
A Wide-Proteome Analysis to Identify Molecular Pathways Involved in Kidney Response to High-Fat Diet in Mice

 , ,
, ,  , ,
, ,  ,
, 
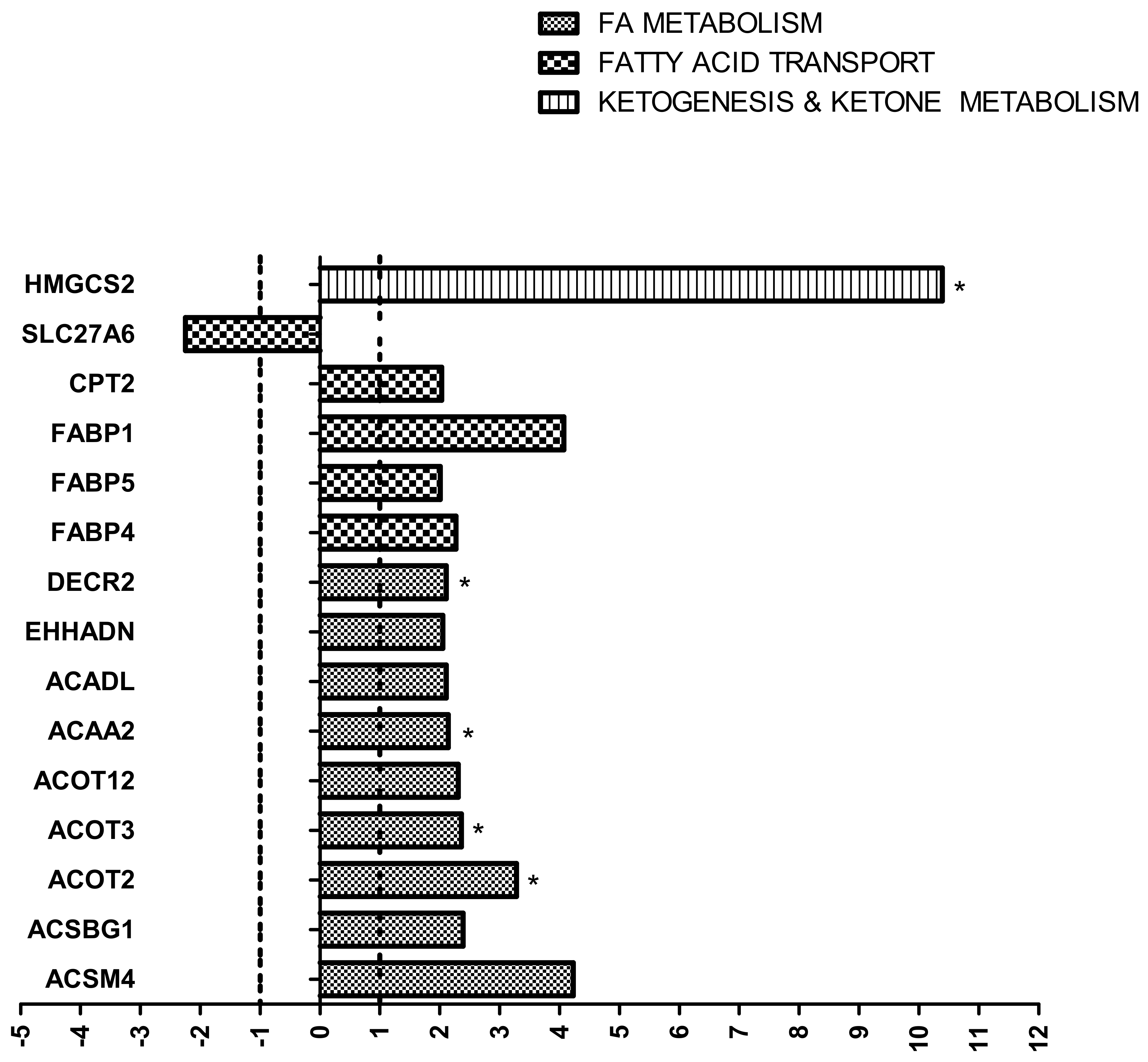{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Impact of HF Diet on Lipid Metabolism-Related Transcriptome in Kidney
2.2. Impact of HF Diet on Fibrosis-Related Transcriptome in Kidney
2.3. Impact of HF Diet on Lipid Content in Kidney
2.4. Impact of HF Diet on Kidney Proteome
2.4.1. Impact of HF Diet on Whole Kidney Proteome
2.4.2. Evaluation of Lipid Metabolism-Related Proteome in Kidney
2.4.3. Evaluation of Fibrosis-Related Proteome in Kidney
2.5. Evaluation of Kidney Morphology, Fibrosis, Amyloidosis, and Other Kidney Lesions
2.5.1. Hematoxylin–Eosin Staining
2.5.2. Masson’s Trichrome Staining
2.5.3. Congo Red
2.5.4. PAS
3. Discussion
3.1. Lipid Metabolism
3.2. Fibrosis
4. Materials and Methods
4.1. Animal Model
4.2. Total RNA Extraction and Reverse Transcription
4.3. RT2 Profiler PCR Arrays
4.4. Analysis of RT2 Profiler PCR Arrays
4.5. Lipid Extraction and Quantification
4.6. Shotgun Mass Spectrometry Analysis for Label-Free Proteomics
4.7. Tissue Preparation, Histological Examination, and Scoring
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gai, Z.; Wang, T.; Visentin, M.; Kullak-Ublick, G.A.; Fu, X.; Wang, Z. Lipid Accumulation and Chronic Kidney Disease. Nutrients 2019, 11, 722. [Google Scholar] [CrossRef] [PubMed]
- Susztak, K.; Ciccone, E.; McCue, P.; Sharma, K.; Bottinger, E.P. Multiple metabolic hits convergeon CD36 as novel mediator of tubular epithelial apoptosis in diabetic nephropathy. PLoS Med. 2005, 2, e45. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.G., Jr.; Burant, C.F. PPAR-gammaaction: It’s all in your head. Nat. Med. 2011, 17, 544–545. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Chen, Y.; Huang, W.; Viterna, J.; Liu, J.; Westfall, K.; Tian, J.; Bartlett, D.J.; Tang, W.W.; Xie, Z.; et al. CD36 and Na/K-ATPase-α1 Form a Proinflammatory Signaling Loop in Kidney. Hypertension 2013, 61, 216–224. [Google Scholar] [CrossRef]
- Herman-Edelstein, M.; Scherzer, P.; Tobar, A.; Levi, M.; Gafter, U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid Res. 2014, 55, 561–572. [Google Scholar] [CrossRef]
- Xu, Y.; Huang, J.; Xin, W.; Chen, L.; Zhao, X.; Lv, Z.; Liu, Y.; Wan, Q. Lipid accumulation is ahead of epithelial-to-mesenchymal transition and therapeutic intervention by acetyl-CoA carboxylase 2 silence in diabetic nephropathy. Metabolism 2014, 63, 716–726. [Google Scholar] [CrossRef]
