
Development of VPC-70619, a Small-Molecule N-Myc Inhibitor as a Potential Therapy for Neuroendocrine Prostate Cancer

 , , and
, , and
Abstract
:1. Introduction
2. Results
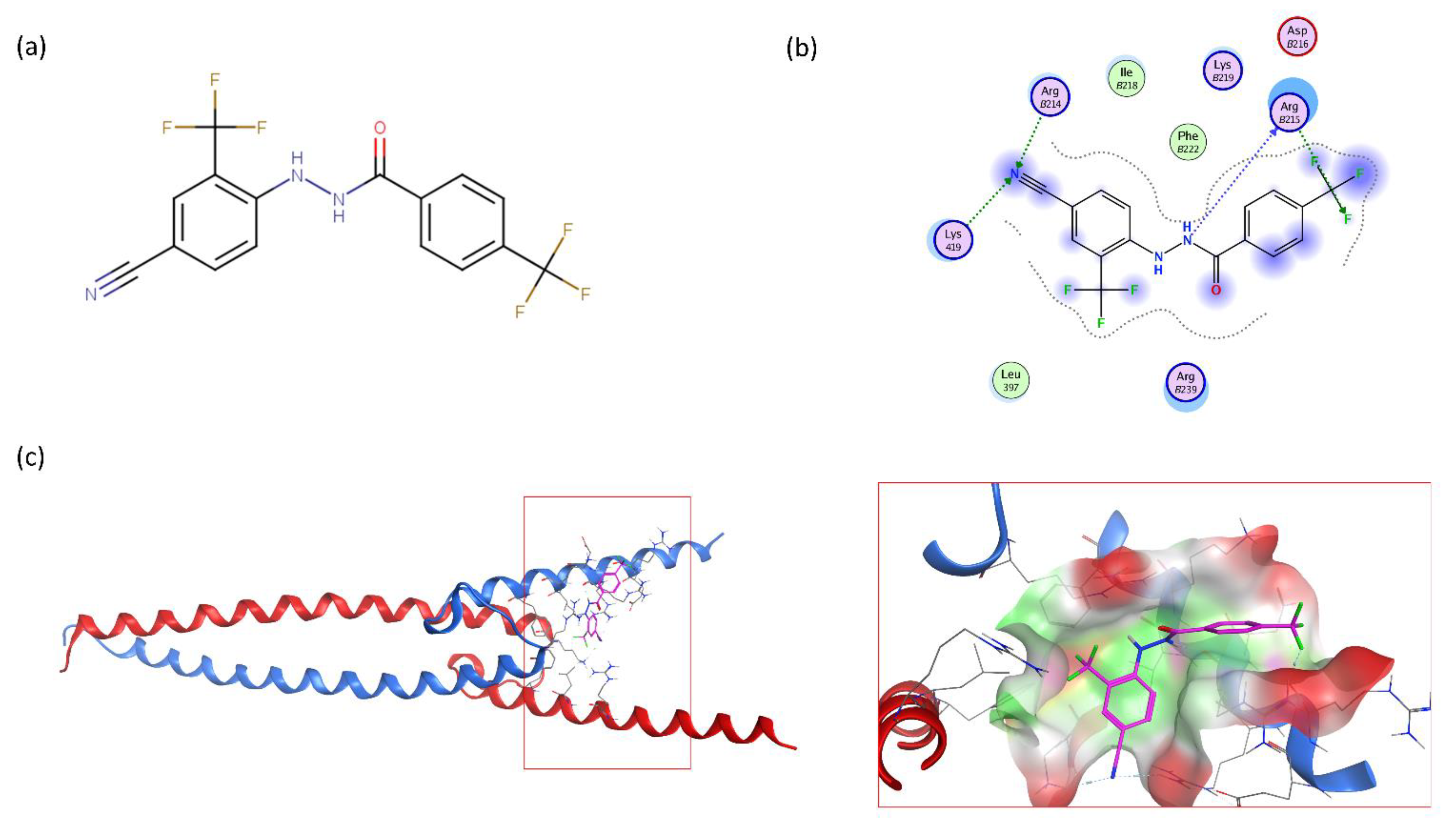
2.1. Large-Scale Structure-Based Similarity Search and Scaffold Tuning
2.2. Extensive N-Myc Specific SAR Exploration
2.2.1. Large, Extended, or Bulky Substitutions Are Detrimental to Anti-N-Myc Activity
2.2.2. Sulfonyl and Oxy-Linkers Did Not Improve Scaffold Potency
2.2.3. Halogenic Substitutions Are Beneficial for N-Myc Transcription Inhibition
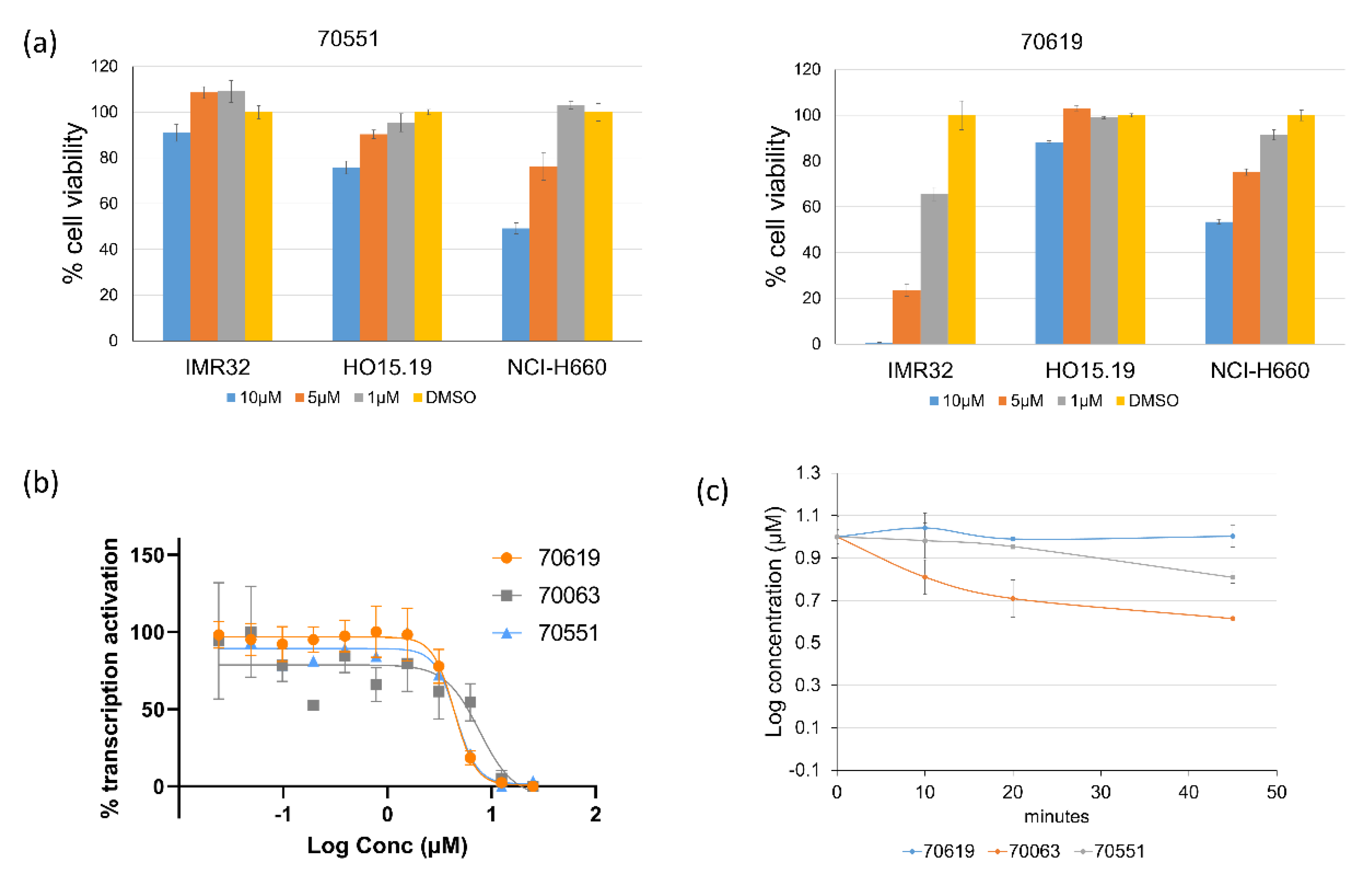
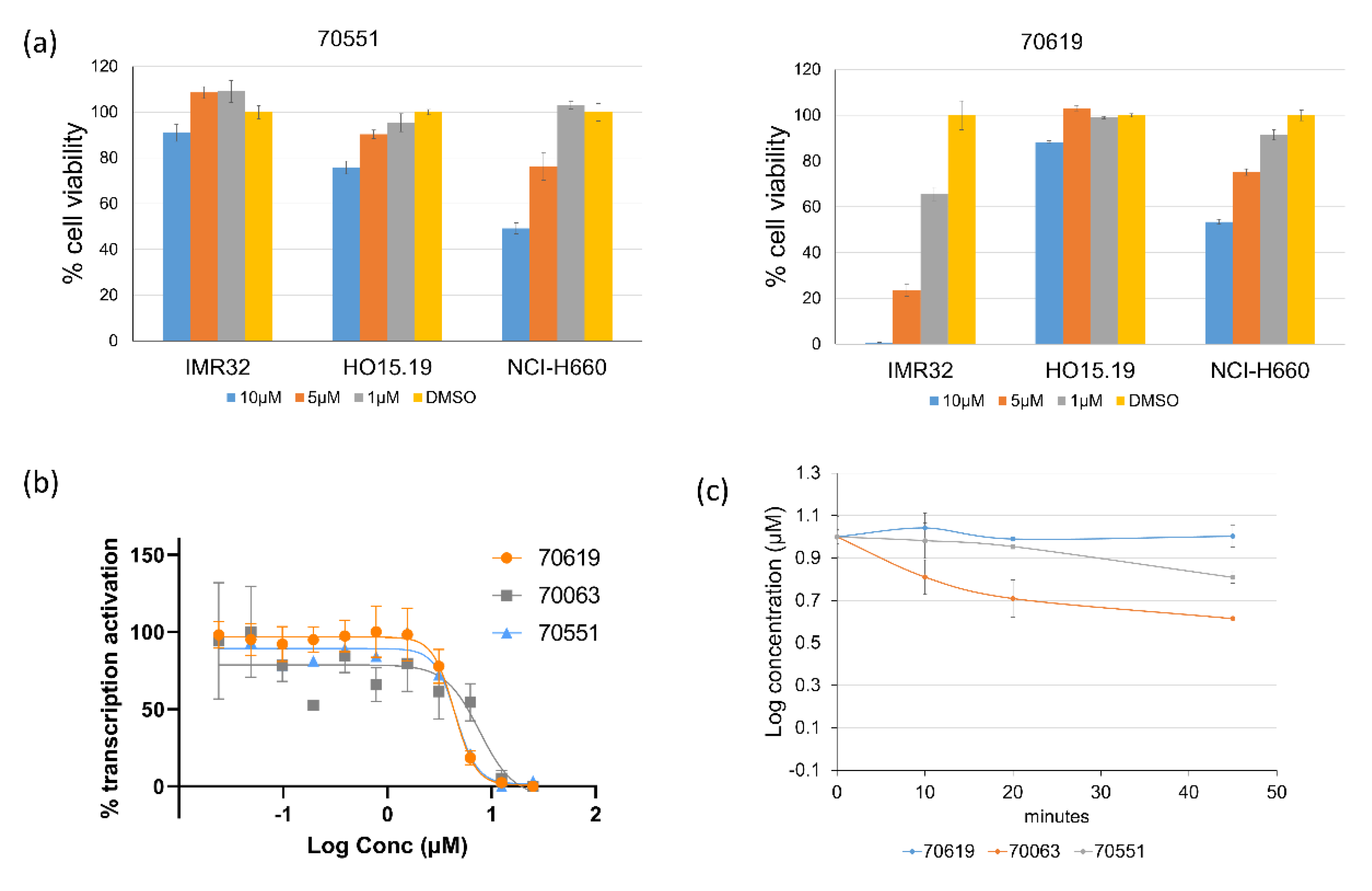
2.3. VPC-70619 Shows a Good Balance of Potency and Viability in Multiple N-Myc Expressing Cell Lines
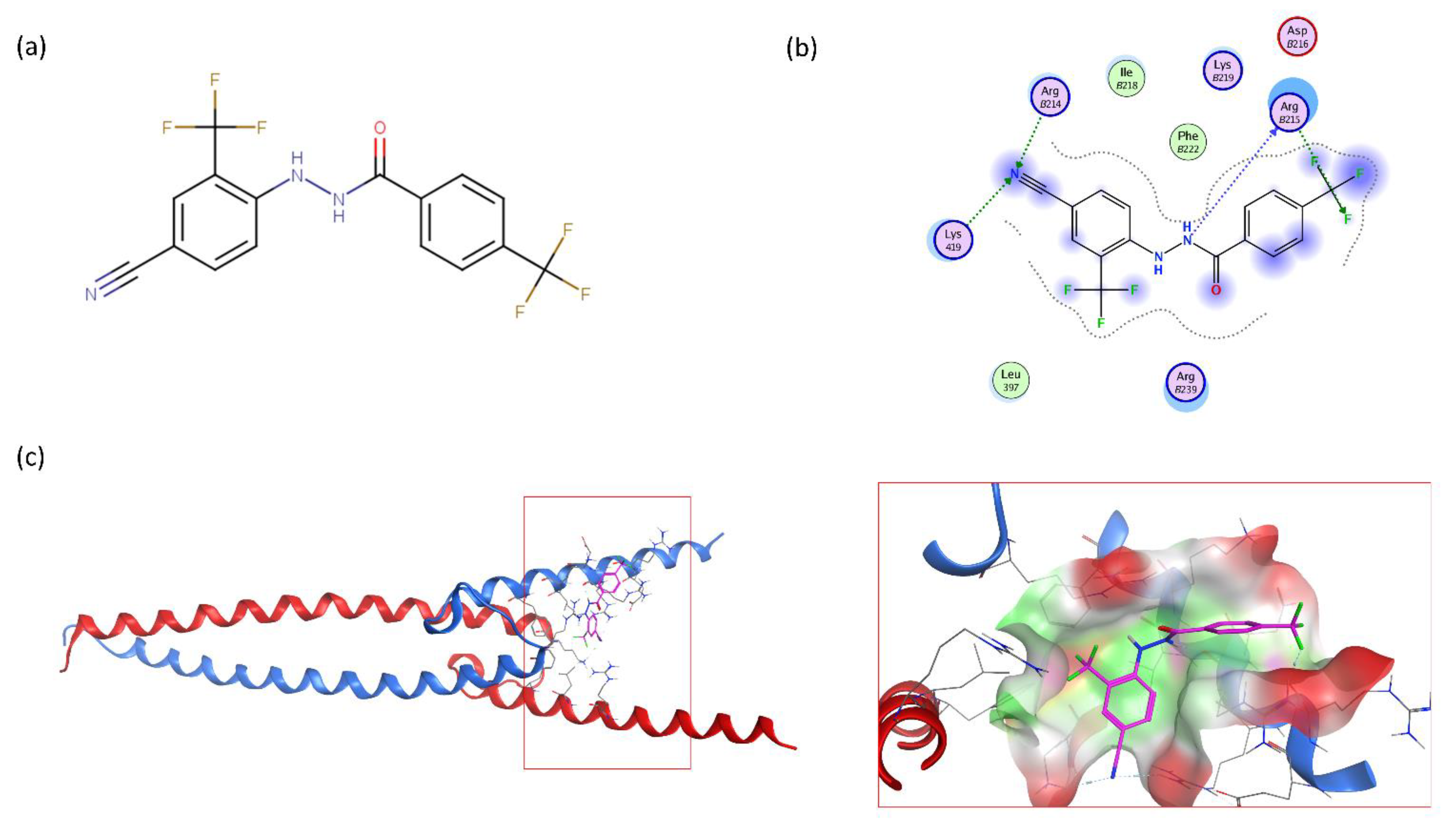
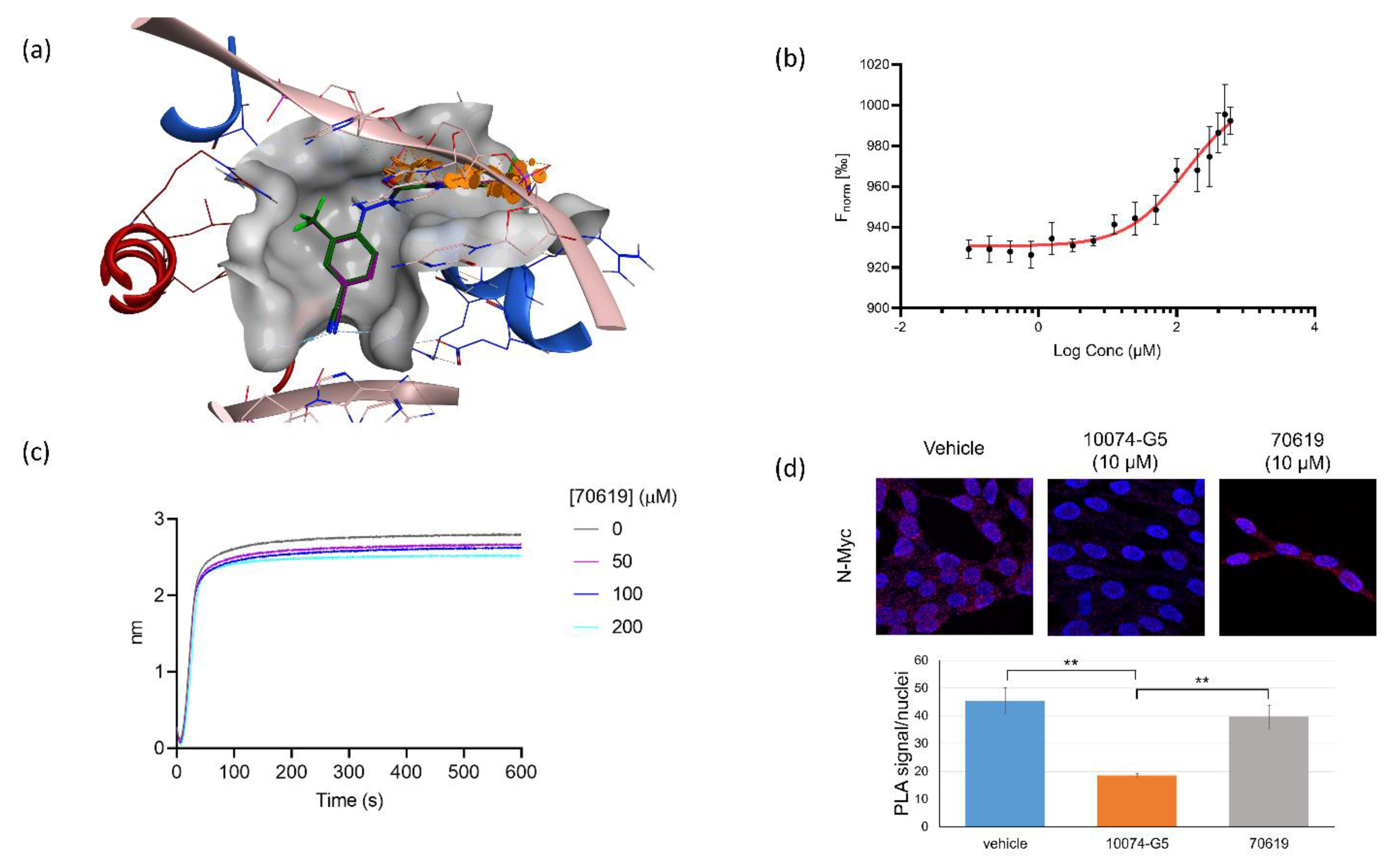
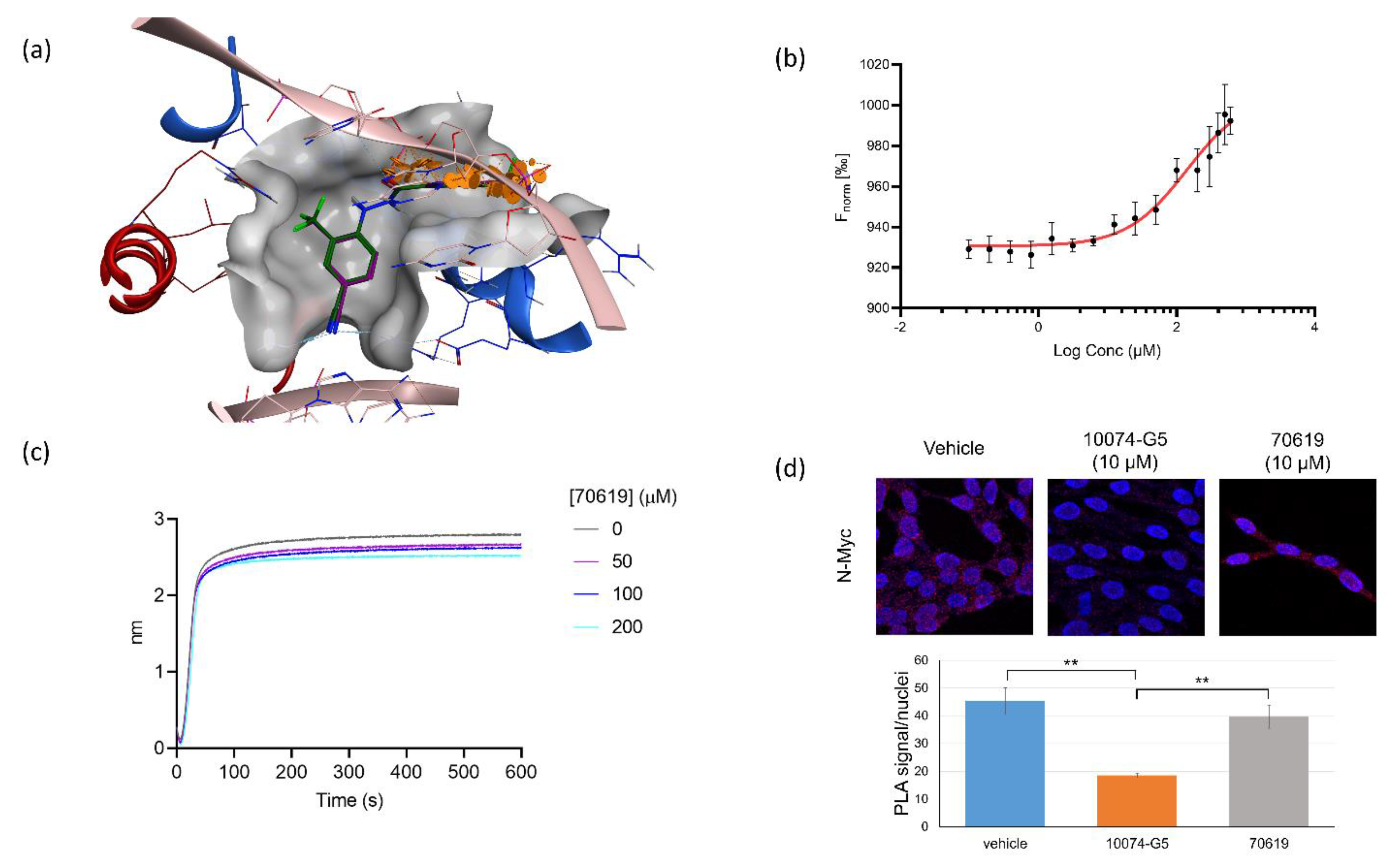
2.4. VPC-70619 Interaction with N-Myc-Max Complex Interferes with Its Binding to the DNA
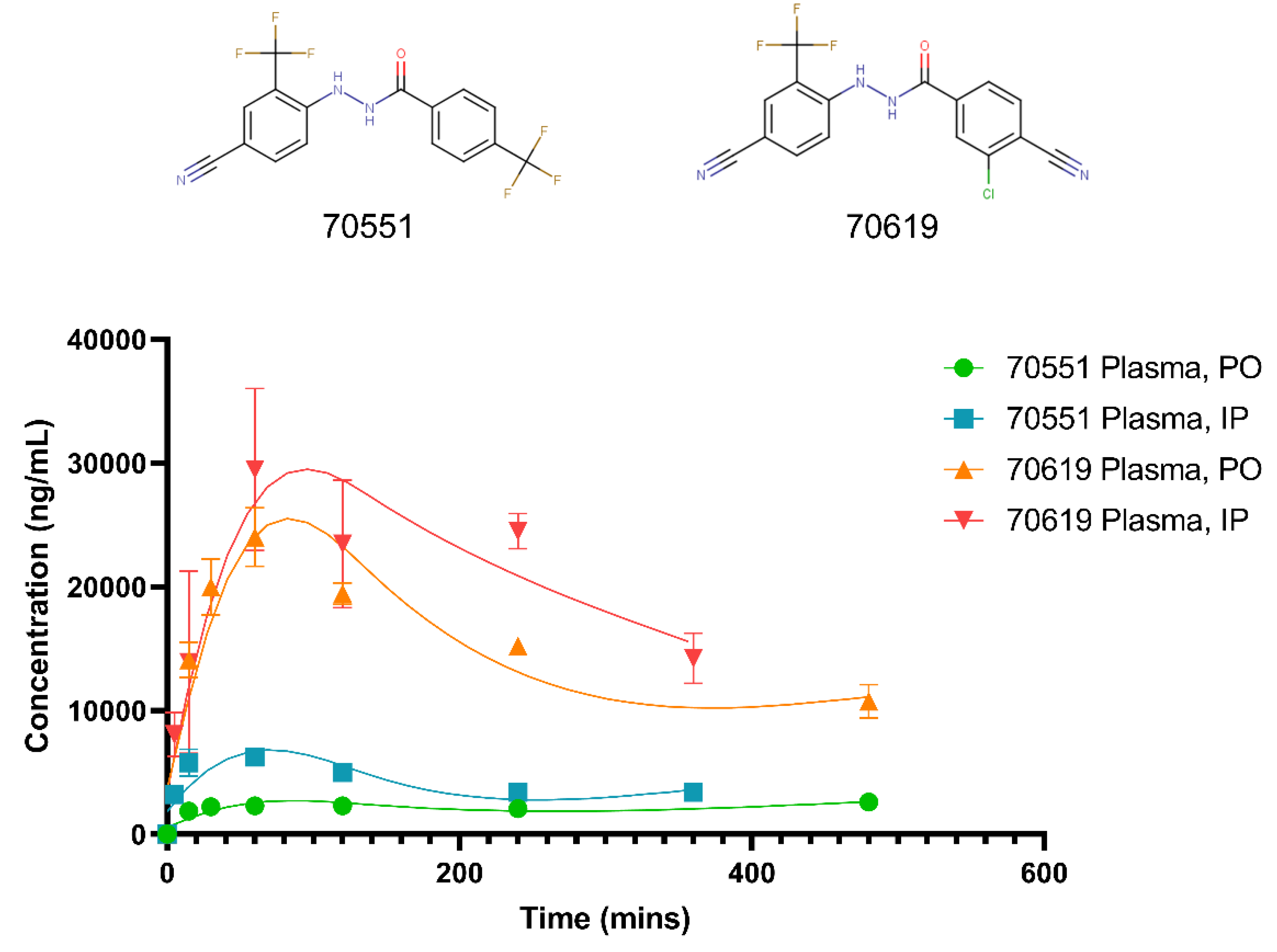
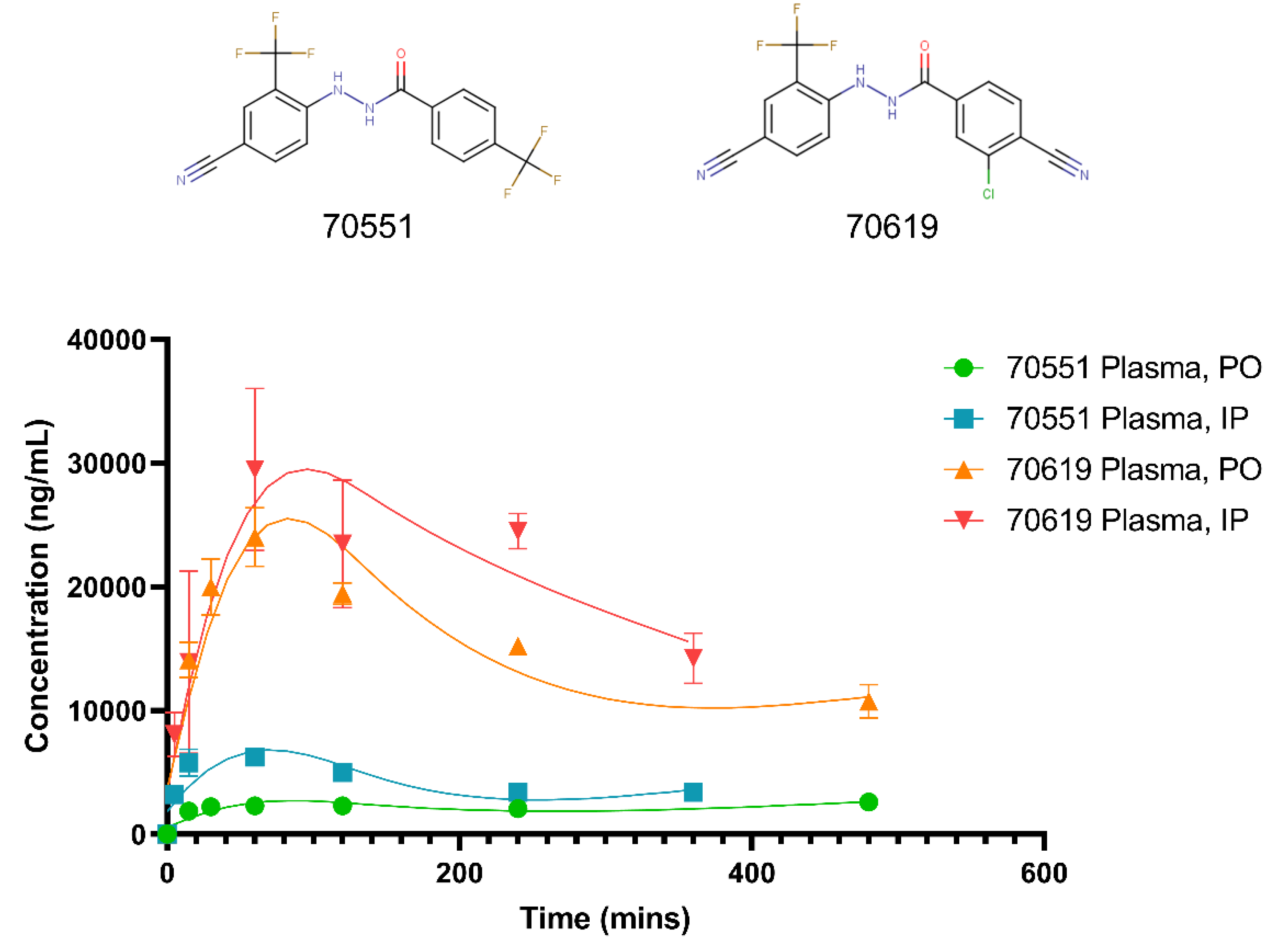
2.5. Pharmacokinetic Study of VPC-70619 Reveals Good Intraperitoneal and Peroral Tolerance
3. Discussion
4. Materials and Methods
4.1. Computational Methods
4.1.1. Chemical Similarity Searches
4.1.2. Protein and Ligand Preparation
4.1.3. Docking
4.2. In Vitro Evaluation of Hit Compounds
4.2.1. Cell Lines
4.2.2. Myc Transcription Assay
4.2.3. Cell Viability
4.2.4. Protein Purification
4.2.5. Biolayer Interferometry Assay
4.2.6. Proximity Ligation Assay (PLA)
4.2.7. Pharmacokinetics Studies
4.2.8. Microsomal Stability Assay
4.2.9. Thermophoresis (MST)
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yin, Y.; Xu, L.; Chang, Y.; Zeng, T.; Chen, X.; Wang, A.; Groth, J.; Foo, W.-C.; Liang, C.; Hu, H.; et al. N-Myc promotes therapeutic resistance development of neuroendocrine prostate cancer by differentially regulating miR-421/ATM pathway. Mol. Cancer 2019, 18, 11. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Hruszkewycz, A.; Scher, H.I.; Hildesheim, J.; Isaacs, J.; Yu, E.Y.; Kelly, K.; Lin, D.; Dicker, A.P.; Arnold, J.T.; et al. The role of lineage plasticity in prostate cancer therapy resistance. Clin. Cancer Res. 2019, 25, 6916–6924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zheng, D.; Zhou, T.; Song, H.; Hulsurkar, M.; Su, N.; Liu, Y.; Wang, Z.; Shao, L.; Ittmann, M.; et al. Androgen deprivation promotes neuroendocrine differentiation and angiogenesis through CREB-EZH2-TSP1 pathway in prostate cancers. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Conteduca, V.; Oromendia, C.; Eng, K.W.; Bareja, R.; Sigouros, M.; Molina, A.; Faltas, B.M.; Sboner, A.; Mosquera, J.M.; Elemento, O.; et al. Clinical features of neuroendocrine prostate cancer. Eur. J. Cancer 2019, 121, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, S.; Moore, C.M.; Chiong, E.; Beltran, H.; Bristow, R.G.; Williams, S.G. Prostate cancer. Lancet 2021, 398, 1075–1090. [Google Scholar] [CrossRef]
- Beltran, H.; Demichelis, F. Therapy considerations in neuroendocrine prostate cancer: What next? Endocrine-Related Cancer 2021, 28, T67–T78. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal. Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Cicenas, J.; Kalyan, K.; Sorokinas, A.; Stankunas, E.; Levy, J.; Meskinyte, I.; Stankevicius, V.; Kaupinis, A.; Valius, M. Roscovitine in cancer and other diseases. Ann. Transl. Med. 2015, 3, 135. [Google Scholar] [CrossRef]
- Brockmann, M.; Poon, E.; Berry, T.; Carstensen, A.; Deubzer, H.E.; Rycak, L.; Jamin, Y.; Thway, K.; Robinson, S.; Roels, F.; et al. Small Molecule Inhibitors of Aurora-A Induce Proteasomal Degradation of N-Myc in Childhood Neuroblastoma. Cancer Cell 2013, 24, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, W.C.; Meyerowitz, J.; Nekritz, E.A.; Chen, J.; Benes, C.; Charron, E.; Simonds, E.; Seeger, R.; Matthay, K.K.; Hertz, N.; et al. Drugging MYCN through an Allosteric Transition in Aurora Kinase, A. Cancer Cell 2014, 26, 414–427. [Google Scholar] [CrossRef] [Green Version]
