
Novel Insights into Autophagy and Prostate Cancer: A Comprehensive Review

 ,
,  , , , , , , , and
, , , , , , , and 
Abstract
:1. Introduction
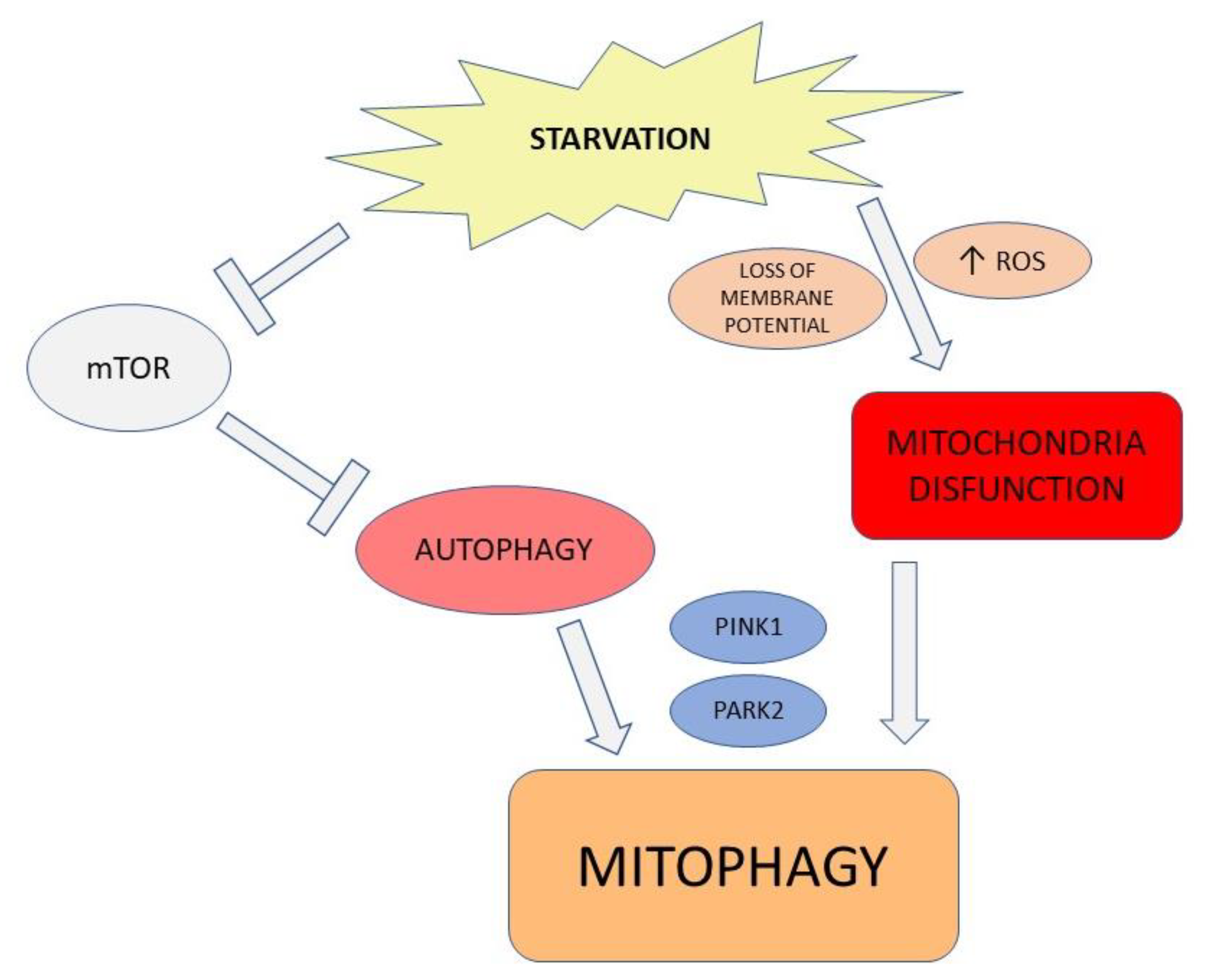
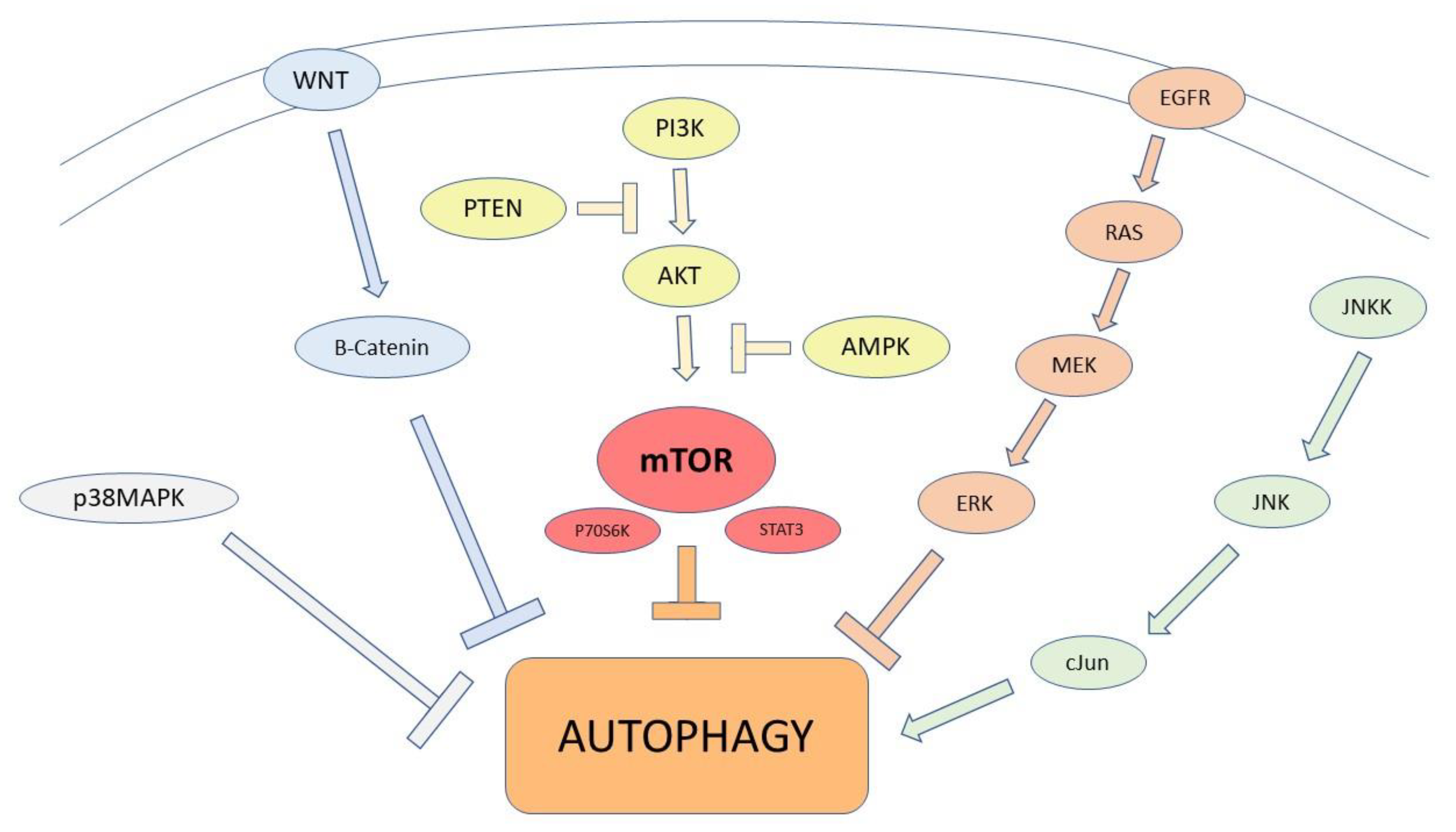
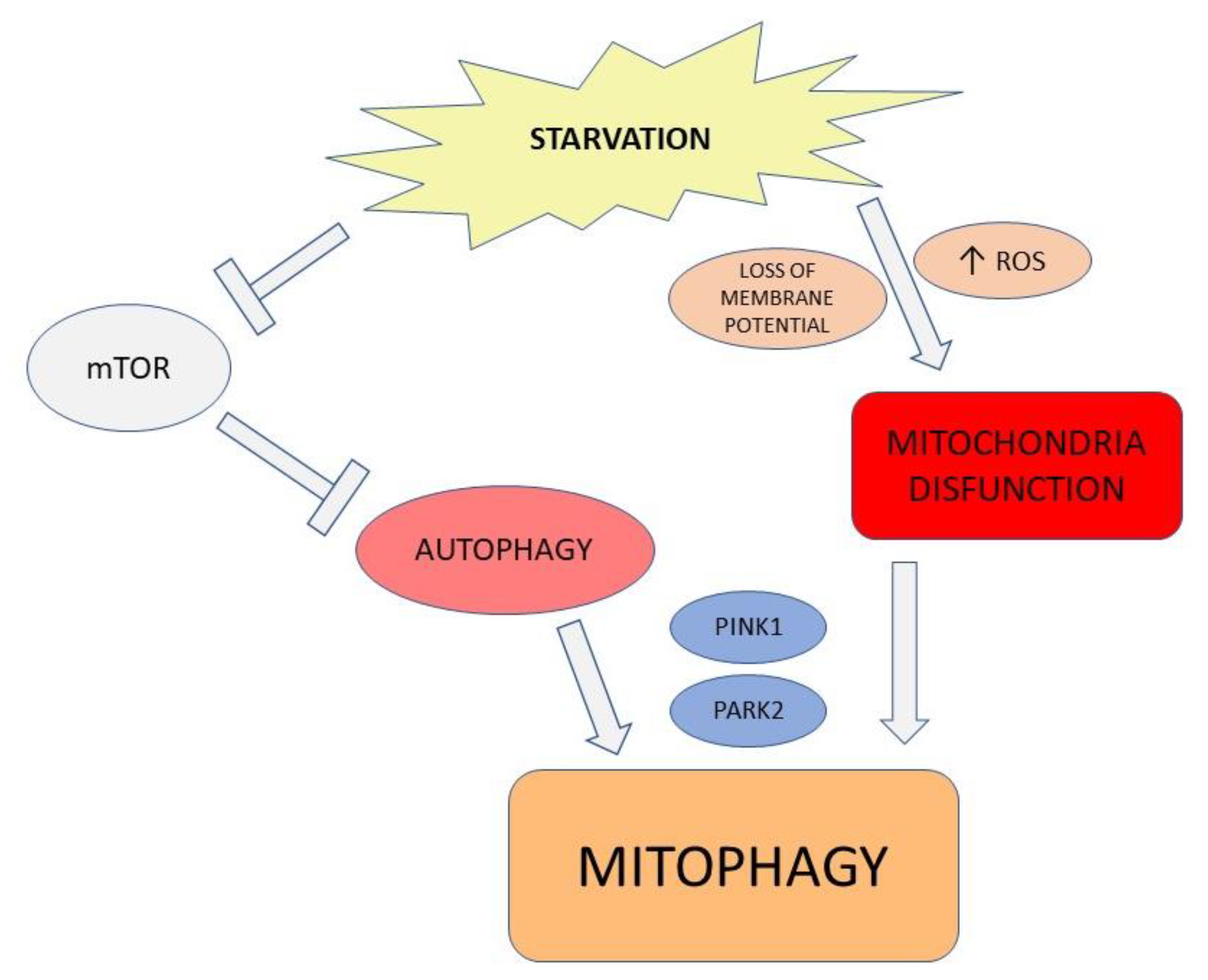
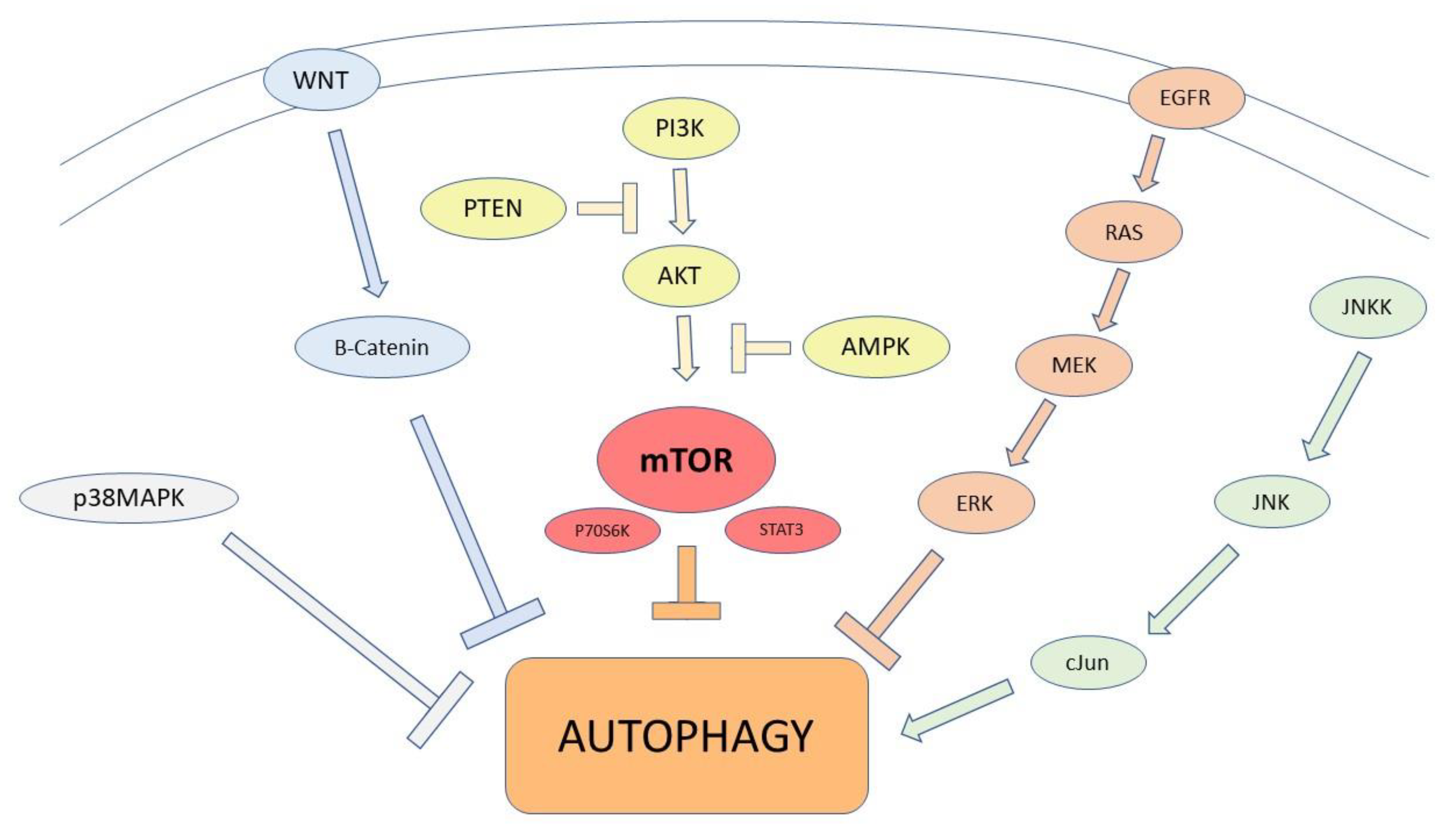
2. Autophagic Machinery: Recent Evidence
2.1. Autophagic Modulation
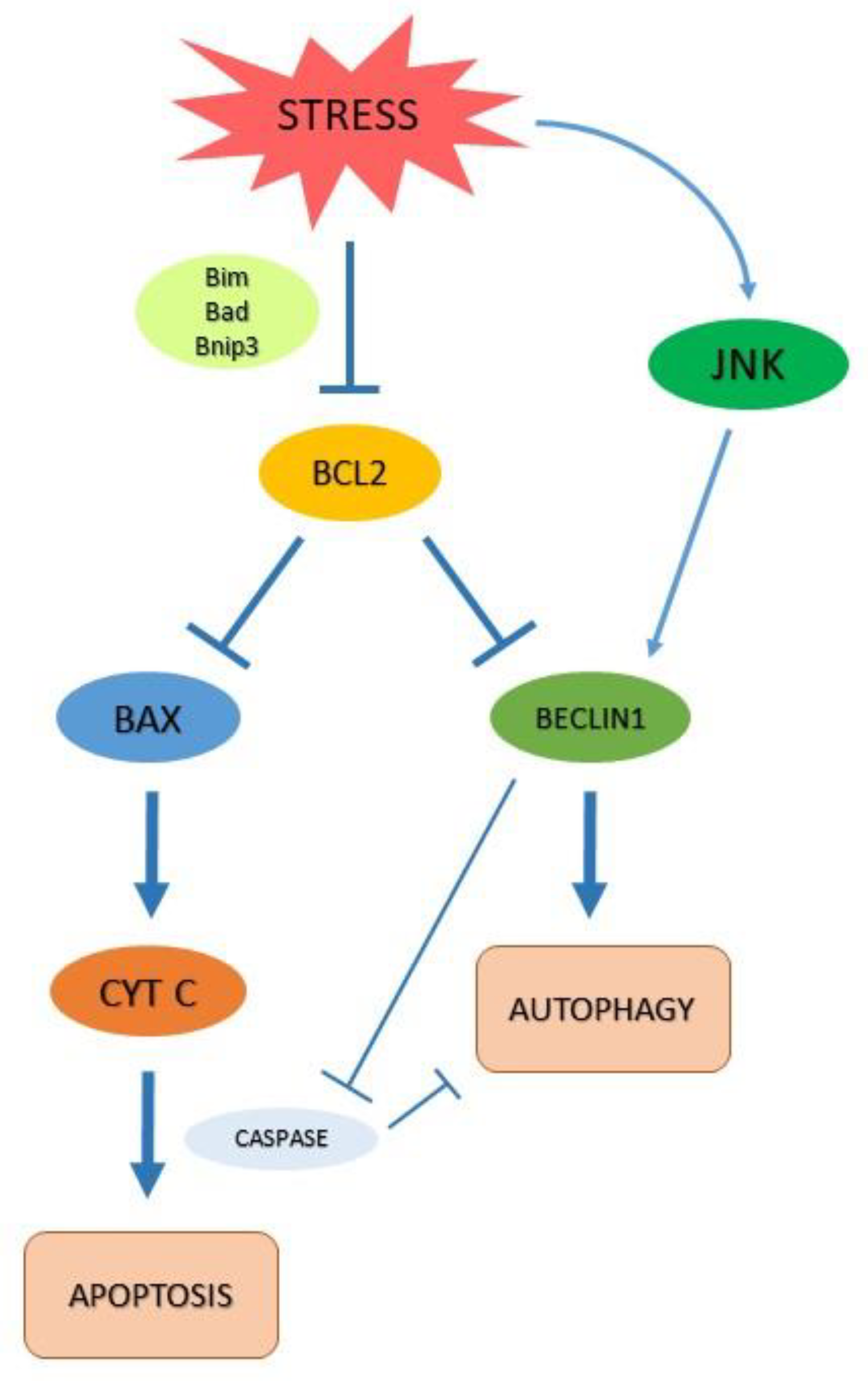
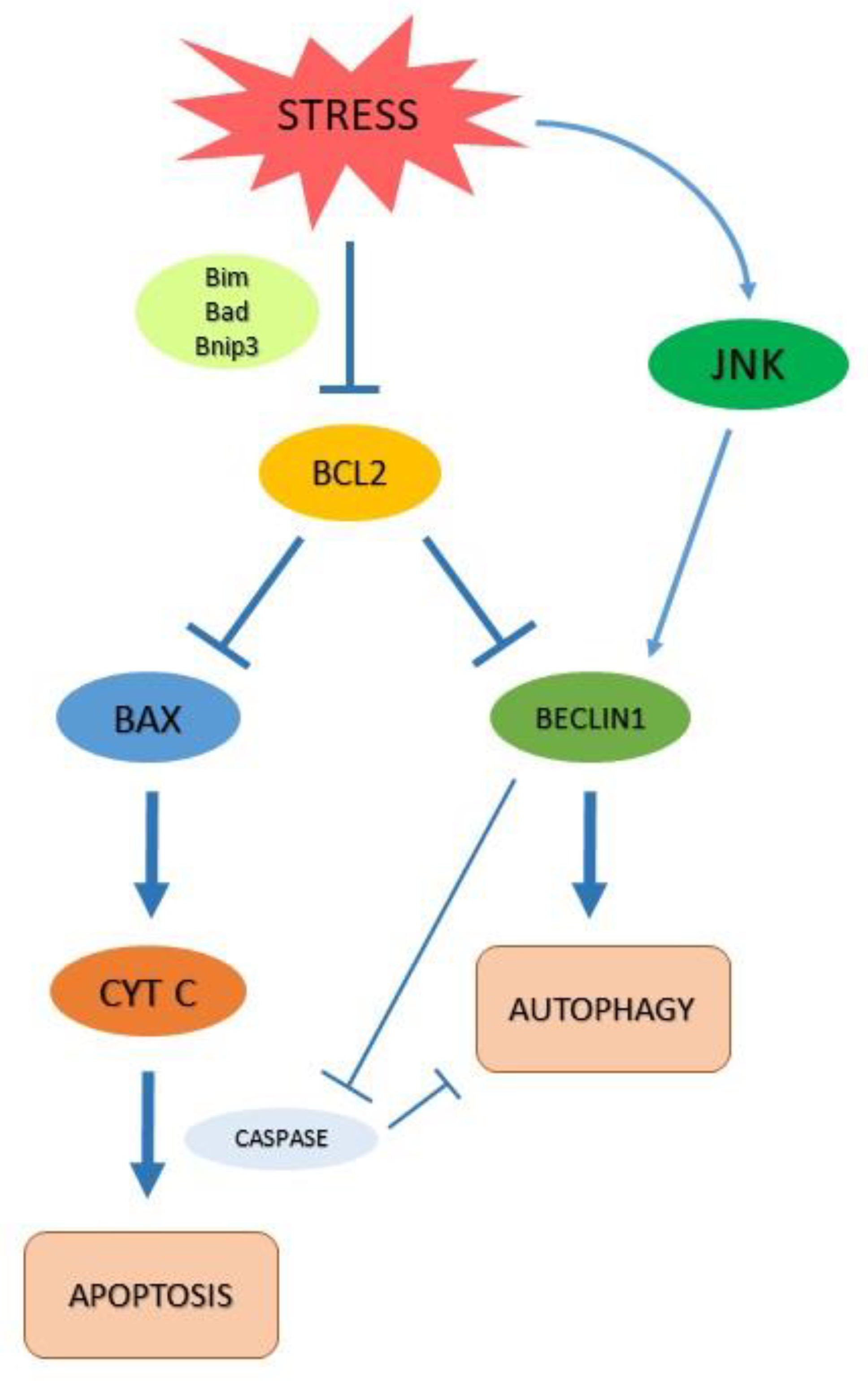
2.2. Autophagy and Apoptosis
3. Autophagy and Prostate Cancer
4. Prostate Cancer Treatment and Autophagy
5. Autophagic Modulation in PCa Treatment
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abreu, S.; Sanchez-Wandelmer, J.; Reggiori, F. The Origin of Autophagosomes: The Beginning of an End. In Autophagy and Cancer; Wang, H.-W., Ed.; Springer: New York, NY, USA, 2013; pp. 47–61. [Google Scholar]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degenhardt KMathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; Nelson, D.A.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, H.L.; Stockley, J.; Fleming, J.T.; Mandal, R.; O’Prey, J.; Ryan, K.M.; Robson, C.N.; Leung, H.Y. Does androgen-ablation therapy (AAT) associated autophagy have a pro-survival effect in LNCaP human prostate cancer cells? BJU Int. 2013, 111, 672–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, G.M.; Tan, Y.; Wang, H. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol. Cancer 2019, 18, 17. [Google Scholar] [CrossRef] [Green Version]
- Kuballa, P.; Nolte, W.; Castoreno, A.; Xavier, R. Autophagy and the Immune System. Annu. Rev. Immunol. 2012, 30, 611–646. [Google Scholar] [CrossRef]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian Autophagy: How Does It Work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef]
- Menon, M.B.; Dhamija, S. Beclin 1 Phosphorylation—At the Center of Autophagy Regulation. Front. Cell Dev. Biol. 2018, 6, 137. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [PubMed] [Green Version]
- Padman BSBach, M.; Lucarelli, G.; Prescott, M.; Ramm, G. The protonophore CCCP interferes with lysosomal degradation of autophagic cargo in yeast and mammalian cells. Autophagy 2013, 9, 1862–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, L.M.; Gerner, L.; Rettel, M.; Stein, F.; Burrows, J.F.; Mills, I.G.; Evergren, E. CaMKK2 facilitates Golgi-associated vesicle trafficking to sustain cancer cell proliferation. Cell Death Dis. 2021, 12, 1040. [Google Scholar] [CrossRef]
