
Natterin-Induced Neutrophilia Is Dependent on cGAS/STING Activation via Type I IFN Signaling Pathway

 ,
, 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
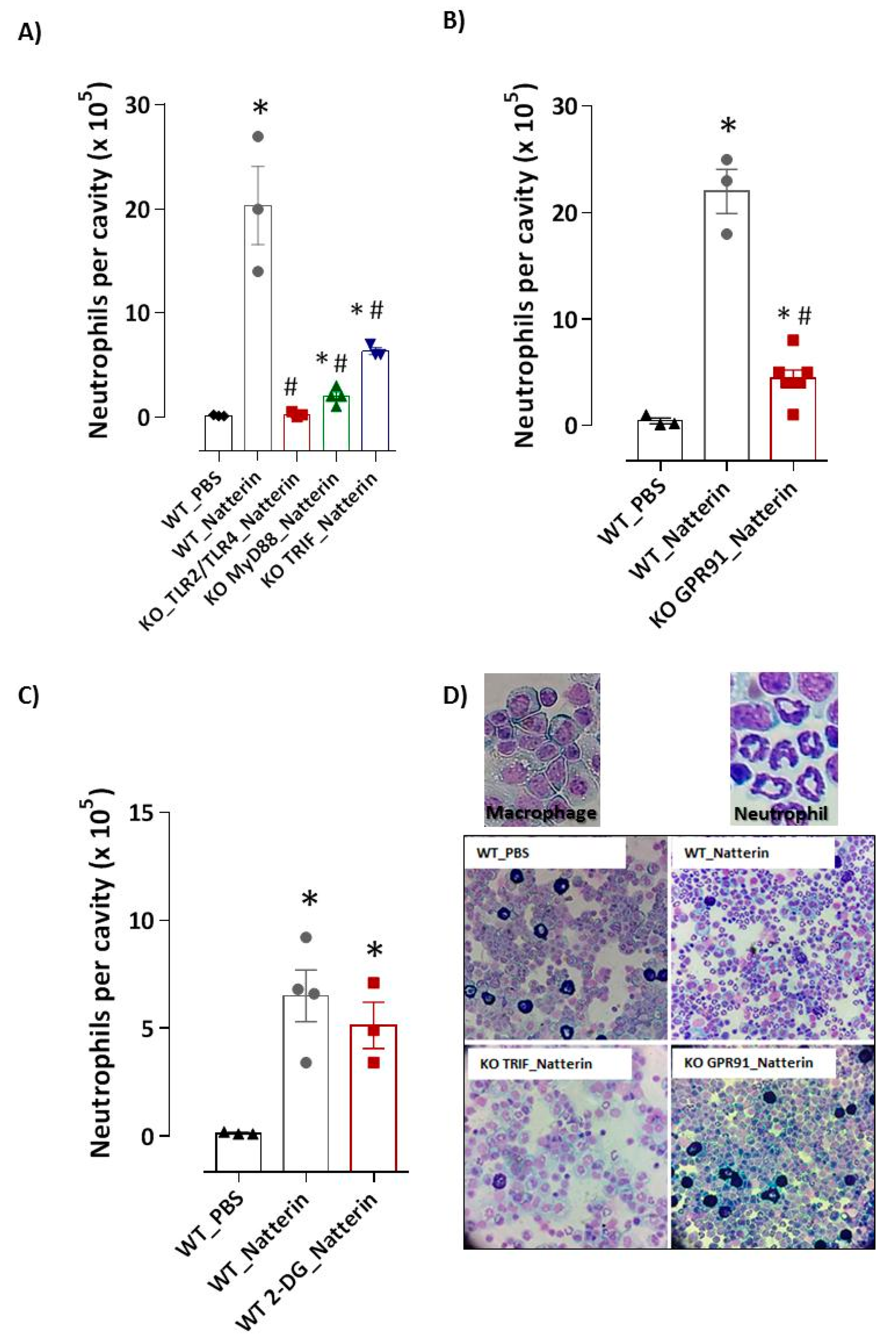
2.1. Natterin Induces Signals through TLR4 and MyD88/TRIF Adaptors
2.2. GPR91 Succinate Sensor Drives Neutrophilic Inflammation
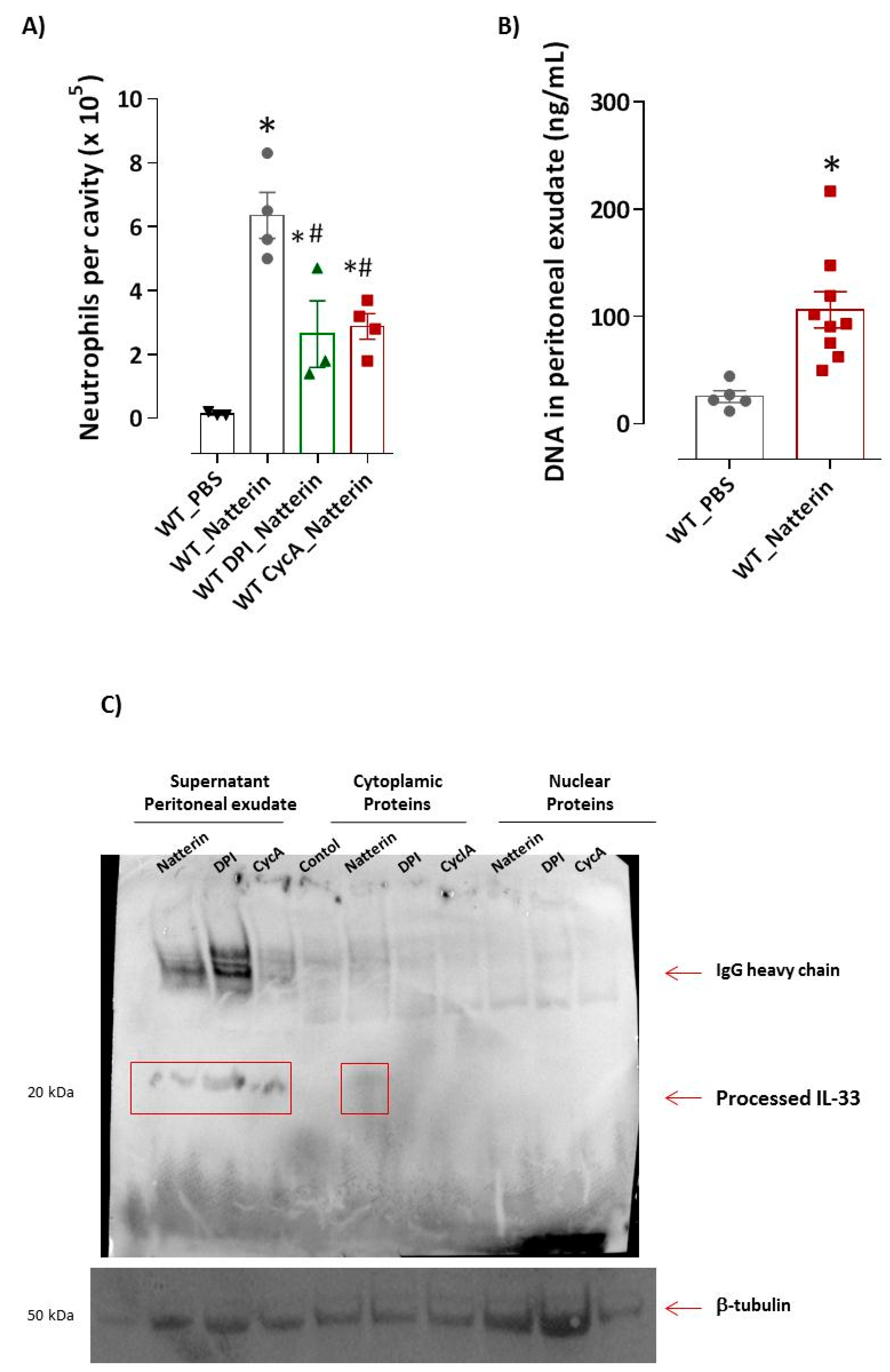
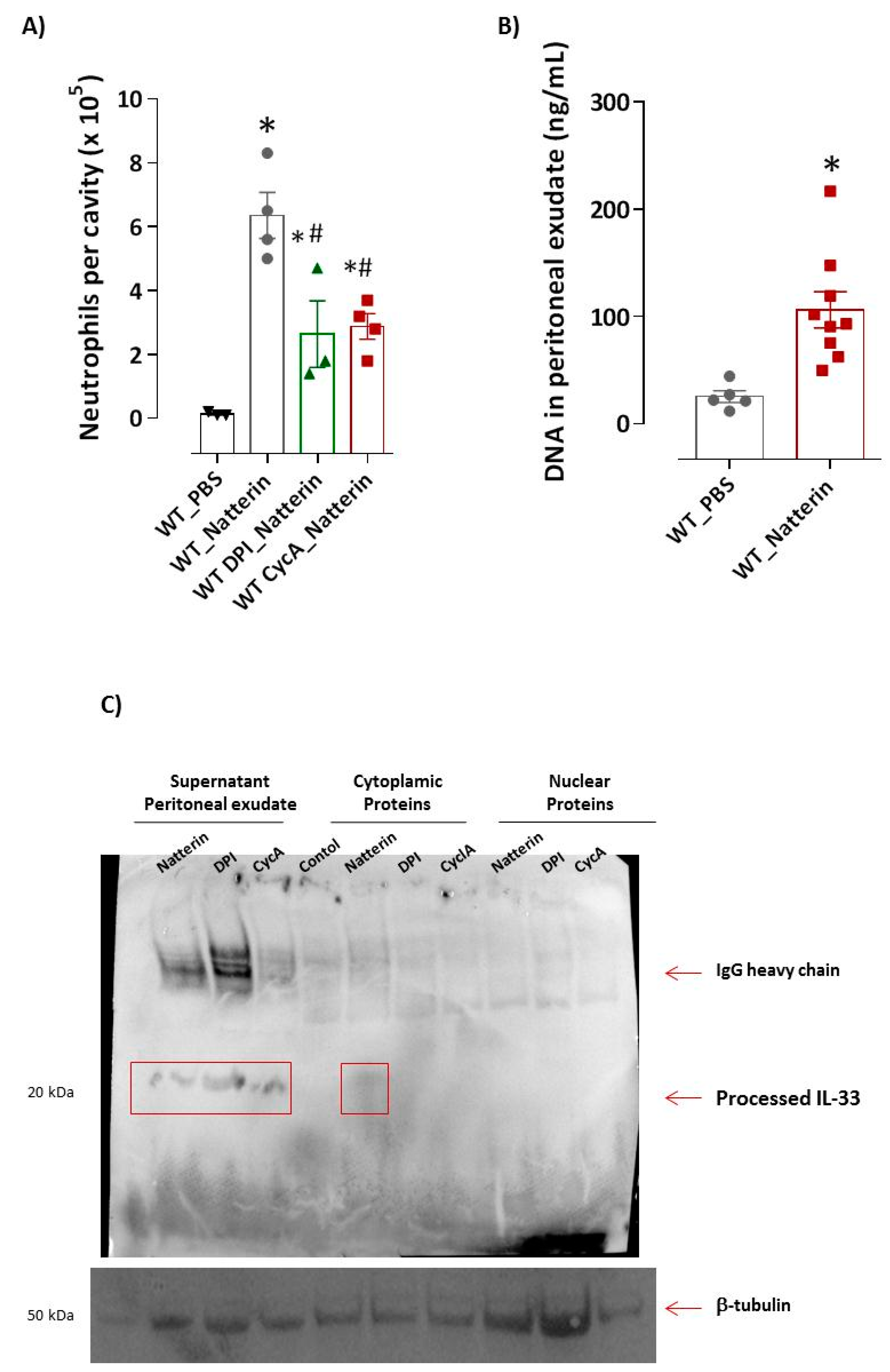
2.3. Mitochondrial Dysfunction Is Important for Natterin-Dependent Neutrophilic Recruitment
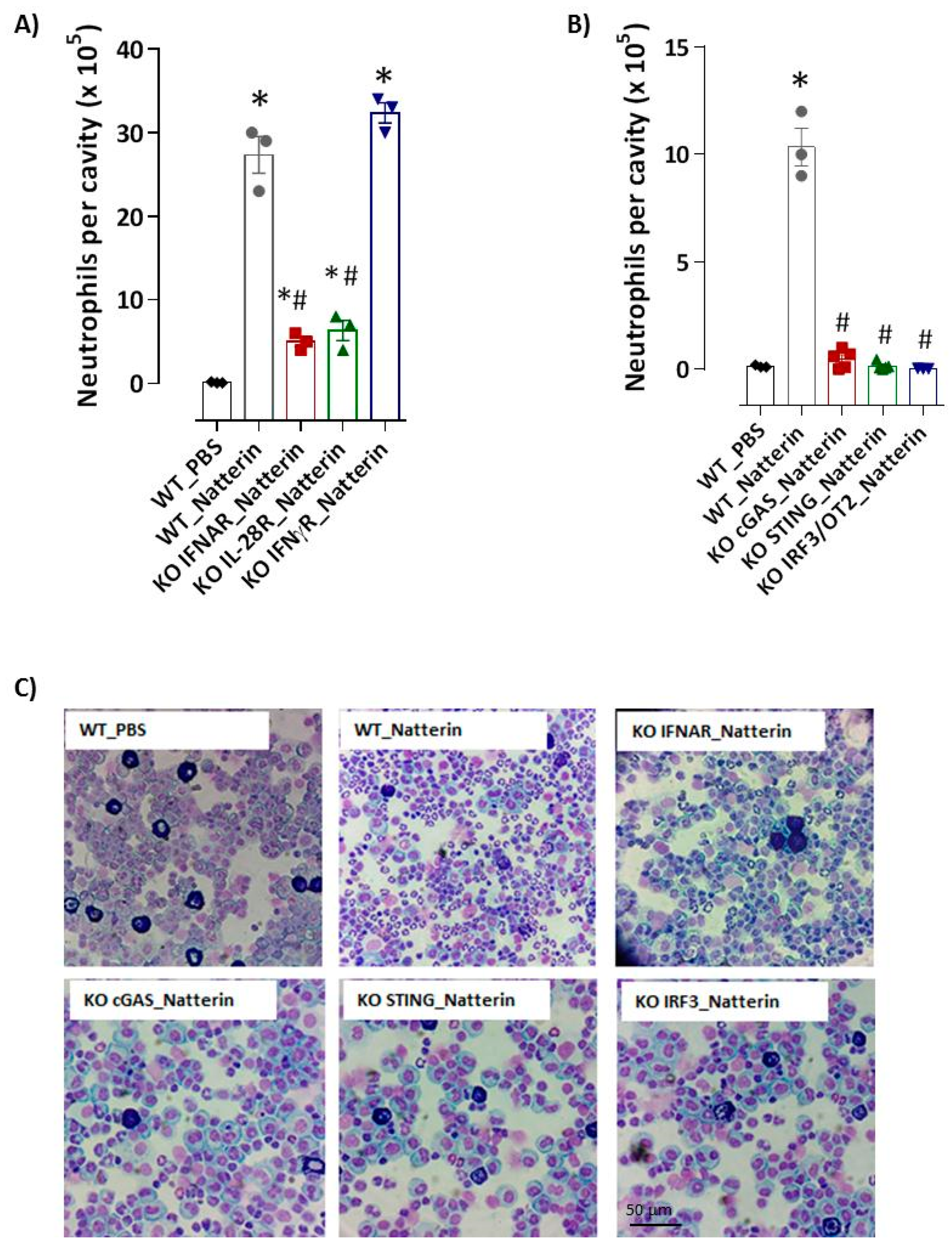
2.4. cGAS/STING/IRF3 via Type I IFN Axis Supports Natterin-Neutrophilic Inflammation
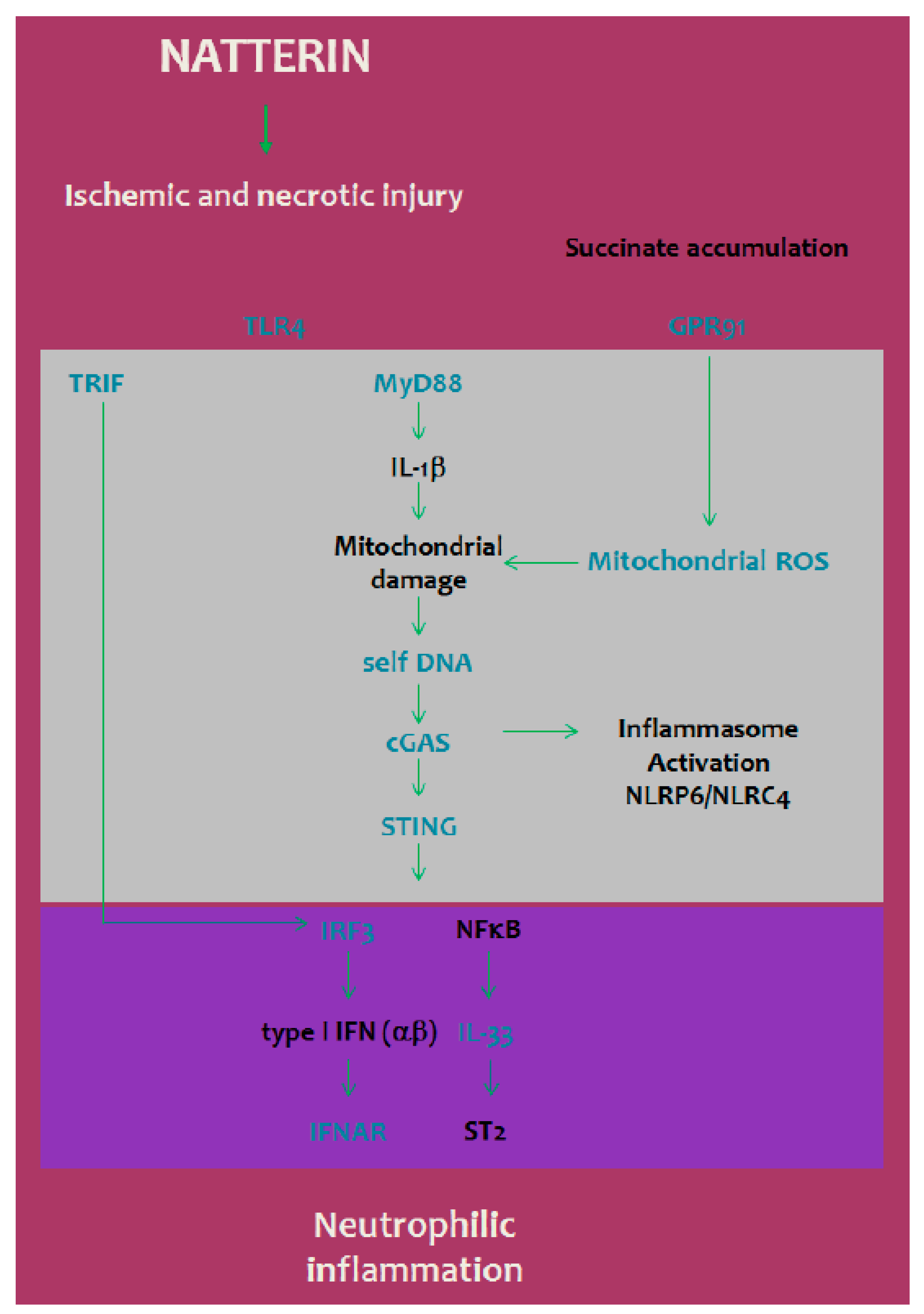
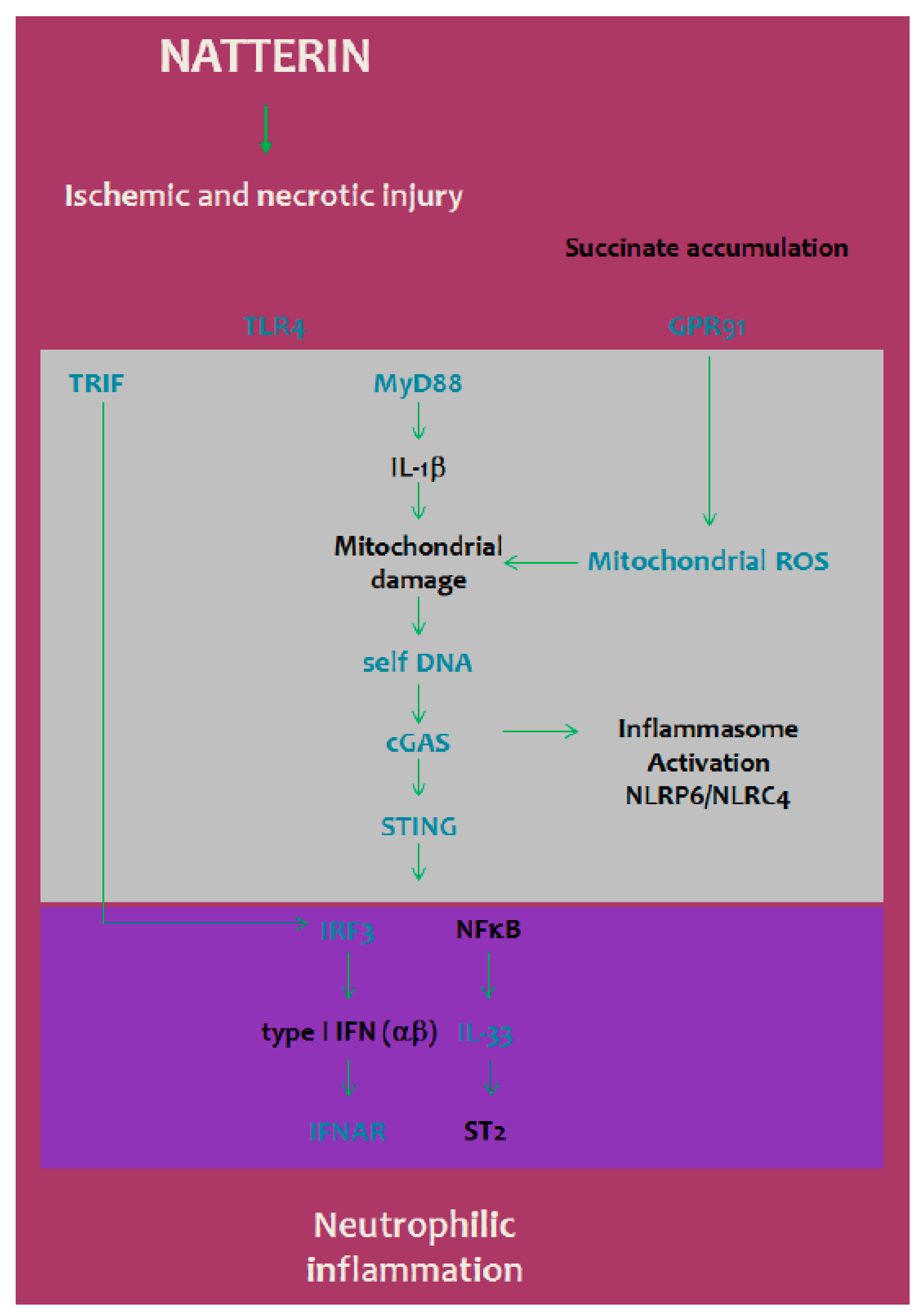
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Natterin Preparation
4.3. Acute Inflammation Induced by Natterin and Pharmacological Treatments
4.4. Peritoneal Cell Suspension Collection
4.5. Double-Stranded DNA Content Measurement
4.6. Western Blot
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Magalhães, G.S.; Junqueira-de-Azevedo, I.L.; Lopes-Ferreira, M.; Lorenzini, D.M.; Ho, P.L.; Moura-da-Silva, A.M. Transcriptome analysis of expressed sequence tags from the venom glands of the fish Thalassophryne nattereri. Biochimie 2006, 88, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, G.S.; Lopes-Ferreira, M.; Junqueira-De-Azevedo, I.L.M.; Spencer, P.J.; Araújo, M.S.; Portaro, F.C.V.; Ma, L.; Valente, R.H.; Juliano, L.; Fox, J.W.; et al. Natterins, a