- Chen, W.; Jiang, Y.; Han, J.; Hu, J.; He, T.; Yan, T.; Huang, N.; Zhang, Q.; Mei, H.; Liao, Y.; et al. Atgl deficiency induces podocyte apoptosis and leads to glomerular filtration barrier damage. FEBS J. 2017, 284, 1070–1081. [Google Scholar] [CrossRef]
- Feng, L.; Gu, C.; Li, Y.; Huang, J. High glucose promotes CD36 expression by upregulating peroxisome proliferator-activated receptor γ levels to exacerbate lipid deposition in renal tubular cells. Biomed. Res. Int. 2017, 2017, 1414070. [Google Scholar] [CrossRef]
- Jang, H.S.; Noh, M.R.; Kim, J.; Padanilam, B.J. Defective mitochondrial fatty acid oxidation and lipotoxicity in kidney diseases. Front. Med. 2020, 7, 65. [Google Scholar] [CrossRef]
- Wahl, P.; Ducasa, G.M.; Fornoni, A. Systemic and renal lipids in kidney disease development and progression. Am. J. Physiol. Renal. Physiol. 2016, 310, F433–F445. [Google Scholar] [CrossRef]
- Arici, M.; Brown, J.; Williams, M.; Harris, K.P.G.; Walls, J.; Brunskill, N.J. Fatty acids carried on albumin modulate proximal tubular cell fibronectin production: A role for protein kinase C. Nephrol. Dial. Transplant. 2002, 17, 1751–1757. [Google Scholar] [CrossRef] [PubMed]
- De Vries, A.P.J.; Ruggenenti, P.; Ruan, X.Z.; Praga, M.; Cruzado, J.M.; Bajema, I.M.; D’Agati, V.D.; Lamb, H.J.; Barlovic, D.P.; Hojs, R.; et al. Fatty kidney: Emerging role of ectopic lipid in obesity-related renal disease. Lancet Diabetes Endocrinol. 2014, 2, 417–426. [Google Scholar] [CrossRef]
- Adeosun, S.O.; Gordon, D.M.; Weeks, M.F.; Moore, K.H.; Hall, J.E.; Hinds, T.D., Jr.; Stec, D.E. Loss of biliverdin reductase-A promotes lipid accumulation and lipotoxicity in mouse proximal tubule cells. Am. J. Physiol. Renal. Physiol. 2018, 315, F323–F331. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Yang, P.; Xiao, Y.; Luo, X.; Zhao, Y.; Zhao, L.; Wang, Y.; Wu, T.; Wei, L.; Chen, Y. Inflammatory stress promotes the development of obesity-related chronic kidney disease via CD36 in mice. J. Lipid Res. 2017, 58, 1417–1427. [Google Scholar] [CrossRef]
- Chagnac, A.; Weinstein, T.; Korzets, A.; Ramadan, E.; Hirsch, J.; Gafter, U. Glomerular hemodynamics in severe obesity. Am. J. Physiol. Renal. Physiol. 2000, 278, F817–F822. [Google Scholar] [CrossRef]
- Chagnac, A.; Herman, M.; Zingerman, B.; Erman, A.; Rozen-Zvi, B.; Hirsh, J.; Gafter, U. Obesity-induced glomerular hyperfiltration: Its involvement in the pathogenesis of tubular sodium reabsorption. Nephrol. Dial. Transplant. 2008, 23, 3946–3952. [Google Scholar] [CrossRef]
- Kriz, W.; Lemley, K.V. A Potential Role for Mechanical Forces in the Detachment of Podocytes and the Progression of CKD. J. Am. Soc. Nephrol. 2014, 26, 258–269. [Google Scholar] [CrossRef]
