
Specialized Pro-Resolving Lipid Mediators: New Therapeutic Approaches for Vascular Remodeling

 ,
,  , ,
, , 
Abstract
1. Introduction
2. Vascular Remodeling
3. Specialized Pro-Resolving Lipid Mediators (SPMs)
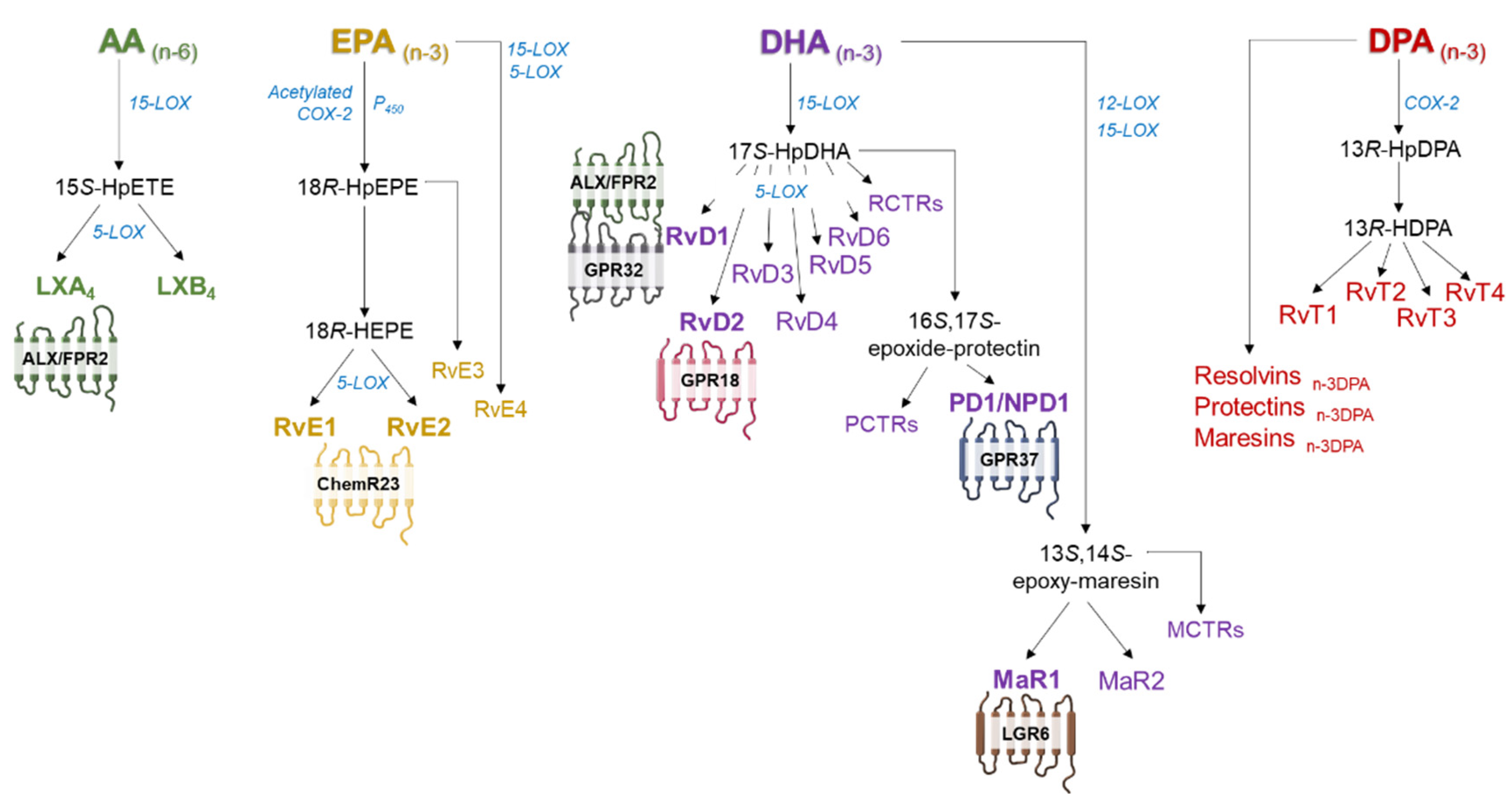
3.1. Biosynthesis of SPMs
3.2. Receptors for SPMs
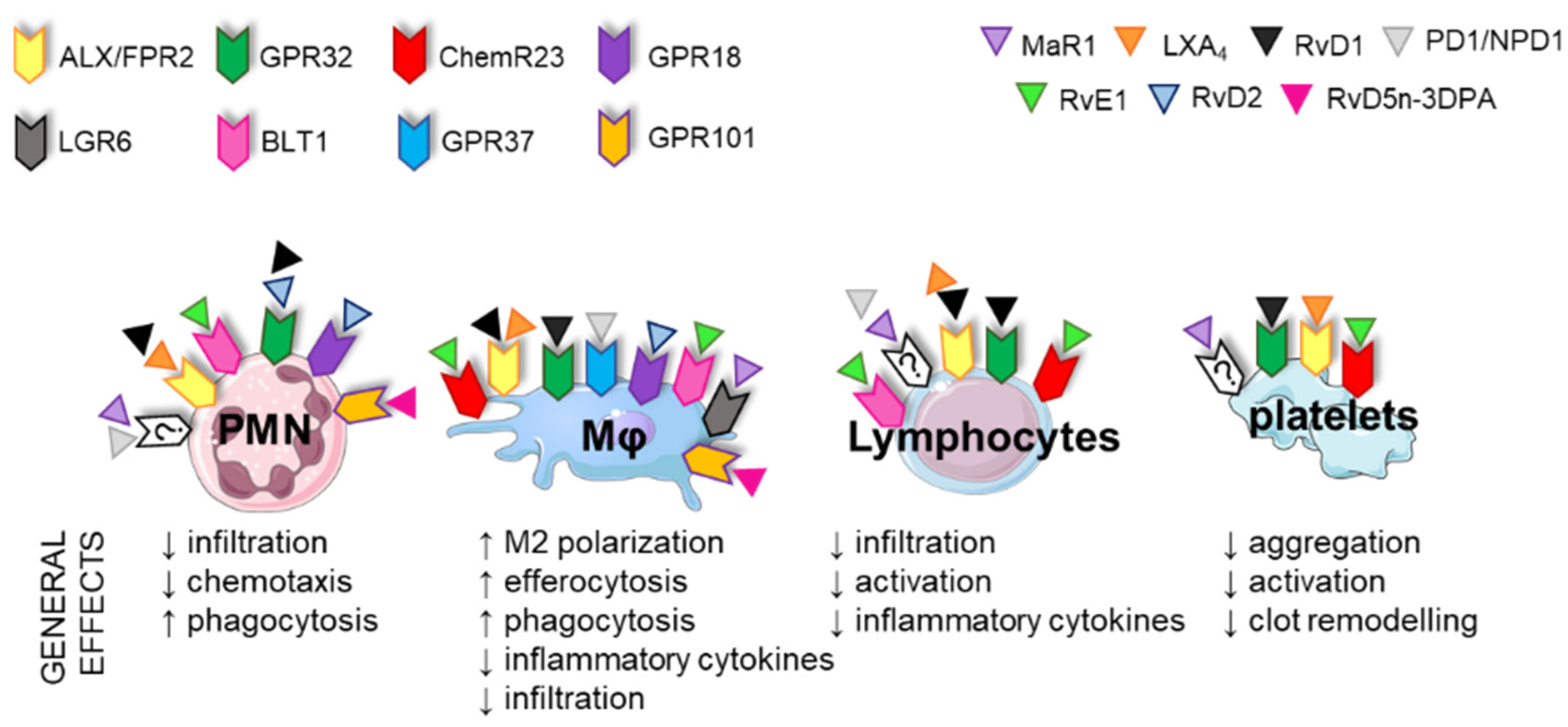
3.3. Immune Functions of SPMs
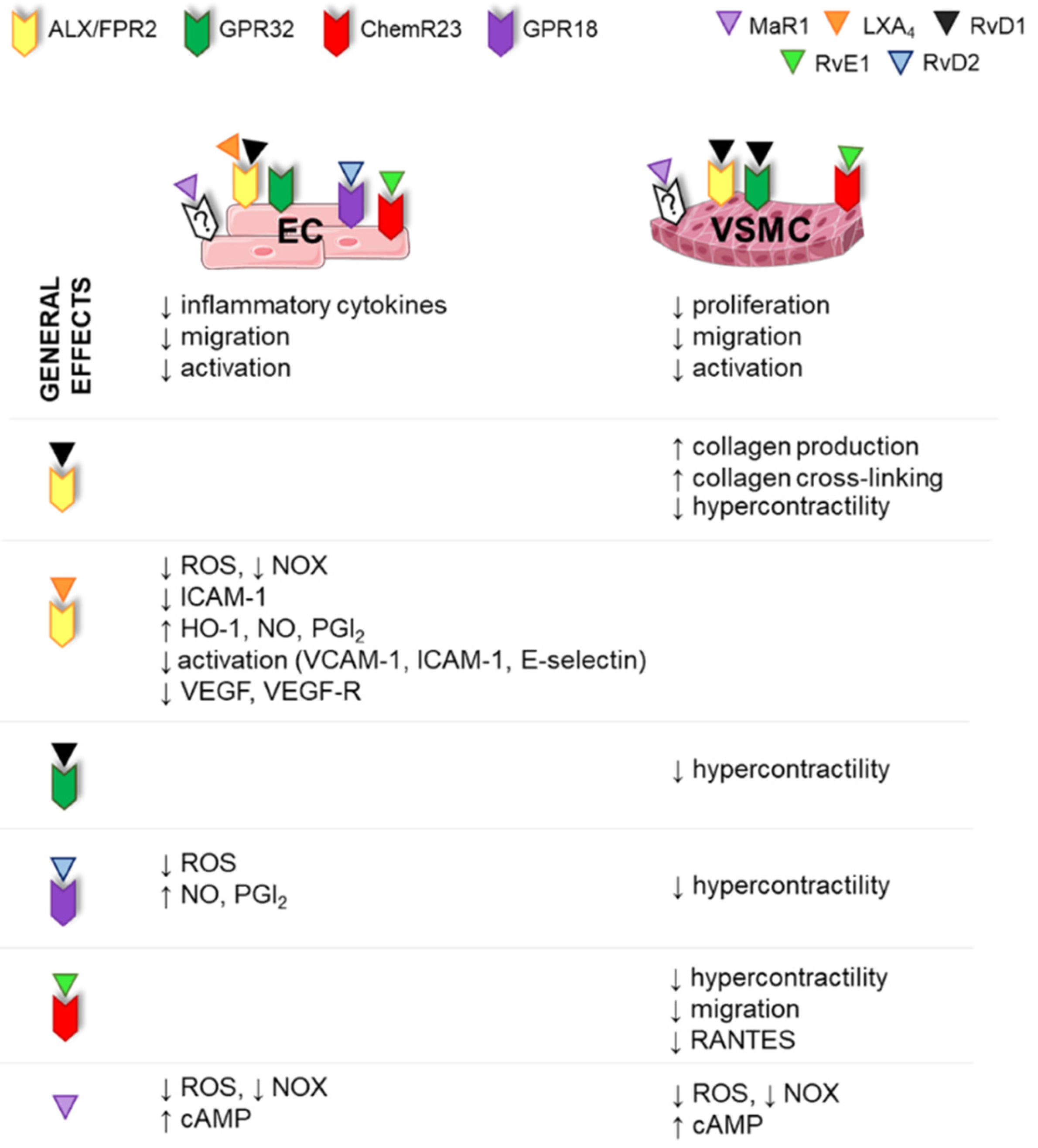
3.4. Vascular Functions of SPMs
3.4.1. SPM Effects in ECs
3.4.2. SPM Effects in VSMCs
4. SPMs in Cardiovascular Diseases
4.1. SPMs in Atherosclerosis
4.2. SPMs in Aneurysm
4.3. SPMs in Hyperplasia

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Approach | Target Cell and Action | Main Effects | Ref. | ||
|---|---|---|---|---|---|---|
| SPM Admin. | Genetic Modification | |||||
| Atherosclerosis | RvD1 | - | MØ | ↑ efferocytosis | Promoted plaque stability, suppressed plaque progression | [107] |
| VSMCs | unaltered No. | |||||
