
VHH Structural Modelling Approaches: A Critical Review

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
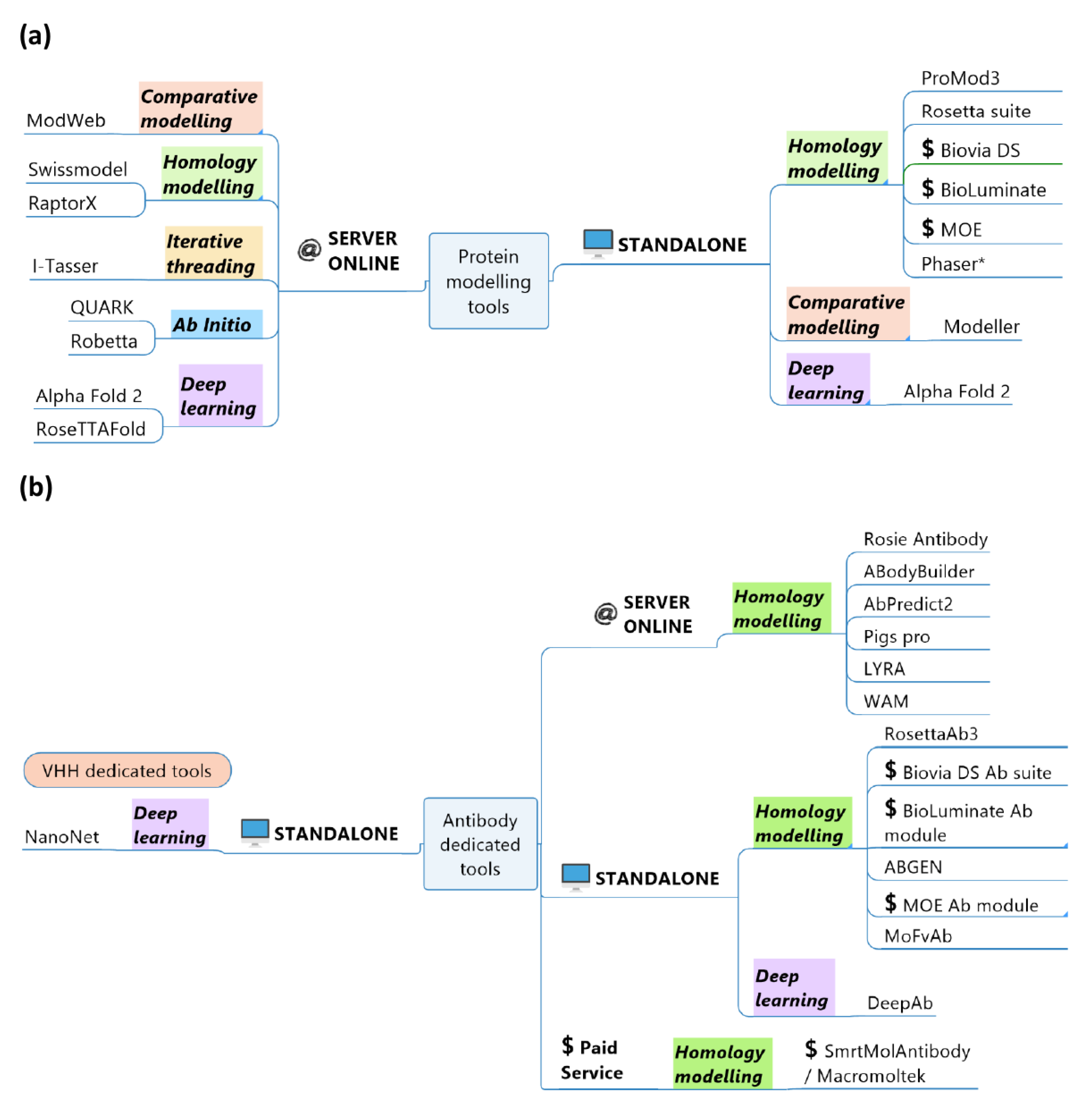
2. VHH Modelling
2.1. General Principle, a Short History
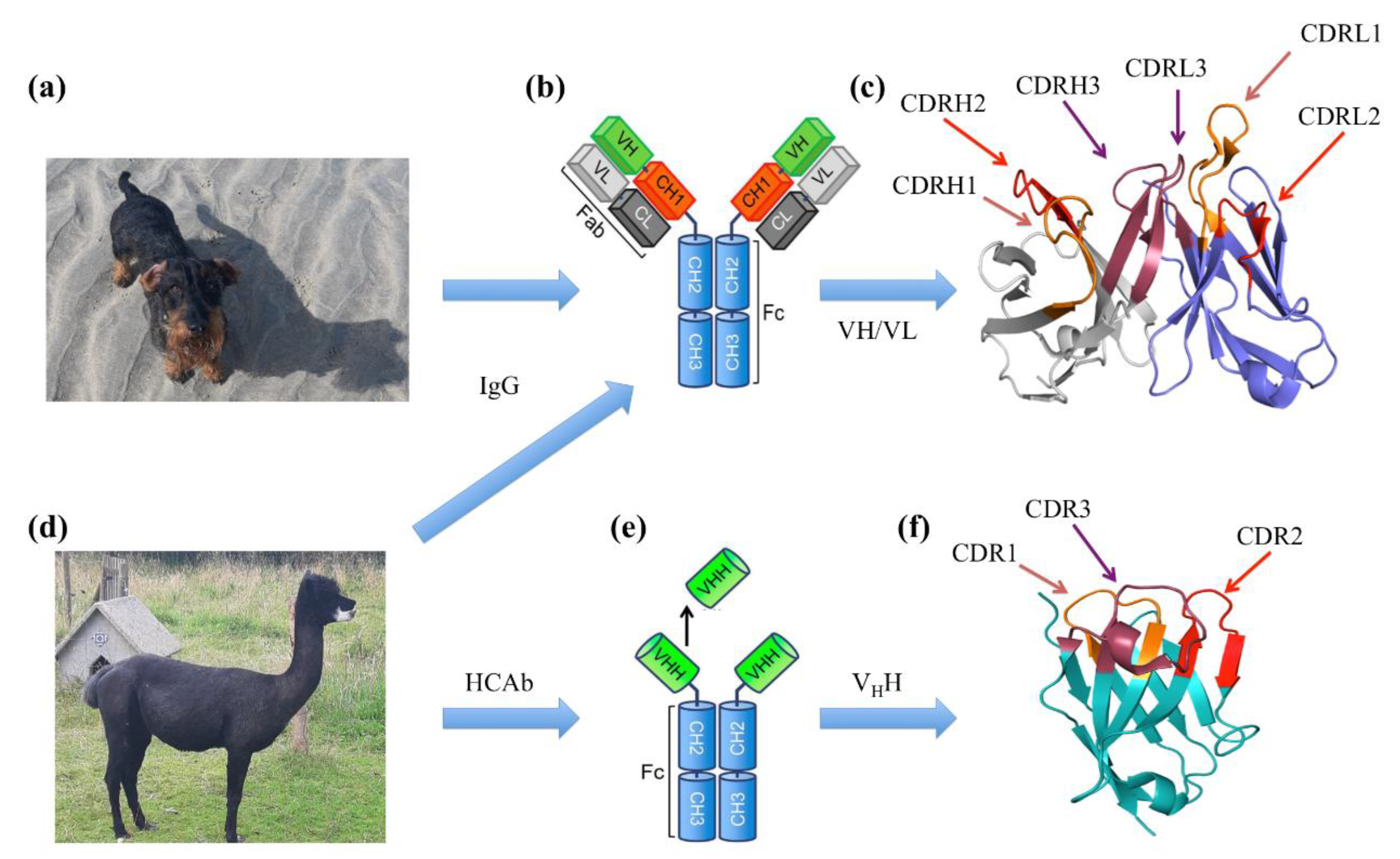
2.2. Abs and VHHs Specificities
2.3. Modeller and ModWeb
2.4. ABGEN
2.5. Web Antibody Modelling
- A similarity search is performed to build the framework (backbone and side chains) and canonical loop backbones with closer structures (in terms of sequence similarity).
- Using CONGEN [131], the canonical loop side chains are constructed using an iterative placement technique searching for the global minimum energy conformation.
- Depending on the loop length, alternative conformations are produced by using directly the PDB or CONGEN again.
- A specific solvent-modified version of the Valence force field is tested to assess the different conformations.
- Finally, the conformation is selected from the five lowest energy conformations. The final model is the conformation with the set of torsion angles closest to the canonical one, as defined in the chosen clustering approach.
2.6. SWISS-MODEL
2.7. MoFvAb
2.8. Prediction of Immunoglobulin Structure
- Frameworks are prepared: Structural templates for the frameworks are selected using sequence identity with protein structures from PDB.
- Five of the six CDRs are built: CDRs L1–L3 and H1 and H2 are modelled by getting conformations from antibodies with the same canonical structures discarding the identity sequence.
- To complete, a CDRH3 loop is proposed using the structural template with the highest sequence identity with the query sequence.
- All is merged: The complex VH–VL is modelled.
- At last, SCWRL is used to optimise the side chain conformations of the predicted model.
2.9. AbodyBuilder
2.10. LYmphocyte Receptor Automated Modelling
2.11. Phyre2
- A specific step is done to optimise the loop modelling where the indels (insertions and deletions) in these models are found.
- The side chains are grafted to build the final Phyre2 model.
2.12. RaptorX
2.13. Rosetta, Robetta, Rosetta Antibody and VHH Modelling Application
- In the initial step, the CDRs are identified using the Kabat CDR definition, and the residues are renumbered using the Chothia scheme. The template selection is then carried out for all the frameworks, and five of the six CDRs (CDRH1 and CDRH2 and CDRL1–CDR3),
- From the selected templates, a preliminary model is created using homology modelling, as it was shown to be more accurate than a completely de novo approach.