- Garofalo, C.; Borrelli, S.; Minutolo, R.; Chiodini, P.; DeNicola, L.; Conte, G. A systematic review and meta-analysis suggests obesity predicts onset of chronic kidney disease in the general population. Kidney Int. 2017, 91, 1224–1235. [Google Scholar] [CrossRef]
- Lodhi, I.J.; Semenkovich, C.F. Peroxisomes: A nexus for lipid metabolism and cellular signaling. Cell Metab. 2014, 19, 380–392. [Google Scholar] [CrossRef]
- Wanders, R.J.; Waterham, H.R.; Ferdinandusse, S. Metabolic interplay between peroxisomes and other subcellular organelles including mitochondria and the endoplasmic reticulum. Front. Cell Dev. Biol. 2016, 3, 83. [Google Scholar] [CrossRef] [PubMed]
- Reiser, G.; Schonfeld, P.; Kahlert, S. Mechanism of toxicity of the branched-chain fatty acid phytanic acid, a marker of Refsum disease, in astrocytes involves mitochondrial impairment. Int. J. Dev. Neurosci. 2006, 24, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Baarine, M.; Andreoletti, P.; Athias, A.; Nury, T.; Zarrouk, A.; Ragot, K.; Vejux, A.; Riedinger, J.M.; Kattan, Z.; Bessede, G.; et al. Evidence of oxidative stress in very long chain fatty acid–treated oligodendrocytes and potentialization of ROS production using RNA interference-directed knockdown of ABCD1 and ACOX1 peroxisomal proteins. Neuroscience 2012, 213, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Lismont, C.; Nordgren, M.; VanVeldhoven, P.P.; Fransen, M. Redox interplay between mitochondria and peroxisomes. Front. Cell Dev. Biol. 2015, 3, 35. [Google Scholar] [CrossRef]
- Waterham, H.R.; Ferdinandusse, S.; Wanders, R.J. Human disorders of peroxisome metabolism and biogenesis. Biochim. Biophys. Acta 2016, 1863, 922–933. [Google Scholar] [CrossRef]
- Decleves, A.E.; Zolkipli, Z.; Satriano, J.; Wang, L.; Nakayama, T.; Rogac, M.; Le, T.P.; Nortier, J.L.; Farquhar, M.G.; Naviaux, R.K.; et al. Regulation of lipid accumulation by AMK-activated kinase [corrected] in high fat diet–induced kidney injury. Kidney Int. 2014, 85, 611–623. [Google Scholar] [CrossRef]
- Deji, N.; Kume, S.; Araki, S.; Soumura, M.; Sugimoto, T.; Isshiki, K.; Chin-Kanasaki, M.; Sakaguchi, M.; Koya, D.; Haneda, M.; et al. Structural and functional changes in the kidneys of high-fat diet-induced obese mice. Am. J. Physiol. Renal. Physiol. 2009, 296, F118–F126. [Google Scholar] [CrossRef]
- Sun, Y.; Ge, X.; Li, X.; He, J.; Wei, X.; Du, J.; Sun, J.; Li, X.; Xun, Z.; Liu, W.; et al. High-fat diet promotes renal injury by inducing oxidative stress and mitochondrial dysfunction. Cell Death Dis. 2020, 11, 914. [Google Scholar] [CrossRef]