- Bushweller, J.H. Targeting transcription factors in cancer—from undruggable to reality. Nat. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef]
- Hurlin, P.J. N-myc functions in transcription and development. Birth Defects Res. Part. C Embryo Today Rev. 2005, 75, 340–352. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC oncogene—the grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 2021, 19, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Carabet, L.A.; Rennie, P.S.; Cherkasov, A. Therapeutic Inhibition of Myc in Cancer. Structural Bases and Computer-Aided Drug Discovery Approaches. Int. J. Mol. Sci. 2018, 20, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radaeva, M.; Ton, A.-T.; Hsing, M.; Ban, F.; Cherkasov, A. Drugging the ‘undruggable’. Therapeutic targeting of protein–DNA interactions with the use of computer-aided drug discovery methods. Drug Discov. Today 2021, 26, 2660–2679. [Google Scholar] [CrossRef]
- Boike, L.; Cioffi, A.G.; Majewski, F.C.; Co, J.; Henning, N.J.; Jones, M.D.; Liu, G.; McKenna, J.M.; Tallarico, J.A.; Schirle, M.; et al. Discovery of a Functional Covalent Ligand Targeting an Intrinsically Disordered Cysteine within MYC. Cell Chem. Biol. 2020, 28, 4–13.e17. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Jain, A.D.; Truica, M.I.; Izquierdo-Ferrer, J.; Anker, J.; Lysy, B.; Sagar, V.; Luan, Y.; Chalmers, Z.R.; Unno, K.; et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell 2019, 36, 483–497.e15. [Google Scholar] [CrossRef]