- CDRH3 de novo loop modelling completes the model prediction, along with the optimisation of the VH–VL interface.
2.14. AbPredict2
2.15. Biovia Discovery Studio/Antibody Modelling
2.16. MOE/Antibody Pipeline
2.17. Schrödinger BioLuminate and Antibody Pipeline
- Frameworks are detected in a curated antibody database, providing structural templates (selected using a sequence identity).
- Next, a set of CDRs are grafted after scanning another custom database containing only CDRs and a selection based on structural clustering, sequence similarity and stem residue geometry matching.
- In the final step, the in-house Prime software repacks the side chains and minimises the antibody model.
- A CDR3 loop is built using the ab initio method.
2.18. Macromoltek’s SmrtMolAntibody
2.19. I-TASSER and C-I-TASSER
2.20. QUARK and C-QUARK
2.21. AlphaFold 2
2.22. RoseTTAFold and DeepAb
2.23. NanoNet
3. Discussion
- (i)
- (ii)
- More recently, as Modeller [84] remains the most used comparative modelling approach, we evaluated the Modeller quality in VHH modelling [74]. Using 100 different VHHs, different template selection strategies for comparative modelling using Modeller have been extensively assessed. This study analysed the conformational changes in both the FRs and CDRs using an original strategy of conformational discretisation based on a structural alphabet [255,256]. It showed that, often, multi-template is the best method to obtain a correct VHH model and that the DOPE score is a relevant measure to select this model [107]. Nonetheless, it remains difficult to propose satisfactory models for some VHHs. Even sometimes, to use the closest VHH in terms of the RMSD is not always the best choice to obtain a good model, underlying the possibility for future improvement.
- (iii)
- Finally, NanoNet has demonstrated its superiority over RosettaAntibody and AlphaFold 2 but with a limited number of examples (see the previous subsection) [253].
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- DeLano, W.L. Pymol, Version 2.5; Delano Scientific: San Carlos, CA, USA, 2002.
- Delano, W.L. The Pymol Molecular Graphics System on World Wide Web. Available online: http://www.pymol.org (accessed on 20 February 2022).
- Schrodinger, L.L.C. The Pymol Molecular Graphics System, Version 1.7.2.2; Delano Scientific: San Carlos, CA, USA, 2015.
- Berger, M.; Shankar, V.; Vafai, A. Therapeutic applications of monoclonal antibodies. Am. J. Med. Sci. 2002, 324, 14–30. [Google Scholar] [CrossRef]
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. mAbs 2015, 7, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Huang, X.; Zhu, Y.; Lv, Z. A brief review of monoclonal antibody technology and its representative applications in immunoassays. J. Immunoass. Immunochem. 2018, 39, 351–364. [Google Scholar] [CrossRef]
- Ma, J.; Mo, Y.; Tang, M.; Shen, J.; Qi, Y.; Zhao, W.; Huang, Y.; Xu, Y.; Qian, C. Bispecific antibodies: From research to clinical application. Front. Immunol. 2021, 12, 626616. [Google Scholar] [CrossRef] [PubMed]
- Linke, R.; Klein, A.; Seimetz, D. Catumaxomab: Clinical development and future directions. mAbs 2010, 2, 129–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dees, S.; Ganesan, R.; Singh, S.; Grewal, I.S. Bispecific antibodies for triple negative breast cancer. Trends Cancer 2021, 7, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Lenting, P.J.; Denis, C.V.; Christophe, O.D. Emicizumab, a bispecific antibody recognizing coagulation factors ix and x: How does it actually compare to factor viii? Blood 2017, 130, 2463–2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samaranayake, H.; Wirth, T.; Schenkwein, D.; Räty, J.K.; Ylä-Herttuala, S. Challenges in monoclonal antibody-based therapies. Ann. Med. 2009, 41, 322–331. [Google Scholar] [CrossRef]
- Sapra, P.; Shor, B. Monoclonal antibody-based therapies in cancer: Advances and challenges. Pharmacol. Ther. 2013, 138, 452–469. [Google Scholar] [CrossRef]
- Sifniotis, V.; Cruz, E.; Eroglu, B.; Kayser, V. Current advancements in addressing key challenges of therapeutic antibody design, manufacture, and formulation. Antibodies 2019, 8, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keyt, B.A.; Baliga, R.; Sinclair, A.M.; Carroll, S.F.; Peterson, M.S. Structure, function, and therapeutic use of igm antibodies. Antibodies 2020, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.C.; Wilson, I.A. Structural insights into the design of novel anti-influenza therapies. Nat. Struct. Mol. Biol. 2018, 25, 115–121. [Google Scholar] [CrossRef]
- Zhao, J.; Nussinov, R.; Wu, W.J.; Ma, B. In silico methods in antibody design. Antibodies 2018, 7, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almagro, J.C.; Beavers, M.P.; Hernandez-Guzman, F.; Maier, J.; Shaulsky, J.; Butenhof, K.; Labute, P.; Thorsteinson, N.; Kelly, K.; Teplyakov, A.; et al. Antibody modeling assessment. Proteins 2011, 79, 3050–3066. [Google Scholar] [CrossRef] [PubMed]
- Almagro, J.C.; Teplyakov, A.; Luo, J.; Sweet, R.W.; Kodangattil, S.; Hernandez-Guzman, F.; Gilliland, G.L. Second antibody modeling assessment (ama-ii). Proteins 2014, 82, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Fasnacht, M.; Butenhof, K.; Goupil-Lamy, A.; Hernandez-Guzman, F.; Huang, H.; Yan, L. Automated antibody structure prediction using accelrys tools: Results and best practices. Proteins 2014, 82, 1583–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcatili, P.; Rosi, A.; Tramontano, A. Pigs: Automatic prediction of antibody structures. Bioinformatics 2008, 24, 1953–1954. [Google Scholar] [CrossRef] [PubMed]
- Weitzner, B.D.; Jeliazkov, J.R.; Lyskov, S.; Marze, N.; Kuroda, D.; Frick, R.; Adolf-Bryfogle, J.; Biswas, N.; Dunbrack, R.L.; Gray, J.J. Modeling and docking of antibody structures with rosetta. Nat. Protoc. 2017, 12, 401–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchalonis, J.J.; Schluter, S.F.; Bernstein, R.M.; Hohman, V.S. Antibodies of sharks: Revolution and evolution. Immunol. Rev. 1998, 166, 103–122. [Google Scholar] [CrossRef]
- Juma, S.N.; Gong, X.; Hu, S.; Lv, Z.; Shao, J.; Liu, L.; Chen, G. Shark new antigen receptor (ignar): Structure, characteristics and potential biomedical applications. Cells 2021, 10, 1140. [Google Scholar] [CrossRef]
- Arbabi Ghahroudi, M.; Desmyter, A.; Wyns, L.; Hamers, R.; Muyldermans, S. Selection and identification of single domain antibody fragments from camel heavy-chain antibodies. FEBS Lett. 1997, 414, 521–526. [Google Scholar] [CrossRef] [Green Version]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- Muyldermans, S.; Cambillau, C.; Wyns, L. Recognition of antigens by single-domain antibody fragments: The superfluous luxury of paired domains. Trends Biochem. Sci. 2001, 26, 230–235. [Google Scholar] [CrossRef]
- De Genst, E.; Saerens, D.; Muyldermans, S.; Conrath, K. Antibody repertoire development in camelids. Dev. Comp. Immunol. 2006, 30, 187–198. [Google Scholar] [CrossRef]
- Omidfar, K.; Rasaee, M.J.; Kashanian, S.; Paknejad, M.; Bathaie, Z. Studies of thermostability in camelus bactrianus (bactrian camel) single-domain antibody specific for the mutant epidermal-growth-factor receptor expressed by pichia. Biotechnol. Appl. Biochem. 2007, 46, 41–49. [Google Scholar]
- Perruchini, C.; Pecorari, F.; Bourgeois, J.P.; Duyckaerts, C.; Rougeon, F.; Lafaye, P. Llama vhh antibody fragments against gfap: Better diffusion in fixed tissues than classical monoclonal antibodies. Acta Neuropathol. 2009, 118, 685–695. [Google Scholar] [CrossRef]
- Tu, Z.; Huang, X.; Fu, J.; Hu, N.; Zheng, W.; Li, Y.; Zhang, Y. Landscape of variable domain of heavy-chain-only antibody repertoire from alpaca. Immunology 2020, 161, 53–65. [Google Scholar] [CrossRef]
- Muyldermans, S. Applications of nanobodies. Annu. Rev. Anim. Biosci. 2021, 9, 401–421. [Google Scholar] [CrossRef] [PubMed]
- Smolarek, D.; Bertrand, O.; Czerwinski, M. Variable fragments of heavy chain antibodies (vhhs): A new magic bullet molecule of medicine? Postepy Hig. I Med. Dosw. (Online) 2012, 66, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Bao, C.; Gao, Q.; Li, L.L.; Han, L.; Zhang, B.; Ding, Y.; Song, Z.; Zhang, R.; Zhang, J.; Wu, X.H. The application of nanobody in car-t therapy. Biomolecules 2021, 11, 238. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, L.; Jin, D.; Liu, Y. Nanobody-a versatile tool for cancer diagnosis and therapeutics. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnology 2021, 13, e1697. [Google Scholar] [CrossRef]
- Hosseindokht, M.; Bakherad, H.; Zare, H. Nanobodies: A tool to open new horizons in diagnosis and treatment of prostate cancer. Cancer Cell Int. 2021, 21, 580. [Google Scholar] [CrossRef]
- Moradi, A.; Pourseif, M.M.; Jafari, B.; Parvizpour, S.; Omidi, Y. Nanobody-based therapeutics against colorectal cancer: Precision therapies based on the personal mutanome profile and tumor neoantigens. Pharmacol. Res. 2020, 156, 104790. [Google Scholar] [CrossRef]