- Nishikawa, S.; Yasoshima, A.; Doi, K.; Nakayama, H.; Uetsuka, K. Involvement of sex, strain and age factors in high fat diet-induced obesity in C57BL/6J and BALB/cA mice. Exp. Anim. 2007, 56, 263–272. [Google Scholar] [CrossRef]
- Rosca, M.G.; Vazquez, E.J.; Chen, Q.; Kerner, J.; Kern, T.S.; Hoppel, C.L. Oxidation of fatty acids is the source of increased mitochondrial reactive oxygen species production in kidney cortical tubules in early diabetes. Diabetes 2012, 61, 2074–2083. [Google Scholar] [CrossRef]
- Fransen, M.; Lismont, C.; Walton, P. The peroxisome-mitochondria connection: How and why? Int. J. Mol. Sci. 2017, 18, 1126. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Miao, N.; Wang, B.; Xu, J.; Gan, X.; Xu, D.; Zhou, L.; Xue, H.; Zhang, W.; Yang, L.; et al. c-Myc promotes renal fibrosis by inducing integrin αv-mediated transforming growth factor-β signaling. Kidney Int. 2017, 92, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhou, Y.; Ko, K.S.; Yang, H. Interactions between Myc and mediators of inflammation in chronic liver diseases. Mediat. Inflamm. 2015, 2015, 276850. [Google Scholar] [CrossRef]
- Qin, H.; Tang, Y.; Mao, Y.; Zhou, X.; Xu, T.; Liu, W.; Su, X. C-MYC induces idiopathic pulmonary fibrosis via modulation of miR-9-5p-mediated TBPL1. Cell. Signal. 2022, 93, 110274. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-SanPedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef] [PubMed]
- Dozio, E.; Vianello, E.; Bandera, F.; Longhi, E.; Brizzola, S.; Nebuloni, M.; Romanelli, M.M.C. Soluble Receptor for Advanced Glycation End Products: A Protective Molecule against Intramyocardial Lipid Accumulation in Obese Zucker Rats? Mediat. Inflamm. 2019, 2019, 2712376. [Google Scholar] [CrossRef] [PubMed]
- Mozzi, A.; Forcella, M.; Riva, A.; Difrancesco, C.; Molinari, F.; Martin, V.; Papini, N.; Bernasconi, B.; Nonnis, S.; Tedeschi, G.; et al. NEU3 activity enhances EGFR activation without affecting EGFR expression and acts on its sialylation levels. Glycobiology 2015, 25, 855–868. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Eberini, I.; Calabresi, L.; Wait, R.; Tedeschi, G.; Pirillo, A.; Puglisi, L.; Sirtori, C.R.; Gianazza, E. Macrophage metalloproteinases degrade high-density-lipoprotein-associated apolipoprotein AI at both the N-and C-termini. Biochem. J. 2002, 362, 627–634. [Google Scholar] [CrossRef]
- Galli, A.; Maffioli, E.; Sogne, E.; Moretti, S.; DiCairano, E.S.; Negri, A.; Nonnis, S.; Norata, G.D.; Bonacina, F.; Borghi, F.; et al. Cluster-assembled zirconia substrates promote long-term differentiation and functioning of human islets of Langerhans. Sci. Rep. 2018, 8, 9979. [Google Scholar] [CrossRef]