| RvD2, MaR1 | - | MØ | polarization to M2 | Reduced atheroprogression and promoted plaque stability | [106] | |
| VSMCs | ↑ collagen synthesis, ↑ No. | |||||
| RvE1 | - | MØ | ↓ foam cell infiltration | Reduced atheroprogression | [109] [110] | |
| - | ChemR23−/− | MØ | ↓ phagocytosis, ↑ oxLDL uptake | Increased atherosclerotic plaque size and necrotic core formation | [113] | |
| VSMCs | ↓ collagen ↓ proliferation | |||||
| Frp2−/− | MØ | ↓ infiltration | Reduced atheroprogression Impaired plaque stability | [95] | ||
| VSMCs | ↓ collagen synthesis | |||||
| - | Frp2−/− | Leukocytes | ↑recruitment | Enhanced atheroprogression | [114] | |
| AT-LXA4 | - | MØ | ↓ infiltration | Reduced atheroprogression ↓ systemic inflammation | [91] | |
| - | hGPR32 × Fpr2−/− | MØ, neutrophils, monocytes | ↓ aortic infiltration | Reduced atheroprogression, necrotic core, aortic inflammation | [115] | |
| Aneurysms | - | Frp2−/− | MØ, neutrophils | ↑ aortic infiltration | ↑ aortic dilation, elastin disruption and ↓ collagen deposition | [123] |