- Park, S.R.; Lee, J.H.; Kim, K.; Kim, T.M.; Lee, S.H.; Choo, Y.K.; Kim, K.S.; Ko, K. Expression and in vitro function of anti-breast cancer llama-based single domain antibody vhh expressed in tobacco plants. Int. J. Mol. Sci. 2020, 21, 1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaar, E.R.; Woodham, A.W.; Ploegh, H.L. Nanobodies in cancer. Semin. Immunol. 2021, 52, 101425. [Google Scholar] [CrossRef]
- Zhai, T.; Wang, C.; Xu, Y.; Huang, W.; Yuan, Z.; Wang, T.; Dai, S.; Peng, S.; Pang, T.; Jiang, W.; et al. Generation of a safe and efficacious llama single-domain antibody fragment (vhh) targeting the membrane-proximal region of 4-1bb for engineering therapeutic bispecific antibodies for cancer. J. Immunother. Cancer 2021, 9, e002131. [Google Scholar] [CrossRef]
- Iezzi, M.E.; Policastro, L.; Werbajh, S.; Podhajcer, O.; Canziani, G.A. Single-domain antibodies and the promise of modular targeting in cancer imaging and treatment. Front. Immunol. 2018, 9, 273. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Mukhtar, H.; Ma, L.; Pang, Q.; Wang, X. Vhh antibodies: Reagents for mycotoxin detection in food products. Sensors 2018, 18, 485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bever, C.S.; Dong, J.X.; Vasylieva, N.; Barnych, B.; Cui, Y.; Xu, Z.L.; Hammock, B.D.; Gee, S.J. Vhh antibodies: Emerging reagents for the analysis of environmental chemicals. Anal. Bioanal. Chem. 2016, 408, 5985–6002. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Zhu, M.; Li, G.; Lu, X.; Wan, Y. Specific determination of influenza h7n2 virus based on biotinylated single-domain antibody from a phage-displayed library. Anal. Biochem. 2016, 500, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Dash, L.; Subramaniam, S.; Khulape, S.A.; Prusty, B.R.; Pargai, K.; Narnaware, S.D.; Patil, N.V.; Pattnaik, B. Development and utilization of vhh antibodies derived from camelus dromedarius against foot-and-mouth disease virus. Anim. Biotechnol. 2019, 30, 57–62. [Google Scholar] [CrossRef]
- Li, H.; Dekker, A.; Sun, S.; Burman, A.; Kortekaas, J.; Harmsen, M.M. Novel capsid-specific single-domain antibodies with broad foot-and-mouth disease strain recognition reveal differences in antigenicity of virions, empty capsids, and virus-like particles. Vaccines 2021, 9, 620. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Wang, Y.; Pei, H.; Sun, Z.; Cao, H.; Zhang, C.; Chen, Q.; Liu, X. Phage-mediated double-nanobody sandwich immunoassay for detecting alpha fetal protein in human serum. Anal. Methods Adv. Methods Appl. 2020, 12, 4742–4748. [Google Scholar] [CrossRef]
- Ji, Y.; Chen, L.; Wang, Y.; Zhang, K.; Wu, H.; Liu, Y.; Wang, Y.; Wang, J. Development of a double nanobody-based sandwich immunoassay for the detecting staphylococcal enterotoxin c in dairy products. Foods 2021, 10, 2426. [Google Scholar] [CrossRef]
- Steeland, S.; Vandenbroucke, R.E.; Libert, C. Nanobodies as therapeutics: Big opportunities for small antibodies. Drug Discov. Today 2016, 21, 1076–1113. [Google Scholar] [CrossRef]
- Rossotti, M.A.; Bélanger, K.; Henry, K.A.; Tanha, J. Immunogenicity and humanization of single-domain antibodies. FEBS J. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Scully, M.; Cataland, S.R.; Peyvandi, F.; Coppo, P.; Knöbl, P.; Kremer Hovinga, J.A.; Metjian, A.; de la Rubia, J.; Pavenski, K.; Callewaert, F.; et al. Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N. Engl. J. Med. 2019, 380, 335–346. [Google Scholar] [CrossRef]
- Jovčevska, I.; Muyldermans, S. The therapeutic potential of nanobodies. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2020, 34, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senolt, L. Emerging therapies in rheumatoid arthritis: Focus on monoclonal antibodies. F1000Research 2019, 8, 1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, J.; Le Bas, A.; Ruza, R.R.; Duyvesteyn, H.M.E.; Mikolajek, H.; Malinauskas, T.; Tan, T.K.; Rijal, P.; Dumoux, M.; Ward, P.N.; et al. Neutralizing nanobodies bind sars-cov-2 spike rbd and block interaction with ace2. Nat. Struct. Mol. Biol. 2020, 27, 846–854. [Google Scholar] [CrossRef]
- Wrapp, D.; De Vlieger, D.; Corbett, K.S.; Torres, G.M.; Wang, N.; Van Breedam, W.; Roose, K.; van Schie, L.; Hoffmann, M.; Pöhlmann, S.; et al. Structural basis for potent neutralization of betacoronaviruses by single-domain camelid antibodies. Cell 2020, 181, 1004–1015.e15. [Google Scholar] [CrossRef]
- Chen, F.; Liu, Z.; Jiang, F. Prospects of neutralizing nanobodies against sars-cov-2. Front. Immunol. 2021, 12, 690742. [Google Scholar] [CrossRef] [PubMed]
- Güttler, T.; Aksu, M.; Dickmanns, A.; Stegmann, K.M.; Gregor, K.; Rees, R.; Taxer, W.; Rymarenko, O.; Schünemann, J.; Dienemann, C.; et al. Neutralization of sars-cov-2 by highly potent, hyperthermostable, and mutation-tolerant nanobodies. EMBO J. 2021, 40, e107985. [Google Scholar] [CrossRef] [PubMed]
- Hanke, L.; Vidakovics Perez, L.; Sheward, D.J.; Das, H.; Schulte, T.; Moliner-Morro, A.; Corcoran, M.; Achour, A.; Karlsson Hedestam, G.B.; Hällberg, B.M.; et al. An alpaca nanobody neutralizes sars-cov-2 by blocking receptor interaction. Nat. Commun. 2020, 11, 4420. [Google Scholar] [CrossRef]
- Koenig, P.A.; Das, H.; Liu, H.; Kümmerer, B.M.; Gohr, F.N.; Jenster, L.M.; Schiffelers, L.D.J.; Tesfamariam, Y.M.; Uchima, M.; Wuerth, J.D.; et al. Structure-guided multivalent nanobodies block sars-cov-2 infection and suppress mutational escape. Science 2021, 371, eabe6230. [Google Scholar] [CrossRef]
- Schoof, M.; Faust, B.; Saunders, R.A.; Sangwan, S.; Rezelj, V.; Hoppe, N.; Boone, M.; Billesbølle, C.B.; Puchades, C.; Azumaya, C.M.; et al. An ultrapotent synthetic nanobody neutralizes sars-cov-2 by stabilizing inactive spike. Science 2020, 370, 1473–1479. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, C.; Xia, S.; Tian, X.; Kong, Y.; Wang, Z.; Gu, C.; Zhang, R.; Tu, C.; Xie, Y.; et al. Identification of human single-domain antibodies against sars-cov-2. Cell Host Microbe 2020, 27, 891–898.e5. [Google Scholar] [CrossRef]
- Xiang, Y.; Nambulli, S.; Xiao, Z.; Liu, H.; Sang, Z.; Duprex, W.P.; Schneidman-Duhovny, D.; Zhang, C.; Shi, Y. Versatile and multivalent nanobodies efficiently neutralize sars-cov-2. Science 2020, 370, 1479–1484. [Google Scholar] [CrossRef]
- Xu, J.; Xu, K.; Jung, S.; Conte, A.; Lieberman, J.; Muecksch, F.; Lorenzi, J.C.C.; Park, S.; Schmidt, F.; Wang, Z.; et al. Nanobodies from camelid mice and llamas neutralize sars-cov-2 variants. Nature 2021, 595, 278–282. [Google Scholar] [CrossRef]
- Favorskaya, I.A.; Shcheblyakov, D.V.; Esmagambetov, I.B.; Dolzhikova, I.V.; Alekseeva, I.A.; Korobkova, A.I.; Voronina, D.V.; Ryabova, E.I.; Derkaev, A.A.; Kovyrshina, A.V.; et al. Single-domain antibodies efficiently neutralize sars-cov-2 variants of concern. Front. Immunol. 2022, 13, 822159. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.B.; Bates, T.A.; Leier, H.C.; McBride, S.K.; Barklis, E.; Tafesse, F.G. A potent alpaca-derived nanobody that neutralizes sars-cov-2 variants. iScience 2022, 25, 103960. [Google Scholar] [CrossRef] [PubMed]
- Verkhivker, G. Allosteric determinants of the sars-cov-2 spike protein binding with nanobodies: Examining mechanisms of mutational escape and sensitivity of the omicron variant. Int. J. Mol. Sci. 2022, 23, 2172. [Google Scholar] [CrossRef]
- Chabrol, E.; Stojko, J.; Nicolas, A.; Botzanowski, T.; Fould, B.; Antoine, M.; Cianférani, S.; Ferry, G.; Boutin, J.A. Vhh characterization.Recombinant vhhs: Production, characterization and affinity. Anal. Biochem. 2020, 589, 113491. [Google Scholar] [CrossRef]
- De Buck, S.; Virdi, V.; De Meyer, T.; De Wilde, K.; Piron, R.; Nolf, J.; Van Lerberge, E.; De Paepe, A.; Depicker, A. Production of camel-like antibodies in plants. Methods Mol. Biol. 2012, 911, 305–324. [Google Scholar]
- Meng, L.; Gao, X.; Liu, X.; Sun, M.; Yan, H.; Li, A.; Yang, Y.; Bai, Z. Enhancement of heterologous protein production in corynebacterium glutamicum via atmospheric and room temperature plasma mutagenesis and high-throughput screening. J. Biotechnol. 2021, 339, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Reader, R.H.; Workman, R.G.; Maddison, B.C.; Gough, K.C. Advances in the production and batch reformatting of phage antibody libraries. Mol. Biotechnol. 2019, 61, 801–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noël, F.; Malpertuy, A.; de Brevern, A.G. Global analysis of vhhs framework regions with a structural alphabet. Biochimie 2016, 131, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Melarkode Vattekatte, A.; Shinada, N.K.; Narwani, T.J.; Noël, F.; Bertrand, O.; Meyniel, J.P.; Malpertuy, A.; Gelly, J.C.; Cadet, F.; de Brevern, A.G. Discrete analysis of camelid variable domains: Sequences, structures, and in-silico structure prediction. PeerJ 2020, 8, e8408. [Google Scholar] [CrossRef]
- Smolarek, D.; Bertrand, O.; Czerwinski, M.; Colin, Y.; Etchebest, C.; de Brevern, A.G. Multiple interests in structural models of darc transmembrane protein. Transfus. Clin. Biol. J. Soc. Fr. Transfus. Sang. 2010, 17, 184–196. [Google Scholar] [CrossRef] [Green Version]