- Singh, A.; Kumar, A.; Kumar, P.; Nayak, N.; Bhardwaj, T.; Giri, R.; Garg, N. A novel inhibitor L755507 efficiently blocks c-Myc–MAX heterodimerization and induces apoptosis in cancer cells. J. Biol. Chem. 2021, 297, 100903. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Cao, S.; Jin, L.; Kobelski, M.; Schouest, B.; Wang, X.; Ungerleider, N.; Baddoo, M.; Zhang, W.; Corey, E.; et al. A positive role of c-Myc in regulating androgen receptor and its splice variants in prostate cancer. Oncogene 2019, 38, 4977–4989. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; Macdonald, T.; et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef] [Green Version]
- Carabet, L.A.; Lallous, N.; Leblanc, E.; Ban, F.; Morin, H.; Lawn, S.; Ghaidi, F.; Lee, J.; Mills, I.G.; Gleave, M.E.; et al. Computer-aided drug discovery of Myc-Max inhibitors as potential therapeutics for prostate cancer. Eur. J. Med. Chem. 2018, 160, 108–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ton, A.-T.; Singh, K.; Morin, H.; Ban, F.; Leblanc, E.; Lee, J.; Lallous, N.; Cherkasov, A. Dual-Inhibitors of N-Myc and AURKA as Potential Therapy for Neuroendocrine Prostate Cancer. Int. J. Mol. Sci. 2020, 21, 8277. [Google Scholar] [CrossRef]
- Lee, J.; Phillips, J.W.; Smith, B.A.; Park, J.W.; Stoyanova, T.; McCaffrey, E.; Baertsch, R.; Sokolov, A.; Meyerowitz, J.; Mathis, C.; et al. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer Cell 2016, 29, 536–547. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Zhang, J.; Yin, J.; Gan, Y.; Xu, S.; Gu, Y.; Huang, W. Alternative approaches to target Myc for cancer treatment. Signal. Transduct. Target. Ther. 2021, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.K.; Ahn, K.-O.; Jung, N.-R.; Shin, J.-S.; Park, W.S.; Lee, K.H.; Lee, S.-J.; Jeong, K.-C. Antitumor activity of the c-Myc inhibitor KSI-3716 in gemcitabine-resistant bladder cancer. Oncotarget 2014, 5, 326–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavi, O. Redundancy: A Critical Obstacle to Improving Cancer Therapy. Cancer Res. 2015, 75, 808–812. [Google Scholar] [CrossRef] [Green Version]