- Fernández, Á.F.; Sebti, S.; Wei, Y.; Zou, Z.; Shi, M.; McMillan, K.L.; He, C.; Ting, T.; Liu, Y.; Chiang, W.C.; et al. Disruption of the beclin 1–BCL2 autophagy regulatory complex promotes longevity in mice. Nature 2018, 558, 136–140. [Google Scholar] [CrossRef]
- Oberstein, A.; Jeffrey, P.D.; Shi, Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J. Biol. Chem. 2007, 282, 13123–13132. [Google Scholar] [CrossRef] [Green Version]
- Thorburn, A. Apoptosis and Autophagy: Regulatory connections between two supposedly different processes. Apoptosis Int. J. Program. Cell Death 2008, 13, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, S.; Kanaseki, T.; Mizushima, N.; Mizuta, T.; Arakawa-Kobayashi, S.; Thompson, C.B.; Tsujimoto, Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 2004, 6, 1221–1228. [Google Scholar] [CrossRef]
- Bildik, G.; Liang, X.; Sutton, M.N.; Bast, R.C.; Lu, Z. DIRAS3: An Imprinted Tumor Suppressor Gene that Regulates RAS and PI3K-driven Cancer Growth, Motility, Autophagy, and Tumor Dormancy. Mol. Cancer Ther. 2022, 21, 25–37. [Google Scholar] [CrossRef]
- Leone, R.D.; Amaravadi, R.K. Autophagy: A targetable linchpin of cancer cell metabolism. Trends Endocrinol. Metab. TEM 2013, 24, 209–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferro, M.; Lucarelli, G.; Crocetto, F.; Dolce, P.; Verde, A.; La Civita, E.; Zappavigna, S.; de Cobelli, O.; Di Lorenzo, G.; Facchini, B.A.; et al. First-line systemic therapy for metastatic castration-sensitive prostate cancer: An updated systematic review with novel findings. Crit. Rev. Oncol. Hematol. 2021, 157, 103198. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, G.; Loizzo, D.; Ferro, M.; Rutigliano, M.; Vartolomei, M.D.; Cantiello, F.; Buonerba, C.; Di Lorenzo, G.; Terracciano, D.; De Cobelli, O.; et al. Metabolomic profiling for the identification of novel diagnostic markers and therapeutic targets in prostate cancer: An update. Expert Rev. Mol. Diagn. 2019, 19, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Fuzio, P.; Lucarelli, G.; Perlino, E.; Battaglia, M.; Bettocchi, C.; Selvaggi, F.P.; Ditonno, P. Androgen deprivation therapy regulation of beta1C integrin expression in prostate cancer. Oncol. Rep. 2009, 22, 327–335. [Google Scholar]
- Fuzio, P.; Ditonno, P.; Lucarelli, G.; Battaglia, M.; Bettocchi, C.; Senia, T.; Perlino, E. Androgen deprivation therapy affects BCL-2 expression in human prostate cancer. Int. J. Oncol. 2011, 39, 1233–1242. [Google Scholar]