new class of proteins with kininogenase activity characterized from Thalassophryne nattereri fish venom. Biochimie 2005, 87, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Ferreira, M.; Emim, J.A.; Oliveira, V.; Puzer, L.; Cezari, M.H.; da Silva, M.; Juliano, L.; Lapa, A.J.; Souccar, C.; Moura-da-Silva, A.M. Kininogenase activity of Thalassophryne nattereri fish venom. Biochem. Pharmacol. 2004, 11, 2151–2157. [Google Scholar] [CrossRef]
- Szczesny, P.; Iacovache, I.; Muszewska, A.; Ginalski, K.; van der Goot, F.G.; Grynberg, M. Extending the aerolysin family: From bacteria to vertebrates. PLoS ONE 2011, 6, e20349. [Google Scholar] [CrossRef] [Green Version]
- Lima, C.; Disner, G.R.; Falcão, M.A.P.; Seni-Silva, A.C.; Maleski, A.L.A.; Marcolino-Souza, M.; Reis, M.C.; Lopes-Ferreira, M. The Natterin protein family diversity: A review on phylogeny, structure, and immune function. Toxins 2021, 13, 538. [Google Scholar] [CrossRef]
- Tamura, S.; Yamakawa, M.; Shiomi, K. Purification, characterization and cDNA cloning of two natterin-like toxins from the skin secretion of oriental catfish Plotosus lineatus. Toxicon 2011, 58, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Lima, C.; Falcao, M.A.P.; Andrade-Barros, A.I.; Seni-Silva, A.C.; Grund, L.Z.; Balogh, E.; Conceiçao, K.; Queniaux, V.F.; Ryffel, B.; Lopes-Ferreira, M. Natterin an aerolysin-like fish toxin drives IL-1β-dependent neutrophilic inflammation mediated by caspase-1 and caspase-11 activated by the inflammasome sensor NLRP6. Int. Immunopharmacol. 2021, 91, 107287. [Google Scholar] [CrossRef]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef]