- Melarkode Vattekatte, A.; Cadet, F.; Gelly, J.C.; de Brevern, A.G. Insights into comparative modeling of v(h)h domains. Int. J. Mol. Sci. 2021, 22, 9771. [Google Scholar] [CrossRef] [PubMed]
- Adolf-Bryfogle, J.; Xu, Q.; North, B.; Lehmann, A.; Dunbrack, R.L. Pyigclassify: A database of antibody cdr structural classifications. Nucleic Acids Res. 2015, 43, D432–D438. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kang, Y.; Luo, J.; Pang, K.; Xu, X.; Wu, J.; Li, X.; Jin, S. Next-generation sequencing reveals the progression of covid-19. Front. Cell. Infect. Microbiol. 2021, 11, 632490. [Google Scholar] [CrossRef]
- Makałowski, W.; Shabardina, V. Bioinformatics of nanopore sequencing. J. Hum. Genet. 2020, 65, 61–67. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.S.; Richardson, D.C.; Goodsell, D.S. Seeing the pdb. J. Biol. Chem. 2021, 296, 100742. [Google Scholar] [CrossRef] [PubMed]
- Garnier, J. Protein structure prediction. Biochimie 1990, 72, 513–524. [Google Scholar] [CrossRef]
- Baker, D.; Sali, A. Protein structure prediction and structural genomics. Science 2001, 294, 93–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gło, N. Statistical mechanics of protein folding, unfolding and fluctuation. Adv. Biophys. 1976, 65–113. [Google Scholar]
- Greer, J. Comparative modeling of homologous proteins. Methods Enzymol. 1991, 202, 239–252. [Google Scholar]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T. Genthreader: An efficient and reliable protein fold recognition method for genomic sequences. J. Mol. Biol. 1999, 287, 797–815. [Google Scholar] [CrossRef] [Green Version]
- Ghouzam, Y.; Postic, G.; de Brevern, A.G.; Gelly, J.C. Improving protein fold recognition with hybrid profiles combining sequence and structure evolution. Bioinformatics 2015, 31, 3782–3789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghouzam, Y.; Postic, G.; Guerin, P.E.; de Brevern, A.G.; Gelly, J.C. Orion: A web server for protein fold recognition and structure prediction using evolutionary hybrid profiles. Sci. Rep. 2016, 6, 28268. [Google Scholar] [CrossRef]
- Bystroff, C.; Shao, Y. Fully automated ab initio protein structure prediction using i-sites, hmmstr and rosetta. Bioinformatics 2002, 18, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Rohl, C.A.; Strauss, C.E.; Misura, K.M.; Baker, D. Protein structure prediction using rosetta. Methods Enzymol. 2004, 383, 66–93. [Google Scholar] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The i-tasser suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with alphafold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Du, Z.; Su, H.; Wang, W.; Ye, L.; Wei, H.; Peng, Z.; Anishchenko, I.; Baker, D.; Yang, J. The trrosetta server for fast and accurate protein structure prediction. Nat. Protoc. 2021, 16, 5634–5651. [Google Scholar] [CrossRef]
- Leman, J.K.; Weitzner, B.D.; Lewis, S.M.; Adolf-Bryfogle, J.; Alam, N.; Alford, R.F.; Aprahamian, M.; Baker, D.; Barlow, K.A.; Barth, P.; et al. Macromolecular modeling and design in rosetta: Recent methods and frameworks. Nat. Methods 2020, 17, 665–680. [Google Scholar] [CrossRef]
- Nguyen, D.D.; Cang, Z.; Wei, G.W. A review of mathematical representations of biomolecular data. Phys. Chem. Chem. Phys. PCCP 2020, 22, 4343–4367. [Google Scholar] [CrossRef] [PubMed]
- Meli, R.; Biggin, P.C. Spyrmsd: Symmetry-corrected rmsd calculations in python. J. Cheminf. 2020, 12, 49. [Google Scholar] [CrossRef]
- Jauch, R.; Yeo, H.C.; Kolatkar, P.R.; Clarke, N.D. Assessment of casp7 structure predictions for template free targets. Proteins 2007, 69, 57–67. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins 2004, 57, 702–710. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. Tm-align: A protein structure alignment algorithm based on the tm-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Gadiyaram, V.; Vishveshwara, S. Validation of protein structure models using network similarity score. Proteins 2017, 85, 1759–1776. [Google Scholar] [CrossRef] [PubMed]
- Lefranc, M.P. Antibody informatics: Imgt, the international immunogenetics information system. Microbiol. Spectr. 2014, 2, 2.2.01. [Google Scholar] [CrossRef] [Green Version]
- Lefranc, M.P.; Lefranc, G. Immunoglobulins or antibodies: Imgt(®) bridging genes, structures and functions. Biomedicines 2020, 8, 319. [Google Scholar] [CrossRef] [PubMed]
- Pallarès, N.; Lefebvre, S.; Contet, V.; Matsuda, F.; Lefranc, M.P. The human immunoglobulin heavy variable genes. Exp. Clin. Immunogenet. 1999, 16, 36–60. [Google Scholar] [CrossRef] [PubMed]
- Mariuzza, R.A.; Phillips, S.E.; Poljak, R.J. The structural basis of antigen-antibody recognition. Annu. Rev. Biophys. Biophys. Chem. 1987, 16, 139–159. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Madrazo, E.; Paz-García, E. An improved model of association for vh-vl immunoglobulin domains: Asymmetries between vh and vl in the packing of some interface residues. J. Mol. Recognit. JMR 2003, 16, 113–120. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Cryst. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, B.; Sali, A. Comparative protein structure modeling using modeller. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. A Publ. Protein Soc. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped blast and psi-blast: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schäffer, A.A.; Wolf, Y.I.; Ponting, C.P.; Koonin, E.V.; Aravind, L.; Altschul, S.F. Impala: Matching a protein sequence against a collection of psi-blast-constructed position-specific score matrices. Bioinformatics 1999, 15, 1000–1011. [Google Scholar] [CrossRef]
- Choong, Y.S.; Lee, Y.V.; Soong, J.X.; Law, C.T.; Lim, Y.Y. Computer-aided antibody design: An overview. Adv. Exp. Med. Biol. 2017, 1053, 221–243. [Google Scholar] [PubMed]
- Smolarek, D.; Hattab, C.; Hassanzadeh-Ghassabeh, G.; Cochet, S.; Gutiérrez, C.; de Brevern, A.G.; Udomsangpetch, R.; Picot, J.; Grodecka, M.; Wasniowska, K.; et al. A recombinant dromedary antibody fragment (vhh or nanobody) directed against human duffy antigen receptor for chemokines. Cell. Mol. Life Sci. CMLS 2010, 67, 3371–3387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolarek, D.; Hattab, C.; Buczkowska, A.; Kaczmarek, R.; Jarząb, A.; Cochet, S.; de Brevern, A.G.; Lukasiewicz, J.; Jachymek, W.; Niedziela, T.; et al. Studies of a murine monoclonal antibody directed against darc: Reappraisal of its specificity. PLoS ONE 2015, 10, e0116472. [Google Scholar]
- Steeland, S.; Puimège, L.; Vandenbroucke, R.E.; Van Hauwermeiren, F.; Haustraete, J.; Devoogdt, N.; Hulpiau, P.; Leroux-Roels, G.; Laukens, D.; Meuleman, P.; et al. Generation and characterization of small single domain antibodies inhibiting human tumor necrosis factor receptor 1. J. Biol. Chem. 2015, 290, 4022–4037. [Google Scholar] [CrossRef] [Green Version]
- Lovell, S.C.; Davis, I.W.; Arendall III, W.B.; de Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by calpha geometry: Phi,psi and cbeta deviation. Proteins 2003, 50, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Comeau, S.R.; Gatchell, D.W.; Vajda, S.; Camacho, C.J. Cluspro: A fully automated algorithm for protein-protein docking. Nucleic Acids Res. 2004, 32, W96–W99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahangian, S.S.; Sajedi, R.H.; Hasannia, S.; Jalili, S.; Mohammadi, M.; Taghdir, M.; Shali, A.; Mansouri, K.; Sariri, R. A conformation-based phage-display panning to screen neutralizing anti-vegf vhhs with vegfr2 mimicry behavior. Int. J. Biol. Macromol. 2015, 77, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Calpe, S.; Wagner, K.; El Khattabi, M.; Rutten, L.; Zimberlin, C.; Dolk, E.; Verrips, C.T.; Medema, J.P.; Spits, H.; Krishnadath, K.K. Effective inhibition of bone morphogenetic protein function by highly specific llama-derived antibodies. Mol. Cancer Ther. 2015, 14, 2527–2540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Wang, J.; Kang, G.; Hu, M.; Yuan, B.; Zhang, Y.; Huang, H. Homology modeling-based in silico affinity maturation improves the affinity of a nanobody. Int. J. Mol. Sci. 2019, 20, 4187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khodabakhsh, F.; Salimian, M.; Ziaee, P.; Kazemi-Lomedasht, F.; Behdani, M.; Ahangari Cohan, R. Designing and development of a tandem bivalent nanobody against vegf(165). Avicenna J. Med. Biotechnol. 2021, 13, 58–64. [Google Scholar] [PubMed]
- Prado, N.D.; Pereira, S.S.; da Silva, M.P.; Morais, M.S.; Kayano, A.M.; Moreira-Dill, L.S.; Luiz, M.B.; Zanchi, F.B.; Fuly, A.L.; Huacca, M.E.; et al. Inhibition of the myotoxicity induced by bothrops jararacussu venom and isolated phospholipases a2 by specific camelid single-domain antibody fragments. PLoS ONE 2016, 11, e0151363. [Google Scholar] [CrossRef] [PubMed]
- Demeestere, D.; Dejonckheere, E.; Steeland, S.; Hulpiau, P.; Haustraete, J.; Devoogdt, N.; Wichert, R.; Becker-Pauly, C.; Van Wonterghem, E.; Dewaele, S.; et al. Development and validation of a small single-domain antibody that effectively inhibits matrix metalloproteinase 8. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 890–902. [Google Scholar] [CrossRef] [PubMed]