- Lucarelli, G.; Fanelli, M.; Larocca, A.M.; Germinario, C.A.; Rutigliano, M.; Vavallo, A.; Selvaggi, F.P.; Bettocchi, C.; Battaglia, M.; Ditonno, P. Serum sarcosine increases the accuracy of prostate cancer detection in patients with total serum PSA less than 4.0 ng/ml. Prostate 2012, 72, 1611–1621. [Google Scholar] [CrossRef]
- Lucarelli, G.; Ditonno, P.; Bettocchi, C.; Spilotros, M.; Rutigliano, M.; Vavallo, A.; Galleggiante, V.; Fanelli, M.; Larocca, A.M.; Germinario, C.A.; et al. Serum sarcosine is a risk factor for progression and survival in patients with metastatic castration-resistant prostate cancer. Future Oncol. 2013, 9, 899–907. [Google Scholar] [CrossRef]
- Lucarelli, G.; Rutigliano, M.; Bettocchi, C.; Palazzo, S.; Vavallo, A.; Galleggiante, V.; Trabucco, S.; Di Clemente, D.; Selvaggi, F.P.; Battaglia, M.; et al. Spondin-2, a secreted extracellular matrix protein, is a novel diagnostic biomarker for prostate cancer. J. Urol. 2013, 190, 2271–2277. [Google Scholar] [CrossRef]
- Lucarelli, G.; Rutigliano, M.; Galleggiante, V.; Giglio, A.; Palazzo, S.; Ferro, M.; Simone, C.; Bettocchi, C.; Battaglia, M.; Ditonno, P. Metabolomic profiling for the identification of novel diagnostic markers in prostate cancer. Expert Rev. Mol. Diagn. 2015, 15, 1211–1224. [Google Scholar] [CrossRef]
- Ferro, M.; Buonerba, C.; Terracciano, D.; Lucarelli, G.; Cosimato, V.; Bottero, D.; Deliu, V.M.; Ditonno, P.; Perdonà, S.; Autorino, R.; et al. Biomarkers in localized prostate cancer. Future Oncol. 2016, 12, 399–411. [Google Scholar] [CrossRef] [Green Version]
- Ferro, M.; Terracciano, D.; Buonerba, C.; Lucarelli, G.; Bottero, D.; Perdonà, S.; Autorino, R.; Serino, A.; Cantiello, F.; Damiano, R.; et al. The emerging role of obesity, diet and lipid metabolism in prostate cancer. Future Oncol. 2017, 13, 285–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferro, M.; Ungaro, P.; Cimmino, A.; Lucarelli, G.; Busetto, G.M.; Cantiello, F.; Damiano, R.; Terracciano, D. Epigenetic Signature: A New Player as Predictor of Clinically Significant Prostate Cancer (PCa) in Patients on Active Surveillance (AS). Int. J. Mol. Sci. 2017, 18, 1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferro, M.; De Cobelli, O.; Lucarelli, G.; Porreca, A.; Busetto, G.M.; Cantiello, F.; Damiano, R.; Autorino, R.; Musi, G.; Vartolomei, M.D.; et al. Beyond PSA: The Role of Prostate Health Index (phi). Int. J. Mol. Sci. 2020, 21, 