossRef]
- Law, M.; Morris, J.; Wald, N. Use of Blood Pressure Lowering Drugs in the Prevention of Cardiovascular Disease: Meta-Analysis of 147 Randomised Trials in the Context of Expectations from Prospective Epidemiological Studies. BMJ 2009, 338, b1665. [Google Scholar] [CrossRef]
- Krikov, M.; Thone-Reineke, C.; Müller, S.; Villringer, A.; Unger, T. Candesartan but Not Ramipril Pretreatment Improves Outcome after Stroke and Stimulates Neurotrophin BNDF/TrkB System in Rats. J. Hypertens. 2008, 26, 544–552. [Google Scholar] [CrossRef]
- Lou, M.; Blume, A.; Zhao, Y.; Gohlke, P.; Deuschl, G.; Herdegen, T.; Culman, J. Sustained Blockade of Brain AT1 Receptors before and after Focal Cerebral Ischemia Alleviates Neurologic Deficits and Reduces Neuronal Injury, Apoptosis, and Inflammatory Responses in the Rat. J. Cereb. Blood Flow Metab. 2004, 24, 536–547. [Google Scholar] [CrossRef]
- Lu, G.-C.; Cheng, J.-W.; Zhu, K.-M.; Ma, X.-J.; Shen, F.-M.; Su, D.-F. A Systematic Review of Angiotensin Receptor Blockers in Preventing Stroke. Stroke 2009, 40, 3876–3878. [Google Scholar] [CrossRef][Green Version]
- Dahlöf, B.; Devereux, R.B.; Kjeldsen, S.E.; Julius, S.; Beevers, G.; de Faire, U.; Fyhrquist, F.; Ibsen, H.; Kristiansson, K.; Lederballe-Pedersen, O. Cardiovascular Morbidity and Mortality in the Losartan Intervention for Endpoint Reduction in Hypertension Study (LIFE): A Randomised Trial against Atenolol. Lancet 2002, 359, 995–1003. [Google Scholar] [CrossRef]
- Schrader, J.; Lüders, S.; Kulschewski, A.; Hammersen, F.; Plate, K.; Berger, J.; Zidek, W.; Dominiak, P.; Diener, H.C.; MOSES Study Group. Morbidity and Mortality after Stroke, Eprosartan Compared with Nitrendipine for Secondary Prevention: Principal Results of a Prospective Randomized Controlled Study (MOSES). Stroke 2005, 36, 1218–1224. [Google Scholar] [CrossRef]
- Selim, M.; Savitz, S.; Linfante, I.; Caplan, L.; Schlaug, G. Effect of Pre-Stroke Use of ACE Inhibitors on Ischemic Stroke Severity. BMC Neurol. 2005, 5, 10. [Google Scholar] [CrossRef]
- Kumar, S.; Savitz, S.; Schlaug, G.; Caplan, L.; Selim, M. Antiplatelets, ACE Inhibitors, and Statins Combination Reduces Stroke Severity and Tissue at Risk. Neurology 2006, 66, 1153–1158. [Google Scholar] [CrossRef]
- Verdecchia, P.; Reboldi, G.; Angeli, F.; Gattobigio, R.; Bentivoglio, M.; Thijs, L.; Staessen, J.A.; Porcellati, C. Angiotensin-Converting Enzyme Inhibitors and Calcium Channel Blockers for Coronary Heart Disease and Stroke Prevention. Hypertension 2005, 46, 386–392. [Google Scholar] [CrossRef]
- Reinecke, K.; Lucius, R.; Reinecke, A.; Rickert, U.; Herdegen, T.; Unger, T. Angiotensin II Accelerates Functional Recovery in the Rat Sciatic Nerve in Vivo: Role of the AT 2 Receptor and the Transcription Factor NF-κB. FASEB J. 2003, 17, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Adams, H.P., Jr.; Del Zoppo, G.; Alberts, M.J.; Bhatt, D.L.; Brass, L.; Furlan, A.; Grubb, R.L.; Higashida, R.T.; Jauch, E.C.; Kidwell, C. Guidelines for the Early Management of Adults with Ischemic Stroke: A Guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups: The American Academy of Neurology Affirms the Value of This Guideline as an Educational Tool for Neurologists. Stroke 2007, 38, 1655–1711. [Google Scholar] [PubMed]