- Pang, Q.; Chen, Y.; Mukhtar, H.; Xiong, J.; Wang, X.; Xu, T.; Hammock, B.D.; Wang, J. Camelization of a murine single-domain antibody against aflatoxin b(1) and its antigen-binding analysis. Mycotoxin Res. 2022, 38, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Lee, S.L.; Russell, A.M.; Huang, R.F.; Batt, M.A.; Chang, S.S.; Ferrante, A.; Verdino, P. A structure-based engineering approach to abrogate pre-existing antibody binding to biotherapeutics. PLoS ONE 2021, 16, e0254944. [Google Scholar] [CrossRef] [PubMed]
- Yerabham, A.S.K.; Müller-Schiffmann, A.; Ziehm, T.; Stadler, A.; Köber, S.; Indurkhya, X.; Marreiros, R.; Trossbach, S.V.; Bradshaw, N.J.; Prikulis, I.; et al. Biophysical insights from a single chain camelid antibody directed against the disrupted-in-schizophrenia 1 protein. PLoS ONE 2018, 13, e0191162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, C.; Kingery, B.D.; Anchin, J.M.; Subramaniam, S.; Linthicum, D.S. Abgen: A knowledge-based automated approach for antibody structure modeling. Nat. Biotechnol. 1996, 14, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Schwieters, C.D.; Bermejo, G.A.; Clore, G.M. Xplor-nih for molecular structure determination from nmr and other data sources. Protein Sci. A Publ. Protein Soc. 2018, 27, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and testing of the gromos force-field versions 54a7 and 54b7. Eur. Biophys. J. EBJ 2011, 40, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Whitelegg, N.R.; Rees, A.R. Wam: An improved algorithm for modelling antibodies on the web. Protein Eng. 2000, 13, 819–824. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.C.; Cheetham, J.C.; Rees, A.R. Modeling antibody hypervariable loops: A combined algorithm. Proc. Natl. Acad. Sci. USA 1989, 86, 9268–9272. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.C.; Cheetham, J.C.; Rees, A.R. Molecular modeling of antibody combining sites. Methods Enzymol. 1991, 203, 121–153. [Google Scholar]
- Bruccoleri, R.E.; Karplus, M. Prediction of the folding of short polypeptide segments by uniform conformational sampling. Biopolymers 1987, 26, 137–168. [Google Scholar] [CrossRef] [PubMed]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. Swiss-model: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef] [Green Version]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Gallo Cassarino, T.; Bertoni, M.; Bordoli, L.; et al. Swiss-model: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. Swiss-model: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. Hhblits: Lightning-fast iterative protein sequence searching by hmm-hmm alignment. Nat. Methods 2011, 9, 173–175. [Google Scholar] [CrossRef]
- Biasini, M.; Schmidt, T.; Bienert, S.; Mariani, V.; Studer, G.; Haas, J.; Johner, N.; Schenk, A.D.; Philippsen, A.; Schwede, T. Openstructure: An integrated software framework for computational structural biology. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 701–709. [Google Scholar] [CrossRef] [Green Version]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, F.; Zhang, C.; Wang, C.; Lu, P.; Zhao, X.; Hao, L.; Ding, J. Immunoinformatics prediction of omp2b and bcsp31 for designing multi-epitope vaccine against brucella. Mol. Immunol. 2019, 114, 651–660. [Google Scholar] [CrossRef]
- Rahman, N.; Ali, F.; Basharat, Z.; Shehroz, M.; Khan, M.K.; Jeandet, P.; Nepovimova, E.; Kuca, K.; Khan, H. Vaccine design from the ensemble of surface glycoprotein epitopes of sars-cov-2: An immunoinformatics approach. Vaccines 2020, 8, 423. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, D.I.; Corgliano, D.M.; Franco, E.J.; Hofstetter, H.; Hofstetter, O. Investigation of the stereoselectivity of an anti-amino acid antibody using molecular modeling and ligand docking. Chirality 2008, 20, 559–570. [Google Scholar] [CrossRef]
- Shen, H.D.; Tam, M.F.; Huang, C.H.; Chou, H.; Tai, H.Y.; Chen, Y.S.; Sheu, S.Y.; Thomas, W.R. Homology modeling and monoclonal antibody binding of the der f 7 dust mite allergen. Immunol. Cell Biol. 2011, 89, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Studer, G.; Tauriello, G.; Bienert, S.; Biasini, M.; Johner, N.; Schwede, T. Promod3-a versatile homology modelling toolbox. PLoS Comput. Biol. 2021, 17, e1008667. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Kumachi, S.; Matsunaga, Y.; Sato, M.; Wakabayashi-Nakao, K.; Masaki, H.; Yonehara, R.; Motohashi, M.; Nemoto, N.; Tsuchiya, M. Construction of a humanized artificial vhh library reproducing structural features of camelid vhhs for therapeutics. Antibodies 2022, 11, 10. [Google Scholar] [CrossRef]
- Hanke, L.; Das, H.; Sheward, D.J.; Perez Vidakovics, L.; Urgard, E.; Moliner-Morro, A.; Kim, C.; Karl, V.; Pankow, A.; Smith, N.L.; et al. A bispecific monomeric nanobody induces spike trimer dimers and neutralizes sars-cov-2 in vivo. Nat. Commun. 2022, 13, 155. [Google Scholar] [CrossRef]
- Nordeen, S.A.; Andersen, K.R.; Knockenhauer, K.E.; Ingram, J.R.; Ploegh, H.L.; Schwartz, T.U. A nanobody suite for yeast scaffold nucleoporins provides details of the nuclear pore complex structure. Nat. Commun. 2020, 11, 6179. [Google Scholar] [CrossRef] [PubMed]
- Thanongsaksrikul, J.; Srimanote, P.; Maneewatch, S.; Choowongkomon, K.; Tapchaisri, P.; Makino, S.I.; Kurazono, H.; Chaicumpa, W. A v h h that neutralizes the zinc metalloproteinase activity of botulinum neurotoxin type a. J. Biol. Chem. 2010, 285, 9657–9666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashida, R.; Matsunaga, Y. Enhanced conformational sampling of nanobody cdr h3 loop by generalized replica-exchange with solute tempering. Life 2021, 11, 1428. [Google Scholar] [CrossRef] [PubMed]
- Orlov, I.; Hemmer, C.; Ackerer, L.; Lorber, B.; Ghannam, A.; Poignavent, V.; Hleibieh, K.; Sauter, C.; Schmitt-Keichinger, C.; Belval, L.; et al. Structural basis of nanobody recognition of grapevine fanleaf virus and of virus resistance loss. Proc. Natl. Acad. Sci. USA 2020, 117, 10848–10855. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.P.; Meksiriporn, B.; Waraho-Zhmayev, D.; Weyant, K.B.; Kocer, I.; Butler, D.C.; Messer, A.; Escobedo, F.A.; DeLisa, M.P. Computational affinity maturation of camelid single-domain intrabodies against the nonamyloid component of alpha-synuclein. Sci. Rep. 2018, 8, 17611. [Google Scholar] [CrossRef] [PubMed]
- Bujotzek, A.; Fuchs, A.; Qu, C.; Benz, J.; Klostermann, S.; Antes, I.; Georges, G. Mofvab: Modeling the fv region of antibodies. mAbs 2015, 7, 838–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bujotzek, A.; Dunbar, J.; Lipsmeier, F.; Schäfer, W.; Antes, I.; Deane, C.M.; Georges, G. Prediction of vh-vl domain orientation for antibody variable domain modeling. Proteins 2015, 83, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Helmer-Citterich, M.; Rovida, E.; Luzzago, A.; Tramontano, A. Modelling antibody-antigen interactions: Ferritin as a case study. Mol. Immunol. 1995, 32, 1001–1010. [Google Scholar] [CrossRef]
- Morea, V.; Lesk, A.M.; Tramontano, A. Antibody modeling: Implications for engineering and design. Methods 2000, 20, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Morea, V.; Tramontano, A.; Rustici, M.; Chothia, C.; Lesk, A.M. Antibody structure, prediction and redesign. Biophys. Chem. 1997, 68, 9–16. [Google Scholar] [CrossRef]
- Tramontano, A.; Janda, K.; Napper, A.D.; Benkovic, S.J.; Lerner, R.A. Catalytic antibodies. Cold Spring Harb. Symp. Quant. Biol. 1987, 52, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Morea, V.; Tramontano, A.; Rustici, M.; Chothia, C.; Lesk, A.M. Conformations of the third hypervariable region in the vh domain of immunoglobulins. J. Mol. Biol. 1998, 275, 269–294. [Google Scholar] [CrossRef] [PubMed]
- Lepore, R.; Olimpieri, P.P.; Messih, M.A.; Tramontano, A. Pigspro: Prediction of immunoglobulin structures v2. Nucleic Acids Res. 2017, 45, W17–W23. [Google Scholar] [CrossRef] [PubMed]
- Leem, J.; Deane, C.M. High-throughput antibody structure modeling and design using abodybuilder. Methods Mol. Biol. 2019, 1851, 367–380. [Google Scholar] [PubMed]
- Schneider, C.; Raybould, M.I.J.; Deane, C.M. Sabdab in the age of biotherapeutics: Updates including sabdab-nano, the nanobody structure tracker. Nucleic Acids Res. 2022, 50, D1368–D1372. [Google Scholar] [CrossRef] [PubMed]
- George, S. Conformational diversity of cdr region during affinity maturation determines the affinity and stability of sars-cov-1 vhh-72 nanobody. bioRxiv 2020. [Google Scholar] [CrossRef]
- Klausen, M.S.; Anderson, M.V.; Jespersen, M.C.; Nielsen, M.; Marcatili, P. Lyra, a webserver for lymphocyte receptor structural modeling. Nucleic Acids Res. 2015, 43, W349–W355. [Google Scholar] [CrossRef] [Green Version]
- Abhinandan, K.R.; Martin, A.C. Analysis and improvements to kabat and structurally correct numbering of antibody variable domains. Mol. Immunol. 2008, 45, 3832–3839. [Google Scholar] [CrossRef] [PubMed]
- Marcatili, P.; Olimpieri, P.P.; Chailyan, A.; Tramontano, A. Antibody modeling using the prediction of immunoglobulin structure (pigs) web server [corrected]. Nat. Protoc. 2014, 9, 2771–2783. [Google Scholar] [CrossRef]