1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buonerba, C.; Ferro, M.; Dolce, P.; Crocetto, F.; Verde, A.; Lucarelli, G.; Scafuri, L.; Facchini, S.; Vaia, A.; Marinelli, A.; et al. Predictors of efficacy of androgen-receptor-axis-targeted therapies in patients with metastatic castration-sensitive prostate cancer: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2020, 151, 102992. [Google Scholar] [CrossRef]
- Ferro, M.; de Cobelli, O.; Vartolomei, M.D.; Lucarelli, G.; Crocetto, F.; Barone, B.; Sciarra, A.; Del Giudice, F.; Muto, M.; Maggi, M.; et al. Prostate Cancer Radiogenomics-From Imaging to Molecular Characterization. Int. J. Mol. Sci. 2021, 22, 9971. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, N.; Jiang, N. Autophagy provides a conceptual therapeutic framework for bone metastasis from prostate cancer. Cell Death Dis. 2021, 12, 909. [Google Scholar] [CrossRef]
- Menon, S.; Yecies, J.L.; Zhang, H.H.; Howell, J.J.; Nicholatos, J.; Harputlugil, E.; Bronson, R.T.; Kwiatkowski, D.J.; Manning, B.D. Chronic activation of mTOR complex 1 is sufficient to cause hepatocellular carcinoma in mice. Sci. Signal. 2012, 5, ra24. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.Y.; White, E. Autophagy, Metabolism, and Cancer. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Chiacchiera, F.; Simone, C. The AMPK-FoxO3A axis as a target for cancer treatment. Cell Cycle Georget. Tex 2010, 9, 1091–1096. [Google Scholar] [CrossRef] [Green Version]
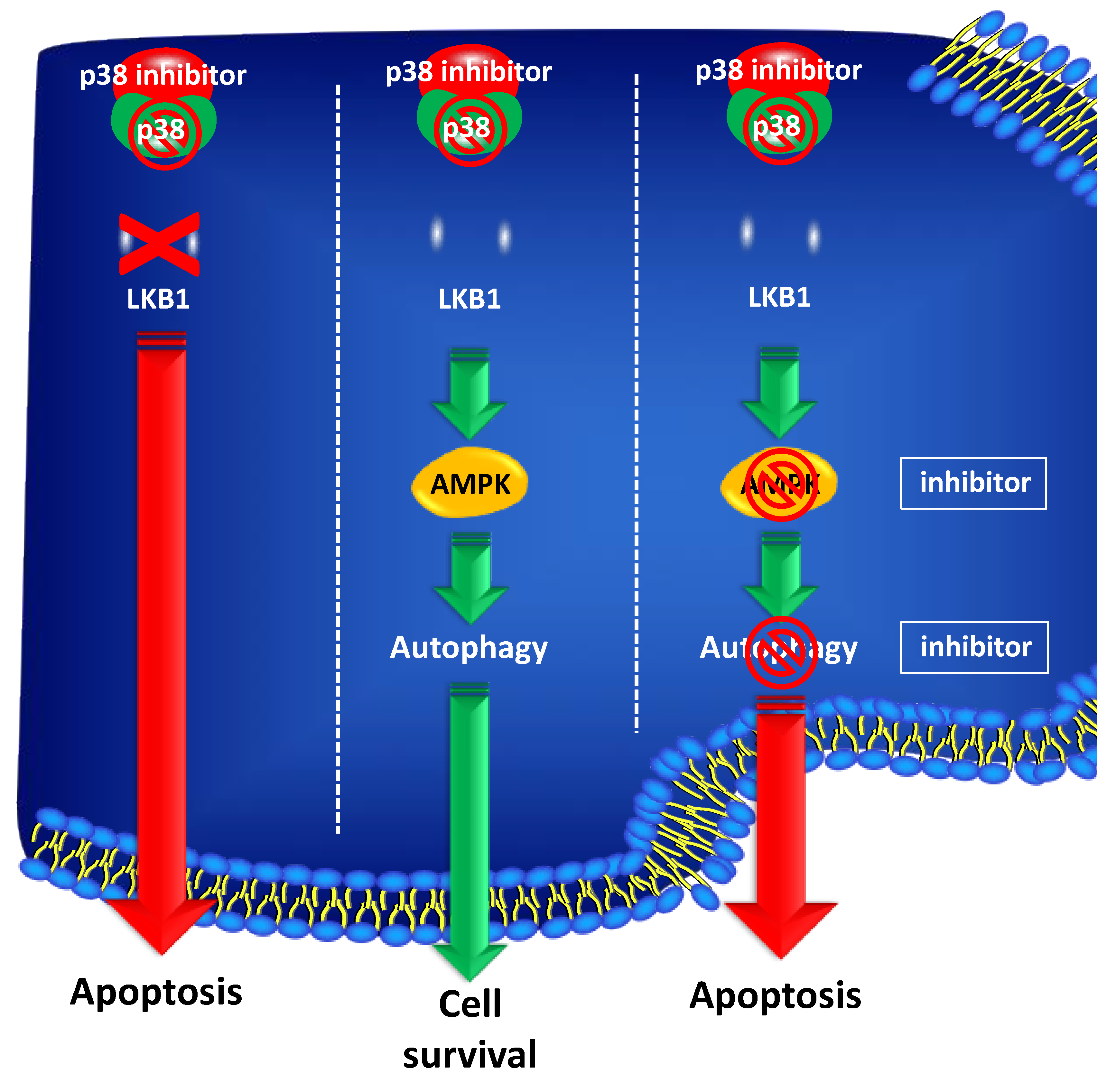
- Grossi, V.; Lucarelli, G.; Forte, G.; Peserico, A.; Matrone, A.; Germani, A.; Rutigliano, M.; Stella, A.; Bagnulo, R.; Loconte, D.; et al. Loss of STK11 expression is an early event in prostate carcinogenesis and predicts therapeutic response to targeted therapy against MAPK/p38. Autophagy 2015, 11, 2102–2113. [Google Scholar] [CrossRef] [Green Version]
- Chiacchiera, F.; Matrone, A.; Ferrari, E.; Ingravallo, G.; Lo Sasso, G.; Murzilli, S.; Petruzzelli, M.; Salvatore, L.; Moschetta, A.; Simone, C. p38alpha blockade inhibits colorectal cancer growth in vivo by inducing a switch from HIF1alpha- to FoxO-dependent transcription. Cell Death Differ. 2009, 16, 1203–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; Blessing, A.M.; Pulliam, T.L.; Shi, Y.; Wilkenfeld, S.R.; Han, J.J.; Murray, M.M.; Pham, A.H.; Duong, K.; Brun, S.N.; et al. Inhibition of CAMKK2 impairs autophagy and castration-resistant prostate cancer via suppression of AMPK-ULK1 signaling. Oncogene 2021, 40, 1690–1705. [Google Scholar] [CrossRef] [PubMed]