- Nussinov, R.; Tsai, C.-J.; Jang, H. Are Parallel Proliferation Pathways Redundant? Trends Biochem. Sci. 2020, 45, 554–563. [Google Scholar] [CrossRef]
- Nero, T.L.; Morton, C.J.; Holien, J.K.; Wielens, J.; Parker, M.W. Oncogenic protein interfaces: Small molecules, big challenges. Nat. Cancer 2014, 14, 248–262. [Google Scholar] [CrossRef]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef] [PubMed]
- Fleming, F.F.; Yao, L.; Ravikumar, P.C.; Funk, L.; Shook, B.C. Nitrile-Containing Pharmaceuticals: Efficacious Roles of the Nitrile Pharmacophore. J. Med. Chem. 2010, 53, 7902–7917. [Google Scholar] [CrossRef] [Green Version]
- Falzone, L.; Salomone, S.; Libra, M. Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murciano-Goroff, Y.R.; Warner, A.B.; Wolchok, J.D. The future of cancer immunotherapy: Microenvironment-targeting combinations. Cell Res. 2020, 30, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Gavhane, Y.N.; Yadav, A.V. Loss of orally administered drugs in GI tract. Saudi Pharm. J. 2012, 20, 331–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.D.; Skillman, A.A.G.; Nicholls, A. Comparison of Shape-Matching and Docking as Virtual Screening Tools. J. Med. Chem. 2006, 50, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Structure | IC50 (µM) | LNCaP-NMYC % Inhibition (25 µM) |
|---|---|---|---|
| 70127 |  | 1 | 106 |
| 70215 | 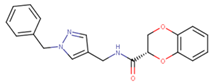 | 20 | 92 |
| 70223 |  | 10 | 103 |
| 70314 | 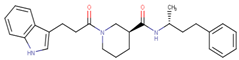 | 15 | 93 |
| 70381 | 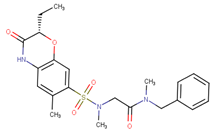 | 20 | 74 |
| 70388 | 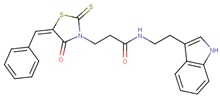 | 20 | 77 |
| 70390 | 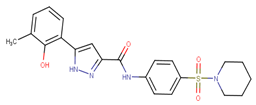 | 9 | 97 |
| 70465 | 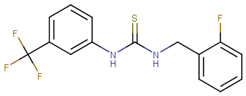 | 10 | 92 |
| 70495 | 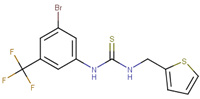 | 5 | 100 |
| 70511 | 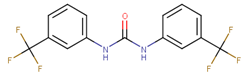 | 2 | 102 |
| 70551 |  | 4 | 98 |