- North, B.; Lehmann, A.; Dunbrack, R.L. A new clustering of antibody cdr loop conformations. J. Mol. Biol. 2011, 406, 228–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, L.A.; Sternberg, M.J. Protein structure prediction on the web: A case study using the phyre server. Nat. Protoc. 2009, 4, 363–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Söding, J. Protein homology detection by hmm-hmm comparison. Bioinformatics 2005, 21, 951–960. [Google Scholar] [CrossRef] [Green Version]
- Jefferys, B.R.; Kelley, L.A.; Sternberg, M.J. Protein folding requires crowd control in a simulated cell. J. Mol. Biol. 2010, 397, 1329–1338. [Google Scholar] [CrossRef] [Green Version]
- Rotkiewicz, P.; Skolnick, J. Fast procedure for reconstruction of full-atom protein models from reduced representations. J. Comput. Chem. 2008, 29, 1460–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Sahinidis, N.V. Residue-rotamer-reduction algorithm for the protein side-chain conformation problem. Bioinformatics 2006, 22, 188–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoseinpoor, R.; Mousavi Gargari, S.L.; Rasooli, I.; Rajabibazl, M.; Shahi, B. Functional mutations in and characterization of vhh against helicobacter pylori urease. Appl. Biochem. Biotechnol. 2014, 172, 3079–3091. [Google Scholar] [CrossRef]
- Payandeh, Z.; Rasooli, I.; Mousavi Gargari, S.L.; Rajabi Bazl, M.; Ebrahimizadeh, W. Immunoreaction of a recombinant nanobody from camelid single domain antibody fragment with acinetobacter baumannii. Trans. R. Soc. Trop. Med. Hyg. 2014, 108, 92–98. [Google Scholar] [CrossRef]
- Chen, C.C.; Hwang, J.K.; Yang, J.M. (ps)2-v2: Template-based protein structure prediction server. BMC Bioinform. 2009, 10, 366. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Zhang, Y. Lomets: A local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007, 35, 3375–3382. [Google Scholar] [CrossRef] [Green Version]
- Sippl, M.J. Recognition of errors in three-dimensional structures of proteins. Proteins 1993, 17, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. Prosa-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Zhang, Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys. J. 2011, 101, 2525–2534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, B.G.; Hourai, Y.; Weng, Z. Accelerating protein docking in zdock using an advanced 3d convolution library. PLoS ONE 2011, 6, e24657. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. Zdock server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef] [PubMed]
- Sefid, F.; Rasooli, I.; Payandeh, Z. Homology modeling of a camelid antibody fragment against a conserved region of acinetobacter baumannii biofilm associated protein (bap). J. Theor. Biol. 2016, 397, 43–51. [Google Scholar] [CrossRef]
- Skottrup, P.D. Structural insights into a high affinity nanobody: Antigen complex by homology modelling. J. Mol. Graph. Model. 2017, 76, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Källberg, M.; Margaryan, G.; Wang, S.; Ma, J.; Xu, J. Raptorx server: A resource for template-based protein structure modeling. Methods Mol. Biol. 2014, 1137, 17–27. [Google Scholar]
- Källberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lu, H.; Xu, J. Template-based protein structure modeling using the raptorx web server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Li, M. Assessment of raptor’s linear programming approach in cafasp3. Proteins 2003, 53, 579–584. [Google Scholar] [CrossRef]
- Xu, J.; Li, M.; Kim, D.; Xu, Y. Raptor: Optimal protein threading by linear programming. J. Bioinform. Comput. Biol. 2003, 1, 95–117. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Li, W.; Liu, S.; Xu, J. Raptorx-property: A web server for protein structure property prediction. Nucleic Acids Res. 2016, 44, W430–W435. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, S.; Li, Z.; Zhang, R.; Xu, J. Accurate de novo prediction of protein contact map by ultra-deep learning model. PLoS Comput. Biol. 2017, 13, e1005324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, J.; Xu, J. Raptorx: Exploiting structure information for protein alignment by statistical inference. Proteins 2011, 79, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Jittavisutthikul, S.; Thanongsaksrikul, J.; Thueng-In, K.; Chulanetra, M.; Srimanote, P.; Seesuay, W.; Malik, A.A.; Chaicumpa, W. Humanized-vhh transbodies that inhibit hcv protease and replication. Viruses 2015, 7, 2030–2056. [Google Scholar] [CrossRef] [Green Version]
- Thueng-in, K.; Thanongsaksrikul, J.; Srimanote, P.; Bangphoomi, K.; Poungpair, O.; Maneewatch, S.; Choowongkomon, K.; Chaicumpa, W. Cell penetrable humanized-vh/v(h)h that inhibit rna dependent rna polymerase (ns5b) of hcv. PLoS ONE 2012, 7, e49254. [Google Scholar] [CrossRef] [Green Version]
- Chavanayarn, C.; Thanongsaksrikul, J.; Thueng-In, K.; Bangphoomi, K.; Sookrung, N.; Chaicumpa, W. Humanized-single domain antibodies (vh/vhh) that bound specifically to naja kaouthia phospholipase a2 and neutralized the enzymatic activity. Toxins 2012, 4, 554–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, A.A.; Imtong, C.; Sookrung, N.; Katzenmeier, G.; Chaicumpa, W.; Angsuthanasombat, C. Structural characterization of humanized nanobodies with neutralizing activity against the bordetella pertussis cyaa-hemolysin: Implications for a potential epitope of toxin-protective antigen. Toxins 2016, 8, 99. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Lee, D.; Park, H.; Coutsias, E.A.; Lee, J.; Seok, C. The falc-loop web server for protein loop modeling. Nucleic Acids Res. 2011, 39, W210–W214. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. Gromacs: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.T.; Bonneau, R.; Ruczinski, I.; Baker, D. Ab initio protein structure prediction of casp iii targets using rosetta. Proteins 1999, 3, 171–176. [Google Scholar] [CrossRef]
- Bystroff, C.; Thorsson, V.; Baker, D. Hmmstr: A hidden markov model for local sequence-structure correlations in proteins. J. Mol. Biol. 2000, 301, 173–190. [Google Scholar] [CrossRef] [Green Version]
- Bradley, P.; Malmström, L.; Qian, B.; Schonbrun, J.; Chivian, D.; Kim, D.E.; Meiler, J.; Misura, K.M.; Baker, D. Free modeling with rosetta in casp6. Proteins 2005, 61, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.; Vernon, R.; Thompson, J.; Tyka, M.; Sadreyev, R.; Pei, J.; Kim, D.; Kellogg, E.; DiMaio, F.; Lange, O.; et al. Structure prediction for casp8 with all-atom refinement using rosetta. Proteins 2009, 77, 89–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; DiMaio, F.; Wang, R.Y.; Kim, D.; Miles, C.; Brunette, T.; Thompson, J.; Baker, D. High-resolution comparative modeling with rosettacm. Structure 2013, 21, 1735–1742. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Kim, D.E.; Ovchinnikov, S.; Baker, D.; DiMaio, F. Automatic structure prediction of oligomeric assemblies using robetta in casp12. Proteins 2018, 86, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, S.; Ertelt, M.; Merkl, R.; Meiler, J. Rosetta design with co-evolutionary information retains protein function. PLoS Comput. Biol. 2021, 17, e1008568. [Google Scholar] [CrossRef]
- Chaudhury, S.; Berrondo, M.; Weitzner, B.D.; Muthu, P.; Bergman, H.; Gray, J.J. Benchmarking and analysis of protein docking performance in rosetta v3.2. PLoS ONE 2011, 6, e22477. [Google Scholar] [CrossRef] [Green Version]
- Tivon, B.; Gabizon, R.; Somsen, B.A.; Cossar, P.J.; Ottmann, C.; London, N. Covalent flexible peptide docking in rosetta. Chem. Sci. 2021, 12, 10836–10847. [Google Scholar] [CrossRef]
- Govaert, J.; Pellis, M.; Deschacht, N.; Vincke, C.; Conrath, K.; Muyldermans, S.; Saerens, D. Dual beneficial effect of interloop disulfide bond for single domain antibody fragments. J. Biol. Chem. 2012, 287, 1970–1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivasubramanian, A.; Sircar, A.; Chaudhury, S.; Gray, J.J. Toward high-resolution homology modeling of antibody fv regions and application to antibody-antigen docking. Proteins 2009, 74, 497–514. [Google Scholar] [CrossRef] [Green Version]
- Sircar, A.; Kim, E.T.; Gray, J.J. Rosettaantibody: Antibody variable region homology modeling server. Nucleic Acids Res. 2009, 37, W474–W479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoeder, C.T.; Schmitz, S.; Adolf-Bryfogle, J.; Sevy, A.M.; Finn, J.A.; Sauer, M.F.; Bozhanova, N.G.; Mueller, B.K.; Sangha, A.K.; Bonet, J.; et al. Modeling immunity with rosetta: Methods for antibody and antigen design. Biochemistry 2021, 60, 825–846. [Google Scholar] [CrossRef]
- Jeliazkov, J.R.; Frick, R.; Zhou, J.; Gray, J.J. Robustification of rosettaantibody and rosetta snugdock. PLoS ONE 2021, 16, e0234282. [Google Scholar] [CrossRef] [PubMed]
- Sircar, A. Methods for the homology modeling of antibody variable regions. Methods Mol. Biol. (Clifton, N.J.) 2012, 857, 301–311. [Google Scholar]