- Swanson, K.V.; Junkins, R.D.; Kurkjian, C.J.; Holley-Guthrie, E.; Pendse, A.A.; El Morabiti, R.; Petrucelli, A.; Barber, G.N.; Benedict, C.A.; Ting, J.P. A noncanonical function of cGAMP in inflammasome priming and activation. J. Exp. Med. 2017, 4, 3611–3626. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Pistollato, F.; Abbadi, S.; Rampazzo, E.; Viola, G.; Della Puppa, A.; Cavallini, L.; Frasson, C.; Persano, L.; Panchision, D.M.; Basso, G. Hypoxia and succinate antagonize 2-deoxyglucose effects on glioblastoma. Biochem. Pharmacol. 2010, 15, 1517–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubic, T.; Lametschwandtner, G.; Jost, S.; Hinteregger, S.; Kund, J.; Carballido-Perrig, N.; Schwärzler, C.; Junt, T.; Voshol, H.; Meingassner, J.G.; et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat. Immunol. 2008, 9, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 11, 238–242. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 20, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Trush, M.A. Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem. Biophys. Res. Commun. 1998, 253, 295–299. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Krysko, D.V.; Agostinis, P.; Krysko, O.; Garg, A.D.; Bachert, C.; Lambrecht, B.N.; Vandenabeele, P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011, 32, 157–164. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Benmerzoug, S.; Ryffel, B.; Togbe, D.; Quesniaux, V.F.J. Self-DNA sensing in lung inflammatory diseases. Trends Immunol. 2019, 40, 719–734. [Google Scholar] [CrossRef]
- Martin, N.T.; Martin, M.U. Interleukin 33 is a guardian of barriers and a local alarmin. Nat. Immunol. 2016, 17, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.; Rochman, M.; Miracle, C.E.; Habel, J.E.; Brusilovsky, M.; Caldwell, J.M.; Rymer, J.K.; Rothenberg, M.E. Chromatin regulates IL-33 release and extracellular cytokine activity. Nat. Commun. 2014, 2018, 3244. [Google Scholar] [CrossRef]
- Georgakis, S.; Gkirtzimanaki, K.; Papadaki, G.; Gakiopoulou, H.; Drakos, E.; Eloranta, M.L.; Makridakis, M.; Kontostathi, G.; Zoidakis, J.; Baira, E.; et al. NETs decorated with bioactive IL-33 infiltrate inflamed tissues and induce IFN-α production in patients with SLE. JCI Insight 2021, 8, e147671. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.S. Caspase-11 Non-Canonical Inflammasome: Emerging Activator and Regulator of Infection-Mediated Inflammatory Responses. Int. J. Mol. Sci. 2020, 21, 2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.; Barber, G.N. STING signaling and host defense against microbial infection. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Majumdar, T.; Kessler, P.; Ozhegov, E.; Zhang, Y.; Chattopadhyay, S.; Barik, S.; Sen, G.C. STING requires the adaptor TRIF to trigger innate immune responses to microbial infection. Cell Host Microbe 2016, 14, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Polumuri, S.K.; Jayakar, G.G.; Shirey, K.A.; Roberts, Z.J.; Perkins, D.J.; Pitha, P.M.; Vogel, S.N. Transcriptional regulation of murine IL-33 by TLR and non-TLR agonists. J. Immunol. 2012, 1, 50–60. [Google Scholar] [CrossRef] [Green Version]
- Ozasa, K.; Temizoz, B.; Kusakabe, T.; Kobari, S.; Momota, M.; Coban, C.; Ito, S.; Kobiyama, K.; Kuroda, E.; Ishii, K.J. Cyclic GMP-AMP Triggers Asthma in an IL-33-Dependent Manner That Is Blocked by Amlexanox, a TBK1 Inhibitor. Front. Immunol. 2019, 26, 2212. [Google Scholar] [CrossRef]
- Nakayama, H.; Otsu, K. Mitochondrial DNA as an inflammatory mediator in cardiovascular diseases. Biochem. J. 2018, 475, 839–852. [Google Scholar] [CrossRef]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [Green Version]
- Härtlova, A.; Erttmann, S.F.; Raffi, F.A.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kröger, A.; Nilsson, J.A.; et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Komegae, E.N.; Grund, L.Z.; Lopes-Ferreira, M.; Lima, C. The longevity of th2 humoral response induced by proteases natterins requires the participation of long-lasting innate-like B cells and plasma cells in spleen. PLoS ONE 2013, 28, e67135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komegae, E.N.; Grund, L.Z.; Lopes-Ferreira, M.; Lima, C. TLR2, TLR4 and the MyD88 signaling are crucial for the in vivo generation and the longevity of long-lived antibody-secreting cells. PLoS ONE 2013, 8, e71185. [Google Scholar] [CrossRef] [PubMed]