- Kischkel, F.C.; Lawrence, D.A.; Chuntharapai, A.; Schow, P.; Kim, K.J.; Ashkenazi, A. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity 2000, 12, 611–620. [Google Scholar] [CrossRef] [Green Version]
- Nazim, U.M.; Yin, H.; Park, S.-Y. Neferine treatment enhances the TRAIL-induced apoptosis of human prostate cancer cells via autophagic flux and the JNK pathway. Int. J. Oncol. 2020, 56, 1152–1161. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Qi, F.; Li, L.; Qin, Z.; Li, X.; Wang, X. Autophagy-related genes are potential diagnostic and prognostic biomarkers in prostate cancer. Transl. Androl. Urol. 2020, 9, 2616–2628. [Google Scholar] [CrossRef]
- Hu, D.; Jiang, L.; Luo, S.; Zhao, X.; Hu, H.; Zhao, G.; Tang, W. Development of an autophagy-related gene expression signature for prognosis prediction in prostate cancer patients. J. Transl. Med. 2020, 18, 160. [Google Scholar] [CrossRef] [Green Version]
- Giatromanolaki, A.; Sivridis, E.; Mendrinos, S.; Koutsopoulos, A.V.; Koukourakis, M.I. Autophagy proteins in prostate cancer: Relation with anaerobic metabolism and Gleason score. Urol. Oncol. 2014, 32, 39.e11–39.e18. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Kepp, O.; Michaud, M.; Martins, I.; Minoux, H.; Métivier, D.; Maiuri, M.C.; Kroemer, R.T.; Kroemer, G. Association and dissociation of autophagy, apoptosis and necrosis by systematic chemical study. Oncogene 2011, 305, 4544–4556. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar]
- Santanam, U.; Banach-Petrosky, W.; Abate-Shen, C.; Shen, M.M.; White, E.; DiPaola, R.S. Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev. 2016, 30, 399–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.G.; Yang, J.C.; Kung, H.J.; Shi, X.B.; Tilki, D.; Lara PNJr DeVere White, R.W.; Gao, A.C.; Evans, C.P. Targeting autophagy overcomes Enzalutamide resistance in castration-resistant prostate cancer cells and improves therapeutic response in a xenograft model. Oncogene 2014, 336, 4521–4530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Zhang, J.; Zhang, W.; Zhang, D.; Li, Y.; Zhang, J.; Zhang, Y.; Diao, T.; Cui, L.; Li, W.; et al. Abiraterone and MDV3100 inhibits the proliferation and promotes the apoptosis of prostate cancer cells through mitophagy. Cancer Cell Int. 2019, 19, 332. [Google Scholar] [CrossRef]
- Mortezavi, A.; Salemi, S.; Kranzbühler, B.; Gross, O.; Sulser, T.; Simon, H.U.; Eberli, D. Inhibition of autophagy significantly increases the antitumor effect of Abiraterone in prostate cancer. World J. Urol. 2019, 37, 351–358. [Google Scholar] [CrossRef]