- Sircar, A.; Sanni, K.A.; Shi, J.; Gray, J.J. Analysis and modeling of the variable region of camelid single-domain antibodies. J. Immunol. 2011, 186, 6357–6367. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, I.; Egloff, P.; Hutter, C.A.; Arnold, F.M.; Stohler, P.; Bocquet, N.; Hug, M.N.; Huber, S.; Siegrist, M.; Hetemann, L.; et al. Synthetic single domain antibodies for the conformational trapping of membrane proteins. eLife 2018, 7, e34317. [Google Scholar] [CrossRef]
- Norn, C.H.; Lapidoth, G.; Fleishman, S.J. High-accuracy modeling of antibody structures by a search for minimum-energy recombination of backbone fragments. Proteins 2017, 85, 30–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapidoth, G.; Parker, J.; Prilusky, J.; Fleishman, S.J. Abpredict 2: A server for accurate and unstrained structure prediction of antibody variable domains. Bioinformatics 2019, 35, 1591–1593. [Google Scholar] [CrossRef] [PubMed]
- Kodali, P.; Schoeder, C.T.; Schmitz, S.; Crowe, J.E.; Meiler, J. Rosettacm for antibodies with very long hcdr3s and low template availability. Proteins 2021, 89, 1458–1472. [Google Scholar] [CrossRef]
- Kemmish, H.; Fasnacht, M.; Yan, L. Fully automated antibody structure prediction using biovia tools: Validation study. PLoS ONE 2017, 12, e0177923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, J.K.; Labute, P. Assessment of fully automated antibody homology modeling protocols in molecular operating environment. Proteins 2014, 82, 1599–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanakieva, D.; Pekar, L.; Evers, A.; Fleischer, M.; Keller, S.; Mueller-Pompalla, D.; Toleikis, L.; Kolmar, H.; Zielonka, S.; Krah, S. Beyond bispecificity: Controlled fab arm exchange for the generation of antibodies with multiple specificities. mAbs 2022, 14, 2018960. [Google Scholar] [CrossRef]
- Berrondo, M.; Kaufmann, S.; Berrondo, M. Automated aufbau of antibody structures from given sequences using macromoltek’s smrtmolantibody. Proteins 2014, 82, 1636–1645. [Google Scholar] [CrossRef]
- Zhang, Y. Interplay of i-tasser and quark for template-based and ab initio protein structure prediction in casp10. Proteins 2014, 82, 175–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-tasser: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Zhang, C.; Li, Y.; Pearce, R.; Bell, E.W.; Zhang, Y. Folding non-homologous proteins by coupling deep-learning contact maps with i-tasser assembly simulations. Cell Rep. Methods 2021, 1, 100014. [Google Scholar] [CrossRef] [PubMed]
- Frenken, L.G.; van der Linden, R.H.; Hermans, P.W.; Bos, J.W.; Ruuls, R.C.; de Geus, B.; Verrips, C.T. Isolation of antigen specific llama vhh antibody fragments and their high level secretion by saccharomyces cerevisiae. J. Biotechnol. 2000, 78, 11–21. [Google Scholar] [CrossRef]
- Fridy, P.C.; Li, Y.; Keegan, S.; Thompson, M.K.; Nudelman, I.; Scheid, J.F.; Oeffinger, M.; Nussenzweig, M.C.; Fenyö, D.; Chait, B.T.; et al. A robust pipeline for rapid production of versatile nanobody repertoires. Nat. Methods 2014, 11, 1253–1260. [Google Scholar] [CrossRef] [Green Version]
- Fridy, P.C.; Thompson, M.K.; Ketaren, N.E.; Rout, M.P. Engineered high-affinity nanobodies recognizing staphylococcal protein a and suitable for native isolation of protein complexes. Anal. Biochem. 2015, 477, 92–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Zhang, Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins 2012, 80, 1715–1735. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Skolnick, J. Spicker: A clustering approach to identify near-native protein folds. J. Comput. Chem. 2004, 25, 865–871. [Google Scholar] [CrossRef]
- Mortuza, S.M.; Zheng, W.; Zhang, C.; Li, Y.; Pearce, R.; Zhang, Y. Improving fragment-based ab initio protein structure assembly using low-accuracy contact-map predictions. Nat. Commun. 2021, 12, 5011. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Li, Y.; Zhang, C.; Pearce, R.; Mortuza, S.M.; Zhang, Y. Deep-learning contact-map guided protein structure prediction in casp13. Proteins 2019, 87, 1149–1164. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Zheng, W.; Mortuza, S.M.; Li, Y.; Zhang, Y. Deepmsa: Constructing deep multiple sequence alignment to improve contact prediction and fold-recognition for distant-homology proteins. Bioinformatics 2020, 36, 2105–2112. [Google Scholar] [CrossRef]
- Kinch, L.N.; Li, W.; Monastyrskyy, B.; Kryshtafovych, A.; Grishin, N.V. Evaluation of free modeling targets in casp11 and roll. Proteins 2016, 84, 51–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, C.H.; Bai, H.; Taylor, T.J.; Lee, B. Assessment of template-free modeling in casp10 and roll. Proteins 2014, 82, 57–83. [Google Scholar] [CrossRef] [PubMed]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef]
- AlQuraishi, M. Alphafold at casp13. Bioinformatics 2019, 35, 4862–4865. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Protein structure prediction using multiple deep neural networks in the 13th critical assessment of protein structure prediction (casp13). Proteins 2019, 87, 1141–1148. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Applying and improving alphafold at casp14. Proteins 2021, 89, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Fersht, A.R. Alphafold-a personal perspective on the impact of machine learning. J. Mol. Biol. 2021, 433, 167088. [Google Scholar] [CrossRef]
- Porta-Pardo, E.; Ruiz-Serra, V.; Valentini, S.; Valencia, A. The structural coverage of the human proteome before and after alphafold. PLoS Comput. Biol. 2022, 18, e1009818. [Google Scholar] [CrossRef]
- Tong, A.B.; Burch, J.D.; McKay, D.; Bustamante, C.; Crackower, M.A.; Wu, H. Could alphafold revolutionize chemical therapeutics? Nat. Struct. Mol. Biol. 2021, 28, 771–772. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. Colabfold-making protein folding accessible to all. bioRxiv 2022. [Google Scholar] [CrossRef]
- Callaway, E. It will change everything: Deepmind’s ai makes gigantic leap in solving protein structures. Nature 2020, 588, 203–204. [Google Scholar] [CrossRef] [PubMed]
- Thornton, J.M.; Laskowski, R.A.; Borkakoti, N. Alphafold heralds a data-driven revolution in biology and medicine. Nat. Med. 2021, 27, 1666–1669. [Google Scholar] [CrossRef]
- Cao, C.; Liu, F.; Tan, H.; Song, D.; Shu, W.; Li, W.; Zhou, Y.; Bo, X.; Xie, Z. Deep learning and its applications in biomedicine. Genom. Proteom. Bioinform. 2018, 16, 17–32. [Google Scholar] [CrossRef]
- Rifaioglu, A.S.; Atas, H.; Martin, M.J.; Cetin-Atalay, R.; Atalay, V.; Doğan, T. Recent applications of deep learning and machine intelligence on in silico drug discovery: Methods, tools and databases. Brief. Bioinform. 2019, 20, 1878–1912. [Google Scholar] [CrossRef]
- Shi, Q.; Chen, W.; Huang, S.; Wang, Y.; Xue, Z. Deep learning for mining protein data. Brief. Bioinform. 2021, 22, 194–218. [Google Scholar] [CrossRef]
- Torrisi, M.; Pollastri, G.; Le, Q. Deep learning methods in protein structure prediction. Comput. Struct. Biotechnol. J. 2020, 18, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Cretin, G.; Galochkina, T.; de Brevern, A.G.; Gelly, J.C. Pythia: Deep learning approach for local protein conformation prediction. Int. J. Mol. Sci. 2021, 22, 8831. [Google Scholar] [CrossRef] [PubMed]
- Vander Meersche, Y.; Cretin, G.; de Brevern, A.G.; Gelly, J.C.; Galochkina, T. Medusa: Prediction of protein flexibility from sequence. J. Mol. Biol. 2021, 433, 166882. [Google Scholar] [CrossRef] [PubMed]
- Alford, R.F.; Leaver-Fay, A.; Jeliazkov, J.R.; O’Meara, M.J.; DiMaio, F.P.; Park, H.; Shapovalov, M.V.; Renfrew, P.D.; Mulligan, V.K.; Kappel, K.; et al. The rosetta all-atom energy function for macromolecular modeling and design. J. Chem. Theory Comput. 2017, 13, 3031–3048. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Ruffolo, J.; Sulam, J.; Gray, J.J. Antibody structure prediction using interpretable deep learning. Patterns 2022, 3, 100406. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.; Halfon, M.; Schneidman-Duhovny, D. Nanonet: Rapid end-to-end nanobody modeling by deep learning at sub angstrom resolution. bioRxiv 2021. [Google Scholar] [CrossRef]
- Sun, D.; Sang, Z.; Kim, Y.J.; Xiang, Y.; Cohen, T.; Belford, A.K.; Huet, A.; Conway, J.F.; Sun, J.; Taylor, D.J.; et al. Potent neutralizing nanobodies resist convergent circulating variants of sars-cov-2 by targeting diverse and conserved epitopes. Nat. Commun. 2021, 12, 4676. [Google Scholar] [CrossRef]
- de Brevern, A.G.; Etchebest, C.; Hazout, S. Bayesian probabilistic approach for predicting backbone structures in terms of protein blocks. Proteins 2000, 41, 271–287. [Google Scholar] [CrossRef]
- Barnoud, J.; Santuz, H.; Craveur, P.; Joseph, A.P.; Jallu, V.; de Brevern, A.G.; Poulain, P. Pbxplore: A tool to analyze local protein structure and deformability with protein blocks. PeerJ 2017, 5, e4013. [Google Scholar] [CrossRef] [Green Version]
- Pompidor, G.; Zimmermann, S.; Loew, C.; Schneider, T. Engineered Nanobodies with a Lanthanide Binding Motif for Crystallographic Phasing. 2021. Available online: https://www.rcsb.org/structure/6XYF (accessed on 20 February 2022).