- Sandset, E.C.; Bath, P.M.; Boysen, G.; Jatuzis, D.; Kõrv, J.; Lüders, S.; Murray, G.D.; Richter, P.S.; Roine, R.O.; Terént, A. The Angiotensin-Receptor Blocker Candesartan for Treatment of Acute Stroke (SCAST): A Randomised, Placebo-Controlled, Double-Blind Trial. Lancet 2011, 377, 741–750. [Google Scholar] [CrossRef]
- Dávalos, A.; Alvarez-Sabín, J.; Castillo, J.; Díez-Tejedor, E.; Ferro, J.; Martínez-Vila, E.; Serena, J.; Segura, T.; Cruz, V.T.; Masjuan, J.; et al. Citicoline in the Treatment of Acute Ischaemic Stroke: An International, Randomised, Multicentre, Placebo-Controlled Study (ICTUS Trial). Lancet 2012, 380, 349–357. [Google Scholar] [CrossRef]
- Fernandez, L.A.; Spencer, D.D.; Kaczmar, T. Angiotensin II Decreases Mortality Rate in Gerbils with Unilateral Carotid Ligation. Stroke 1986, 17, 82–85. [Google Scholar] [CrossRef]
- Fernandez, L.A.; Caride, V.J.; Strömberg, C.; Näveri, L.; Wicke, J.D. Angiotensin AT2 Receptor Stimulation Increases Survival in Gerbils with Abrupt Unilateral Carotid Ligation. J. Cardiovasc. Pharmacol. 1994, 24, 937–940. [Google Scholar] [CrossRef]
- Haberl, R.L. Role of Angiotensin Receptor Subtypes in the Response of Rabbit Brain Arterioles to Angiotensin. Stroke 1994, 25, 1476–1479. [Google Scholar] [CrossRef]
- Regenhardt, R.W.; Desland, F.; Mecca, A.P.; Pioquinto, D.J.; Afzal, A.; Mocco, J.; Sumners, C. Anti-Inflammatory Effects of Angiotensin-(1-7) in Ischemic Stroke. Neuropharmacology 2013, 71, 154–163. [Google Scholar] [CrossRef]
- Bennion, D.M.; Jones, C.H.; Donnangelo, L.L.; Graham, J.T.; Isenberg, J.D.; Dang, A.N.; Rodriguez, V.; Sinisterra, R.D.M.; Sousa, F.B.; Santos, R.A.S.; et al. Neuroprotection by Post-Stroke Administration of an Oral Formulation of Angiotensin-(1-7) in Ischaemic Stroke. Exp. Physiol. 2018, 103, 916–923. [Google Scholar] [CrossRef]
- Regenhardt, R.W.; Mecca, A.P.; Desland, F.; Ritucci-Chinni, P.F.; Ludin, J.A.; Greenstein, D.; Banuelos, C.; Bizon, J.L.; Reinhard, M.K.; Sumners, C. Centrally Administered Angiotensin-(1-7) Increases the Survival of Stroke-Prone Spontaneously Hypertensive Rats: Angiotensin-(1-7) and Hypertension-Induced Stroke. Exp. Physiol. 2014, 99, 442–453. [Google Scholar] [CrossRef]
- Engelhorn, T.; Goerike, S.; Doerfler, A.; Okorn, C.; Forsting, M.; Heusch, G.; Schulz, R. The Angiotensin II Type 1—Receptor Blocker Candesartan Increases Cerebral Blood Flow, Reduces Infarct Size, and Improves Neurologic Outcome after Transient Cerebral Ischemia in Rats. J. Cereb. Blood Flow Metab. 2004, 24, 467–474. [Google Scholar] [CrossRef]
- Nishimura, Y.; Ito, T.; Saavedra, J.M. Angiotensin II AT 1 Blockade Normalizes Cerebrovascular Autoregulation and Reduces Cerebral Ischemia in Spontaneously Hypertensive Rats. Stroke 2000, 31, 2478–2486. [Google Scholar] [CrossRef]
- Dai, W.-J.; Funk, A.; Herdegen, T.; Unger, T.; Culman, J. Blockade of Central Angiotensin AT 1 Receptors Improves Neurological Outcome and Reduces Expression of AP-1 Transcription Factors After Focal Brain Ischemia in Rats. Stroke 1999, 30, 2391–2399. [Google Scholar] [CrossRef]
- Alhusban, A.; Kozak, A.; Ergul, A.; Fagan, S.C. AT1 Receptor Antagonism Is Proangiogenic in the Brain: BDNF a Novel Mediator. J. Pharmacol. Exp. Ther. 2013, 344, 348–359. [Google Scholar] [CrossRef]
- Joseph, J.P.; Mecca, A.P.; Regenhardt, R.W.; Bennion, D.M.; Rodríguez, V.; Desland, F.; Patel, N.A.; Pioquinto, D.J.; Unger, T.; Katovich, M.J.; et al. The Angiotensin Type 2 Receptor Agonist Compound 21 Elicits Cerebroprotection in Endothelin-1 Induced Ischemic Stroke. Neuropharmacology 2014, 81, 134–141. [Google Scholar] [CrossRef]