- Eberli, D.; Kranzbühler, B.; Mortezavi, A.; Sulser, T.; Salemi, S. Apalutamide in combination with autophagy inhibitors improves treatment effects in prostate cancer cells. Urol. Oncol. 2020, 38, e19–e683. [Google Scholar] [CrossRef] [PubMed]
- Veldscholte, J.; Berrevoets, C.A.; Ris-Stalpers, C.; Kuiper, J.J.G.M.; Jenster, G.; Trapman, J.; Brinkmann, A.O.; Mulder, E. The androgen receptor in LNCaP cells contains a mutation in the ligand binding domain which affects steroid binding characteristics and response to antiandrogens. J. Steroid Biochem. Mol. Biol. 1992, 41, 665–669. [Google Scholar] [CrossRef]
- Farrow, J.M.; Yang, J.C.; Evans, C.P. Autophagy as a modulator and target in prostate cancer. Nat. Rev. Urol. 2014, 11, 508–516. [Google Scholar] [CrossRef]
- Veldhoen, R.A.; Banman, S.L.; Hemmerling, D.R.; Odsen, R.; Simmen, T.; Simmonds, A.J.; Underhill, D.A.; Goping, I.S. The chemotherapeutic agent paclitaxel inhibits autophagy through two distinct mechanisms that regulate apoptosis. Oncogene 2013, 32, 736–746. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.Z.; Wang, W.W.; Hu, T.T.; Zhu, G.Y.; Li, L.N.; Zhang, C.Y.; Xu, Z.; Yu, H.B.; Wu, H.F.; Zhu, J.G. FOXM1 contributes to docetaxel resistance in castration-resistant prostate cancer by inducing AMPK/mTOR-mediated autophagy. Cancer Lett. 2020, 469, 481–489. [Google Scholar] [CrossRef]
- Hofman, M.S.; Violet, J.; Hicks, R.J.; Ferdinandus, J.; Thang, S.P.; Akhurst, T.; Iravani, A.; Kong, G.; Ravi Kumar, A.; Murphy, D.G.; et al. [177Lu]-PSMA-617 radionuclide treatment in patients with metastatic castration-resistant prostate cancer (LuPSMA trial): A single-centre, single-arm, phase 2 study. Lancet. Oncol. 2018, 19, 825–833. [Google Scholar] [CrossRef]
- Kratochwil, C.; Bruchertseifer, F.; Rathke, H.; Hohenfellner, M.; Giesel, F.L.; Haberkorn, U.; Morgenstern, A. Targeted α-Therapy of Metastatic Castration-Resistant Prostate Cancer with 225Ac-PSMA-617: Swimmer-Plot Analysis Suggests Efficacy Regarding Duration of Tumor Control. J. Nucl. Med. 2018, 59, 795–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukha, A.; Kahya, U.; Linge, A.; Chen, O.; Löck, S.; Lukiyanchuk, V.; Richter, S.; Alves, T.C.; Peitzsch, M.; Telychko, V.; et al. GLS-driven glutamine catabolism contributes to prostate cancer radiosensitivity by regulating the redox state, stemness and ATG5-mediated autophagy. Theranostics 2021, 11, 7844–7868. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.G.; O’Neill, E.; Habjan, C.; Cornelissen, B. Combination Strategies to Improve Targeted Radionuclide Therapy. J. Nucl. Med. 2020, 61, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- Erkisa, M.; Aydinlik, S.; Cevatemre, B.; Aztopal, N.; Akar, R.O.; Celikler, S.; Yilmaz, V.T.; Ari, F.; Ulukaya, E. A promising therapeutic combination for metastatic prostate cancer: Chloroquine as autophagy inhibitor and palladium(II) barbiturate complex. Biochimie 2020, 175, 159–172. [Google Scholar] [CrossRef]
- Ben Sahra, I.; Laurent, K.; Giuliano, S.; Larbret, F.; Ponzio, G.; Gounon, P.; Le Marchand-Brustel, Y.; Giorgetti-Peraldi, S.; Cormont, M.; Bertolotto, C.; et al. Targeting cancer cell metabolism: The combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010, 70, 2465–2475. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Wang, H.; Geng, X.; Zhang, D.; Zhu, Z.; Zhang, G.; Hou, J. Metformin exerts anti-AR-negative prostate cancer activity via AMPK/autophagy signaling pathway. Cancer Cell Int. 2021, 21, 404. [Google Scholar] [CrossRef]
- Luty, M.; Piwowarczyk, K.; Łabędź-Masłowska, A.; Wróbel, T.; Szczygieł, M.; Catapano, J.; Drabik, G.; Ryszawy, D.; Kędracka-Krok, S.; Madeja, Z.; et al. Fenofibrate Augments the Sensitivity of Drug-Resistant Prostate Cancer Cells to Docetaxel. Cancers 2019, 11, 77. [Google Scholar] [CrossRef] [Green Version]
- Tan, Q.; Joshua, A.M.; Wang, M.; Bristow, R.G.; Wouters, B.G.; Allen, C.J.; Tannock, I.F. Up-regulation of autophagy is a mechanism of resistance to chemotherapy and can be inhibited by pantoprazole to increase drug sensitivity. Cancer Chemother. Pharmacol. 2017, 79, 959–969. [Google Scholar] [CrossRef]
- Hansen, A.R.; Tannock, I.F.; Templeton, A.; Chen, E.; Evans, A.; Knox, J.; Prawira, A.; Sridhar, S.S.; Tan, S.; Vera-Badillo, F.; et al. Pantoprazole Affecting Docetaxel Resistance Pathways via Autophagy (PANDORA): Phase II Trial of High Dose Pantoprazole (Autophagy Inhibitor) with Docetaxel in Metastatic Castration-Resistant Prostate Cancer (mCRPC). Oncologist 2019, 24, 1188–1194. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functional Step | Gene | Function |
|---|---|---|
| Induction of autophagy | mTOR | Negative regulator of autophagy |
| ULK1 | Part of the complex ULK1 kinase | |
| ATG13 | ||
| RB1CC1 | ||
| Autophagosome formation | ATG 2 | Components of ATG9-WIPI complex and Vps34-beclin1 class III PI3-kinase complex |