- Pieper, U.; Eswar, N.; Stuart, A.C.; Ilyin, V.A.; Sali, A. Modbase, a database of annotated comparative protein structure models. Nucleic Acids Res. 2002, 30, 255–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieper, U.; Webb, B.M.; Dong, G.Q.; Schneidman-Duhovny, D.; Fan, H.; Kim, S.J.; Khuri, N.; Spill, Y.G.; Weinkam, P.; Hammel, M.; et al. Modbase, a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res. 2014, 42, D336–D346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duhoo, Y.; Roche, J.; Trinh, T.T.N.; Desmyter, A.; Gaubert, A.; Kellenberger, C.; Cambillau, C.; Roussel, A.; Leone, P. Camelid nanobodies used as crystallization chaperones for different constructs of porm, a component of the type ix secretion system from porphyromonas gingivalis. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2017, 73, 286–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; McLellan, J.; Kwon, Y.; Schmidt, S.; Wu, X.; Zhou, T.; Yang, Y.; Zhang, B.; Forsman, A.; Weiss, R.; et al. Single-Headed Immunoglobulins Efficiently Penetrate Cd4-Binding Site and Effectively Neutralize Hiv-1. 2012. Available online: https://www.rcsb.org/structure/3r0m (accessed on 20 February 2022).
- Hinz, A.; Lutje Hulsik, D.; Forsman, A.; Koh, W.W.; Belrhali, H.; Gorlani, A.; de Haard, H.; Weiss, R.A.; Verrips, T.; Weissenhorn, W. Crystal structure of the neutralizing llama v(hh) d7 and its mode of hiv-1 gp120 interaction. PLoS ONE 2010, 5, e10482. [Google Scholar] [CrossRef] [Green Version]
- Studer, G.; Rempfer, C.; Waterhouse, A.M.; Gumienny, R.; Haas, J.; Schwede, T. Qmeandisco-distance constraints applied on model quality estimation. Bioinformatics 2020, 36, 1765–1771. [Google Scholar] [CrossRef]
- Mitchell, L.S.; Colwell, L.J. Comparative analysis of nanobody sequence and structure data. Proteins 2018, 86, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.M.; Brooks, C.L. X-ray crystal structure analysis of vhh-protein antigen complexes. Methods Mol. Biol. 2022, 2446, 513–530. [Google Scholar] [PubMed]
- Zuo, J.; Li, J.; Zhang, R.; Xu, L.; Chen, H.; Jia, X.; Su, Z.; Zhao, L.; Huang, X.; Xie, W. Institute collection and analysis of nanobodies (ican): A comprehensive database and analysis platform for nanobodies. BMC Genom. 2017, 18, 797. [Google Scholar] [CrossRef] [Green Version]
- Sang, Z.; Xiang, Y.; Bahar, I.; Shi, Y. Llamanade: An open-source computational pipeline for robust nanobody humanization. Structure 2022, 30, 418–429.e3. [Google Scholar] [CrossRef] [PubMed]
- Deszyński, P.; Młokosiewicz, J.; Volanakis, A.; Jaszczyszyn, I.; Castellana, N.; Bonissone, S.; Ganesan, R.; Krawczyk, K. Indi-integrated nanobody database for immunoinformatics. Nucleic Acids Res. 2022, 50, D1273–D1281. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.; Kumar, A.; Zhang, K.Y.J. Nbx: Machine learning-guided re-ranking of nanobody-antigen binding poses. Pharmaceuticals 2021, 14, 968. [Google Scholar] [CrossRef] [PubMed]
- Tahir, S.; Bourquard, T.; Musnier, A.; Jullian, Y.; Corde, Y.; Omahdi, Z.; Mathias, L.; Reiter, E.; Crépieux, P.; Bruneau, G.; et al. Accurate determination of epitope for antibodies with unknown 3d structures. mAbs 2021, 13, 1961349. [Google Scholar] [CrossRef]
- Bekker, G.J.; Ma, B.; Kamiya, N. Thermal stability of single-domain antibodies estimated by molecular dynamics simulations. Protein Sci. A Publ. Protein Soc. 2019, 28, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Che, T.; Majumdar, S.; Zaidi, S.A.; Ondachi, P.; McCorvy, J.D.; Wang, S.; Mosier, P.D.; Uprety, R.; Vardy, E.; Krumm, B.E.; et al. Structure of the nanobody-stabilized active state of the kappa opioid receptor. Cell 2018, 172, 55–67.e15. [Google Scholar] [CrossRef] [Green Version]
- Mohseni, A.; Molakarimi, M.; Taghdir, M.; Sajedi, R.H.; Hasannia, S. Exploring single-domain antibody thermostability by molecular dynamics simulation. J. Biomol. Struct. Dyn. 2019, 37, 3686–3696. [Google Scholar] [CrossRef]
- Soler, M.A.; Fortuna, S.; de Marco, A.; Laio, A. Binding affinity prediction of nanobody-protein complexes by scoring of molecular dynamics trajectories. Phys. Chem. Chem. Phys. PCCP 2018, 20, 3438–3444. [Google Scholar] [CrossRef]
- Zabetakis, D.; Shriver-Lake, L.C.; Olson, M.A.; Goldman, E.R.; Anderson, G.P. Experimental evaluation of single-domain antibodies predicted by molecular dynamics simulations to have elevated thermal stability. Protein Sci. A Publ. Protein Soc. 2019, 28, 1909–1912. [Google Scholar] [CrossRef] [PubMed]
- Narwani, T.J.; Craveur, P.; Shinada, N.K.; Floch, A.; Santuz, H.; Vattekatte, A.M.; Srinivasan, N.; Rebehmed, J.; Gelly, J.C.; Etchebest, C.; et al. Discrete analyses of protein dynamics. J. Biomol. Struct. Dyn. 2020, 38, 2988–3002. [Google Scholar] [CrossRef]
- McCammon, J.A.; Karplus, M. Internal motions of antibody molecules. Nature 1977, 268, 765–766. [Google Scholar] [CrossRef] [PubMed]
- Novotný, J.; Bruccoleri, R.; Newell, J.; Murphy, D.; Haber, E.; Karplus, M. Molecular anatomy of the antibody binding site. J. Biol. Chem. 1983, 258, 14433–14437. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vishwakarma, P.; Vattekatte, A.M.; Shinada, N.; Diharce, J.; Martins, C.; Cadet, F.; Gardebien, F.; Etchebest, C.; Nadaradjane, A.A.; de Brevern, A.G. VHH Structural Modelling Approaches: A Critical Review. Int. J. Mol. Sci. 2022, 23, 3721. https://doi.org/10.3390/ijms23073721
Vishwakarma P, Vattekatte AM, Shinada N, Diharce J, Martins C, Cadet F, Gardebien F, Etchebest C, Nadaradjane AA, de Brevern AG. VHH Structural Modelling Approaches: A Critical Review. International Journal of Molecular Sciences. 2022; 23(7):3721. https://doi.org/10.3390/ijms23073721
Chicago/Turabian StyleVishwakarma, Poonam, Akhila Melarkode Vattekatte, Nicolas Shinada, Julien Diharce, Carla Martins, Frédéric Cadet, Fabrice Gardebien, Catherine Etchebest, Aravindan Arun Nadaradjane, and Alexandre G. de Brevern. 2022. "VHH Structural Modelling Approaches: A Critical Review" International Journal of Molecular Sciences 23, no. 7: 3721. https://doi.org/10.3390/ijms23073721
APA StyleVishwakarma, P., Vattekatte, A. M., Shinada, N., Diharce, J., Martins, C., Cadet, F., Gardebien, F., Etchebest, C., Nadaradjane, A. A., & de Brevern, A. G. (2022). VHH Structural Modelling Approaches: A Critical Review. International Journal of Molecular Sciences, 23(7), 3721. https://doi.org/10.3390/ijms23073721