 | |||||||
|---|---|---|---|---|---|---|---|
| ID | Ring A | Ring B | % Transcript Inhibition (10 µM) | % Transcript Inhibition (5 µM) | IC50 (µM) | ||
| R1 | R2 | R1 | R2 | ||||
| 70582 | C≡N | CF3 | C≡CH | H | 84 | 44 | 8 |
| 70583 | CF3 | H | CF3 | H | 28 | 8 | >20 |
| 70584 | CF3 | H | C≡N | H | 30 | 34 | >20 |
| 70585 | H | F | CF3 | H | 24 | 16 | >20 |
| 70586 | C≡N | H | CF3 | H | 34 | 28 | >20 |
| 70587 | C≡N | H | C≡N | H | 14 | −58 | |
| 70588 | C≡N | CF3 | O–CH–F2 | H | 46 | 32 | |
| 70589 | C≡N | CF3 | I | H | 94 | 64 | 3 |
| 70590 | C≡N | CF3 | CH=CH2 | H | 62 | 38 | 15 |
| 70591 | C≡N | CF3 | F | H | 64 | 18 | 25 |
| 70592 | C≡N | CF3 | CO–CH2 | H | 8 | 4 | |
| 70593 | C≡N | CF3 | C≡N | H | 70 | −26 | 12 |
| 70594 | C≡N | CF3 | C–CH3–CH3-CN | H | 10 | 22 | |
| 70595 | C≡N | CF3 | CH–CH3–CH3 | H | 70 | 30 | 12 |
| 70596 | C≡N | CF3 | H | CF3 | 94 | 96 | 2 |
| 70597 | C≡N | CF3 | CF3 | H | 82 | 26 | 12 |
| 70598 | C≡N | CF3 | CH–CF2 | H | 60 | −4 | 15 |
| 70599 | C≡N | CF3 | Cl | H | 78 | 96 | 3 |
| 70600 | C≡N | CF3 | CH3 | H | 10 | −22 | |
| 70601 | C≡N | CF3 | CO–NH2 | H | −12 | 16 | |
| 70602 | C≡N | CF3 | Br | H | 48 | 12 | |
| 70603 | C≡N | CF3 | H | H | −12 | −30 | >20 |
| 70604 | C≡N | CF3 | C–C3H6 | H | 78 | 24 | 8 |
| 70605 | C≡N | CF3 | N–C2H6 | H | 26 | −74 | |
| 70606 | H | CF3 | C–CH3–F2 | H | −10 | 0 | |
| 70607 | C≡N | H | C–CH3–F2 | H | −8 | −20 | |
| 70608 | C≡N | CF3 | C–CH3–F2 | H | 38 | −14 | |
| 70609 | H | CF3 | F | H | 12 | 20 | |
| 70610 | H | CF3 | C–C3H6 | H | 36 | 24 | |
| 70611 | H | CF3 | CF3 | H | 4 | −28 | >20 |
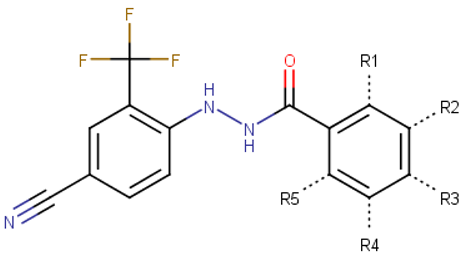 | |||||||
|---|---|---|---|---|---|---|---|
| ID | Substitutions | % Inhibition in IMR32 (10 µM) | % Inhibition in HO15.19 (10 µM) | ||||
| R1 | R2 | R3 | R4 | R5 | |||
| 70619 | H | Cl | C≡N | H | H | 99.4 | 14.1 |
| 70621 | H | F | Cl | H | H | 98.8 | 68.6 |
| 70622 | H | Br | Cl | H | H | 99.4 | 36.4 |