- Chopra, A.K.; Xu, X.; Ribardo, D.; Gonzalez, M.; Kuhl, K.; Peterson, J.W.; Houston, C.W. The cytotoxic enterotoxin of Aeromonas hydrophila induces pro-inflammatory cytokine production and activates arachidonic acid metabolism in macrophages. Infect. Immun. 2000, 68, 2808–2818. [Google Scholar] [CrossRef] [Green Version]
- Abrami, L.; Fivaz, M.; Glauser, P.E.; Sugimoto, N.; Zurzolo, C.; van der Goot, F.G. Sensitivity of polarized epithelial cells to the pore-forming toxin aerolysin. Infect. Immun. 2003, 71, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 1, 640–643. [Google Scholar] [CrossRef]
- Chen, K.; Cagliani, J.; Aziz, M.; Tan, C.; Brenner, M.; Wang, P. Extracellular CIRP activates STING to exacerbate hemorrhagic shock. JCI Insight 2021, 22, e143715. [Google Scholar] [CrossRef]
- Chan, T.K.; Loh, X.Y.; Peh, H.Y.; Tan, W.N.F.; Tan, W.S.D.; Li, N.; Tay, I.J.J.; Wong, W.S.F.; Engelward, B.P. House dust mite-induced asthma causes oxidative damage and DNA double-strand breaks in the lungs. J. Allergy Clin. Immunol. 2016, 138, 84–96.e1. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Chen, L.; Liu, H.; Jin, Z.; Wu, Y.; Wu, Y.; Li, W.; Ying, S.; Chen, Z.; Shen, H.; et al. Airway Epithelial cGAS Is Critical for Induction of Experimental Allergic Airway Inflammation. J. Immunol. 2020, 15, 1437–1447. [Google Scholar] [CrossRef]