- McCarthy, C.A.; Vinh, A.; Miller, A.A.; Hallberg, A.; Alterman, M.; Callaway, J.K.; Widdop, R.E. Direct Angiotensin AT2 Receptor Stimulation Using a Novel AT2 Receptor Agonist, Compound 21, Evokes Neuroprotection in Conscious Hypertensive Rats. PLoS ONE 2014, 9, e95762. [Google Scholar] [CrossRef]
- Schwengel, K.; Namsolleck, P.; Lucht, K.; Clausen, B.H.; Lambertsen, K.L.; Valero-Esquitino, V.; Thöne-Reineke, C.; Müller, S.; Widdop, R.E.; Denton, K.M.; et al. Angiotensin AT2-Receptor Stimulation Improves Survival and Neurological Outcome after Experimental Stroke in Mice. J. Mol. Med. 2016, 94, 957–966. [Google Scholar] [CrossRef]
- Min, L.-J.; Mogi, M.; Tsukuda, K.; Jing, F.; Ohshima, K.; Nakaoka, H.; Kan-no, H.; Wang, X.-L.; Chisaka, T.; Bai, H.-Y.; et al. Direct Stimulation of Angiotensin II Type 2 Receptor Initiated After Stroke Ameliorates Ischemic Brain Damage. Am. J. Hypertens. 2014, 27, 1036–1044. [Google Scholar] [CrossRef][Green Version]
- Ishrat, T.; Fouda, A.Y.; Pillai, B.; Eldahshan, W.; Ahmed, H.; Waller, J.L.; Ergul, A.; Fagan, S.C. Dose–Response, Therapeutic Time-Window and TPA-Combinatorial Efficacy of Compound 21: A Randomized, Blinded Preclinical Trial in a Rat Model of Thromboembolic Stroke. J. Cereb. Blood Flow Metab. 2019, 39, 1635–1647. [Google Scholar] [CrossRef]
- Hill, M.D.; Goyal, M.; Menon, B.K.; Nogueira, R.G.; McTaggart, R.A.; Demchuk, A.M.; Poppe, A.Y.; Buck, B.H.; Field, T.S.; Dowlatshahi, D.; et al. Efficacy and Safety of Nerinetide for the Treatment of Acute Ischaemic Stroke (ESCAPE-NA1): A Multicentre, Double-Blind, Randomised Controlled Trial. Lancet 2020, 395, 878–887. [Google Scholar] [CrossRef]
- Iwanami, J.; Mogi, M.; Tsukuda, K.; Jing, F.; Ohshima, K.; Wang, X.-L.; Nakaoka, H.; Kan-no, H.; Chisaka, T.; Bai, H.-Y.; et al. Possible Synergistic Effect of Direct Angiotensin II Type 2 Receptor Stimulation by Compound 21 with Memantine on Prevention of Cognitive Decline in Type 2 Diabetic Mice. Eur. J. Pharmacol. 2014, 724, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Jackson, L.; Dong, G.; Althomali, W.; Sayed, M.A.; Eldahshan, W.; Baban, B.; Johnson, M.H.; Filosa, J.; Fagan, S.C.; Ergul, A. Delayed Administration of Angiotensin II Type 2 Receptor (AT2R) Agonist Compound 21 Prevents the Development of Post-Stroke Cognitive Impairment in Diabetes Through the Modulation of Microglia Polarization. Transl. Stroke Res. 2020, 11, 762–775. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, C.A.; Vinh, A.; Callaway, J.K.; Widdop, R.E. Angiotensin AT 2 Receptor Stimulation Causes Neuroprotection in a Conscious Rat Model of Stroke. Stroke 2009, 40, 1482–1489. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, C.A.; Vinh, A.; Broughton, B.R.S.; Sobey, C.G.; Callaway, J.K.; Widdop, R.E. Angiotensin II Type 2 Receptor Stimulation Initiated After Stroke Causes Neuroprotection in Conscious Rats. Hypertension 2012, 60, 1531–1537. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Intervention Performed | Effects Observed | Source |
|---|---|---|
| Angiotensin 2 infusion | Reduced post-stroke mortality Enhanced cerebral perfusion | Fernandez et al. [55] |
| Ang-(1-7) treatment with ischemic stroke | Decreased microglial activation and neuronal-derived chemokines | Haberl et al. [57] |