| ATG 9 | ||
| WIPI1/2 | ||
| PI3KC3 | ||
| PI3KR4 | ||
| BECN1 | ||
| ATG14 | ||
| ATG12 | ||
| ATG5 | ||
| ATG7 | ||
| Autophagosome maturation | ATG16 | LC3/ATG8 conjugation |
| ATG10 | ||
| CAMKK2 | ||
| MAP1LC3B | ||
| ATG8 | ||
| Autophagosome/lysosome fusion and degradation | ATG3 | Ubiquitin-like conjugation systems |
| ATG4 | ||
| TFEB | Regulation of lysosomal genes and fusion with autophagosome | |
| RAB7 |
| Drug | Mechanism of Action | Effect on Autophagy |
|---|---|---|
| Abiraterone | Blocking testosterone biosynthesis (CYP17) | Triggering autophagy and mitophagy |
| Enzalutamide | Blocking testosterone binding to AR | Triggering autophagy via mTOR inhibition |
| Preventing AR nuclear translocation | ||
| Apalutamide | Blocking testosterone binding to AR | Triggering autophagy |
| Preventing AR nuclear translocation | ||
| Docetaxel | Inhibition of microtubular depolymerization | Triggering autophagy via upregulation FOXM1 expression |
| Phosphorylation and inactivation of the bcl-2 protein |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loizzo, D.; Pandolfo, S.D.; Rogers, D.; Cerrato, C.; di Meo, N.A.; Autorino, R.; Mirone, V.; Ferro, M.; Porta, C.; Stella, A.; et al. Novel Insights into Autophagy and Prostate Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2022, 23, 3826. https://doi.org/10.3390/ijms23073826
Loizzo D, Pandolfo SD, Rogers D, Cerrato C, di Meo NA, Autorino R, Mirone V, Ferro M, Porta C, Stella A, et al. Novel Insights into Autophagy and Prostate Cancer: A Comprehensive Review. International Journal of Molecular Sciences. 2022; 23(7):3826. https://doi.org/10.3390/ijms23073826
Chicago/Turabian StyleLoizzo, Davide, Savio Domenico Pandolfo, Devin Rogers, Clara Cerrato, Nicola Antonio di Meo, Riccardo Autorino, Vincenzo Mirone, Matteo Ferro, Camillo Porta, Alessandro Stella, and et al. 2022. "Novel Insights into Autophagy and Prostate Cancer: A Comprehensive Review" International Journal of Molecular Sciences 23, no. 7: 3826. https://doi.org/10.3390/ijms23073826
APA StyleLoizzo, D., Pandolfo, S. D., Rogers, D., Cerrato, C., di Meo, N. A., Autorino, R., Mirone, V., Ferro, M., Porta, C., Stella, A., Bizzoca, C., Vincenti, L., Spilotros, M., Rutigliano, M., Battaglia, M., Ditonno, P., & Lucarelli, G. (2022). Novel Insights into Autophagy and Prostate Cancer: A Comprehensive Review. International Journal of Molecular Sciences, 23(7), 3826. https://doi.org/10.3390/ijms23073826