- Saikh, K.U. MyD88 and beyond: A perspective on MyD88-targeted therapeutic approach for modulation of host immunity. Immunol. Res. 2021, 69, 117–128. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aarreberg, L.D.; Esser-Nobis, K.; Driscoll, C.; Shuvarikov, A.; Roby, J.A.; Gale, M., Jr. Interleukin-1beta induces mtDNA release to activate innate immune signaling via cGAS-STING. Mol. Cell 2019, 74, 801–815.e6. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, H.; Inoue, T.; Ouchi, H.; Jao, T.M.; Inoue, R.; Nishi, H.; Fujii, R.; Ishidate, F.; Tanaka, T.; Tanaka, Y.; et al. Mitochondrial Damage Causes Inflammation via cGAS-STING Signaling in Acute Kidney Injury. Cell Rep. 2019, 29, 1261–1273.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, J.C.; Grund, L.Z.; Seibert, C.S.; Marques, E.E.; Soares, A.B.; Quesniaux, V.F.; Ryffel, B.; Lopes-Ferreira, M.; Lima, C. Stingray venom activates IL-33 producing cardiomyocytes, but not mast cell, to promote acute neutrophil-mediated injury. Sci. Rep. 2017, 11, 7912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nascimento, M.; Gombault, A.; Lacerda-Queiroz, N.; Panek, C.; Savigny, F.; Sbeity, M.; Bourinet, M.; Le Bert, M.; Riteau, N.; Ryffel, B.; et al. Self-DNA release and STING-dependent sensing drives inflammation to cigarette smoke in mice. Sci. Rep. 2019, 16, 14848. [Google Scholar] [CrossRef] [PubMed]