- Schulte, C.; Podesta, A.; Lenardi, C.; Tedeschi, G.; Milani, P. Quantitative control of protein and cell interaction with nanostructured surfaces by cluster assembling. Acc. Chem. Res. 2017, 50, 231–239. [Google Scholar] [CrossRef]
- Schulte, C.; Rodighiero, S.; Cappelluti, M.A.; Puricelli, L.; Maffioli, E.; Borghi, F.; Negri, A.; Sogne, E.; Galluzzi, M.; Piazzoni, C.; et al. Conversion of nanoscale topographical information of cluster-assembled zirconia surfaces into mechanotransductive events promotes neuronal differentiation. J. Nano Biotechnol. 2016, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.P.; Mushayamaha, T.; Thomas, P.D. PANTHER version 16: A revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 2021, 49, D394–D403. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Vizcaino, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef] [PubMed]
- Glastras, S.J.; Chen, H.; Teh, R.; McGrath, R.T.; Chen, J.; Pollock, C.A.; Wong, M.G.; Saad, S. Mouse models of diabetes, obesity and related kidney disease. PLoS ONE 2016, 11, e0162131. [Google Scholar] [CrossRef]
- Meuten, D.J.; Moore, F.M.; George, J.W. Mitotic count and the field of view area: Time to standardize. Vet. Pathol. 2016, 53, 7–9. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dozio, E.; Maffioli, E.; Vianello, E.; Nonnis, S.; Grassi Scalvini, F.; Spatola, L.; Roccabianca, P.; Tedeschi, G.; Corsi Romanelli, M.M. A Wide-Proteome Analysis to Identify Molecular Pathways Involved in Kidney Response to High-Fat Diet in Mice. Int. J. Mol. Sci. 2022, 23, 3809. https://doi.org/10.3390/ijms23073809
Dozio E, Maffioli E, Vianello E, Nonnis S, Grassi Scalvini F, Spatola L, Roccabianca P, Tedeschi G, Corsi Romanelli MM. A Wide-Proteome Analysis to Identify Molecular Pathways Involved in Kidney Response to High-Fat Diet in Mice. International Journal of Molecular Sciences. 2022; 23(7):3809. https://doi.org/10.3390/ijms23073809
Chicago/Turabian StyleDozio, Elena, Elisa Maffioli, Elena Vianello, Simona Nonnis, Francesca Grassi Scalvini, Leonardo Spatola, Paola Roccabianca, Gabriella Tedeschi, and Massimiliano Marco Corsi Romanelli. 2022. "A Wide-Proteome Analysis to Identify Molecular Pathways Involved in Kidney Response to High-Fat Diet in Mice" International Journal of Molecular Sciences 23, no. 7: 3809. https://doi.org/10.3390/ijms23073809
APA StyleDozio, E., Maffioli, E., Vianello, E., Nonnis, S., Grassi Scalvini, F., Spatola, L., Roccabianca, P., Tedeschi, G., & Corsi Romanelli, M. M. (2022). A Wide-Proteome Analysis to Identify Molecular Pathways Involved in Kidney Response to High-Fat Diet in Mice. International Journal of Molecular Sciences, 23(7), 3809. https://doi.org/10.3390/ijms23073809