| Ang-(1-7) treatment with ischemic stroke | Reduced nitric oxide secretion Decreased level of CXCL12, IL1-beta, IL6, CD11B | Regenhardt et al. [58] |
| Ang-(1-7) treatment with ischemic stroke | Reduction of cerebral infarction volume Improvement of neurological function No effect on blood pressure, heart rate, or cerebral blood flow | Bennion et al. [59] |
| Ang-(1-7) treatment with ischemic stroke | Reduction of cerebral infarction volume Effect was blocked by A779 co-infusion | Meca et al. [34] |
| Ang-(1-7) treatment with hemoraghic stroke | Increased life expectancy Improved neurological function Decreased microglical activation | Regenhardt et al. [60] |
| Candesartan treatment before and after MCA occlusion | Increased cerebral blood flow in affected hemisphere Reduction of cerebral infarction volume | Engelhorn et al. [61] |
| Candesartan post-stroke treatment | No effect on arterial blood pressure Raised expression of mRNA of brain-derived neurotrophic factor | Dai et al. [63] |
| Candesartan treatment in hypertensive rats with MCA oclusion | Raised expression of mRNA of brain-derived neurotrophic factor | Alhusban et al. [64] |
| Candesartan chronic treatment | Improved cerebral vascular self-regulation Reduction of cerebral infarction size | Nishimura et al. [62] |
| C21 treatment before and after MCA oclusion | Reduction of cerebral infarction volume Improved neurological function | Joseph et al. [65] |
| C21 treatment 5 days after MCA occlusion | Reduction of cerebral infarction volume | McCarthy et al. [66] |
| C21 treatment after MCA occlusion and reperfusion | Improved survival rate Improved neurological function Decreased neuron apoptosis rate | Schwengel et al. [67] |
| C21 treatment in MCA occlusion | Reduction of cerebral infarction volume Improved neurological function Reduced production of TNF-alpha and anionic superoxides | Min et al. [68] |
| C21 treatment after embolic stroke | Improved neurological function Reduction of hemorrhagic transformation | Ishrat et al. [69] Hill et al. [70] |
| C21 treatment in diabetes mellitus post-stroke model | Reduced mortality rate Improved cognitive function | Jackson et al. [72] |
| CGP42112 treatment in MCA occlusion | Improved neurological function Reduced lesion progression | McCarthy et al. [73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andone, S.; Bajko, Z.; Motataianu, A.; Maier, S.; Barcutean, L.; Balasa, R. Neuroprotection in Stroke—Focus on the Renin-Angiotensin System: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 3876. https://doi.org/10.3390/ijms23073876
Andone S, Bajko Z, Motataianu A, Maier S, Barcutean L, Balasa R. Neuroprotection in Stroke—Focus on the Renin-Angiotensin System: A Systematic Review. International Journal of Molecular Sciences. 2022; 23(7):3876. https://doi.org/10.3390/ijms23073876
Chicago/Turabian StyleAndone, Sebastian, Zoltan Bajko, Anca Motataianu, Smaranda Maier, Laura Barcutean, and Rodica Balasa. 2022. "Neuroprotection in Stroke—Focus on the Renin-Angiotensin System: A Systematic Review" International Journal of Molecular Sciences 23, no. 7: 3876. https://doi.org/10.3390/ijms23073876
APA StyleAndone, S., Bajko, Z., Motataianu, A., Maier, S., Barcutean, L., & Balasa, R. (2022). Neuroprotection in Stroke—Focus on the Renin-Angiotensin System: A Systematic Review. International Journal of Molecular Sciences, 23(7), 3876. https://doi.org/10.3390/ijms23073876