- Grund, L.Z.; Novaski, I.; Quesniaux, V.F.; Ryffel, B.; Lopes-Ferreira, M.; Lima, C. Neutrophils releasing IL-17A into NETs are essential to plasma cell differentiation in inflamed tissue dependent on IL-1R. Autoimmunity 2017, 50, 86–101. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lima, C.; Andrade-Barros, A.I.; Bernardo, J.T.G.; Balogh, E.; Quesniaux, V.F.; Ryffel, B.; Lopes-Ferreira, M. Natterin-Induced Neutrophilia Is Dependent on cGAS/STING Activation via Type I IFN Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 3600. https://doi.org/10.3390/ijms23073600
Lima C, Andrade-Barros AI, Bernardo JTG, Balogh E, Quesniaux VF, Ryffel B, Lopes-Ferreira M. Natterin-Induced Neutrophilia Is Dependent on cGAS/STING Activation via Type I IFN Signaling Pathway. International Journal of Molecular Sciences. 2022; 23(7):3600. https://doi.org/10.3390/ijms23073600
Chicago/Turabian StyleLima, Carla, Aline Ingrid Andrade-Barros, Jefferson Thiago Gonçalves Bernardo, Eniko Balogh, Valerie F. Quesniaux, Bernhard Ryffel, and Monica Lopes-Ferreira. 2022. "Natterin-Induced Neutrophilia Is Dependent on cGAS/STING Activation via Type I IFN Signaling Pathway" International Journal of Molecular Sciences 23, no. 7: 3600. https://doi.org/10.3390/ijms23073600
APA StyleLima, C., Andrade-Barros, A. I., Bernardo, J. T. G., Balogh, E., Quesniaux, V. F., Ryffel, B., & Lopes-Ferreira, M. (2022). Natterin-Induced Neutrophilia Is Dependent on cGAS/STING Activation via Type I IFN Signaling Pathway. International Journal of Molecular Sciences, 23(7), 3600. https://doi.org/10.3390/ijms23073600