| 70633 | OH | H | Cl | H | H | 93.6 | 77.0 |
| 70635 | H | Cl | Br | H | H | 7.5 | 11.8 |
| 70639 | H | F | H | F | H | 60.2 | 66.1 |
| 70640 | OH | H | H | Cl | H | 93.5 | 76.7 |
| 70644 | H | Cl | H | F | H | 88.3 | 69.5 |
| 70647 | H | CF3 | F | H | H | 88.6 | 70.9 |
| 70650 | H | Br | H | Cl | H | 92.1 | 80.2 |
| 70652 | H | Cl | H | Cl | H | 92.5 | 80.7 |
| 70654 | H | H | CO–O–C–C–C3H6 | H | H | 17.0 | 13.9 |
| 70658 | H | H | S-CF3 | H | H | 46.9 | 57.2 |
| 70659 | Cl | H | F | F | H | 31.0 | 31.5 |
| 70662 | H | C≡N | Cl | H | H | 11.0 | 9.9 |
| 70663 | H | H | NO2 | H | H | 28.9 | 7.9 |
| 70672 | H | C≡N | H | H | H | 18.0 | 8.1 |
| 70675 | H | Cl | H | CH3 | H | 65.4 | 65.9 |
| 70679 | H | CH3 | F | CH3 | H | 1.1 | 7.3 |
| 70680 | H | Cl | F | H | H | 52.9 | 64.0 |
| 70682 | H | H | S–CF2 | H | H | 11.0 | 22.9 |
| 70685 | H | O–CH3 | Cl | H | H | 24.7 | 15.2 |
| 70686 | H | F | F | F | H | 82.2 | 75.8 |
| 70693 | H | F | CF3 | H | H | 22.4 | 0.2 |
| 70695 | H | Cl | NH2 | Cl | H | 77.1 | 41.2 |
| 70696 | H | C≡N | F | H | H | 15.1 | 13.2 |
| 70698 | Cl | H | H | C≡N | H | 35.4 | 27.3 |
| 70700 | H | O–CH3 | H | Cl | H | 60.5 | 42.0 |
| 70703 | H | I | H | H | H | 18.5 | 13.5 |
| 70704 | Cl | H | CF3 | H | H | 10.6 | 13.6 |
| 70705 | H | Cl | CF3 | H | H | 60.7 | 61.8 |
| 70706 | CF3 | H | Cl | H | H | 54.3 | 65.6 |
| 70715 | F | F | F | H | H | 14.4 | 39.9 |
| 70719 | OH | H | Cl | H | Cl | 95.0 | 76.5 |
| 70722 | H | CF3 | H | Cl | H | 94.7 | 79.3 |
| 70726 | H | I | F | H | H | 75.6 | 66.2 |
| 70730 | Cl | H | Cl | F | H | 87.5 | 77.1 |
| 70732 | H | H | O–phenyl–CO–CH3 | H | H | 98.9 | 98.9 |
| 70733 | H | H | C–C2H6–CF3 | H | H | 57.4 | 57.8 |
| 70734 | H | Br | F | H | H | 86.5 | 77.8 |
| 70739 | H | Br | H | F | H | 87.2 | 75.8 |
| 70741 | F | H | Cl | F | H | 16.4 | 14.5 |
| 70742 | H | F | I | H | H | 26.1 | 71.8 |
| 70743 | H | F | Br | H | H | 38.7 | 74.9 |
| 70756 | H | CF3 | H | CF3 | H | 95.7 | 84.1 |
| 70761 | H | NO2 | H | Cl | H | 93.3 | 81.0 |
| 70767 | H | F | Cl | F | H | 31.6 | 40.3 |
| 70768 | F | Br | H | Cl | H | 35.3 | 39.8 |
| 70769 | H | CF3 | H | H | F | 32.2 | 28.2 |
| 70775 | H | F | Br | Cl | H | 62.4 | 65.1 |
| 70776 | Br | F | H | Cl | H | 27.4 | 26.1 |
| 70792 | H | O–CF2 | H | O–CF2 | H | 84.9 | 79.5 |
| 70794 | H | CF3 | H | F | H | 89.2 | 80.7 |
| 70797 | OH | H | CF3 | H | H | 24.7 | 34.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ton, A.-T.; Foo, J.; Singh, K.; Lee, J.; Kalyta, A.; Morin, H.; Perez, C.; Ban, F.; Leblanc, E.; Lallous, N.; et al. Development of VPC-70619, a Small-Molecule N-Myc Inhibitor as a Potential Therapy for Neuroendocrine Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 2588. https://doi.org/10.3390/ijms23052588
Ton A-T, Foo J, Singh K, Lee J, Kalyta A, Morin H, Perez C, Ban F, Leblanc E, Lallous N, et al. Development of VPC-70619, a Small-Molecule N-Myc Inhibitor as a Potential Therapy for Neuroendocrine Prostate Cancer. International Journal of Molecular Sciences. 2022; 23(5):2588. https://doi.org/10.3390/ijms23052588
Chicago/Turabian StyleTon, Anh-Tien, Jane Foo, Kriti Singh, Joseph Lee, Anastasia Kalyta, Helene Morin, Carl Perez, Fuqiang Ban, Eric Leblanc, Nada Lallous, and et al. 2022. "Development of VPC-70619, a Small-Molecule N-Myc Inhibitor as a Potential Therapy for Neuroendocrine Prostate Cancer" International Journal of Molecular Sciences 23, no. 5: 2588. https://doi.org/10.3390/ijms23052588
APA StyleTon, A.-T., Foo, J., Singh, K., Lee, J., Kalyta, A., Morin, H., Perez, C., Ban, F., Leblanc, E., Lallous, N., & Cherkasov, A. (2022). Development of VPC-70619, a Small-Molecule N-Myc Inhibitor as a Potential Therapy for Neuroendocrine Prostate Cancer. International Journal of Molecular Sciences, 23(5), 2588. https://doi.org/10.3390/ijms23052588