| RvD1, RvD2 | - | MØ | polarization to M2, ↓ pro-inflammatory cytokines | Decreased AAA formation | [124] | |
| Neutrophils | ↓ infiltration, ↓ NETosis | [125] | ||||
| MaR1 | - | MØ | ↑ efferocytosis | Decreased AAA formation | [126] | |
| Hyperplasia | RvD1, RvD2, RvE1, MaR1, PD1 | - | MØ, neutrophils | ↓ infiltration, polarization to M2 | Decreased neointima formation | [89] [90] [92,93,94] [132] |
| VSMCs | ↓ proliferation, ↓ migration | |||||
| AT-LXA4 | - | VSMCs | ↓ migration | Decreased neointima formation | [130] | |
| - | ChemR23−/− | Peritoneal MØ | ↑ inflammatory cytokines | Increased intimal hyperplasia | [131] | |
| VSMCs | ↓ proliferation | |||||
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Simionescu, M. Implications of early structural-functional changes in the endothelium for vascular disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Godo, S.; Shimokawa, H. Endothelial functions. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.G.; Coffman, T.M.; Wilcox, C.S. Pathophysiology of Hypertension: The Mosaic Theory and Beyond. Circ. Res. 2021, 128, 847–863. [Google Scholar] [CrossRef] [PubMed]
- Watson, T.; Goon, P.K.; Lip, G.Y. Endothelial progenitor cells, endothelial dysfunction, inflammation, and oxidative stress in hypertension. Antioxid. Redox Signal. 2008, 10, 1079–1088. [Google Scholar] [CrossRef]
- Kim, A.S.; Conte, M.S. Specialized pro-resolving lipid mediators in cardiovascular disease, diagnosis, and therapy. Adv. Drug Deliv. Rev. 2020, 159, 170–179. [Google Scholar] [CrossRef]
- Libby, P. Inflammation during the life cycle of the atherosclerotic plaque. Cardiovasc. Res. 2021, 117, 2525–2536. [Google Scholar] [CrossRef]
- Soehnlein, O.; Libby, P. Targeting inflammation in atherosclerosis—From experimental insights to the clinic. Nat. Rev. Drug Discov. 2021, 20, 589–610. [Google Scholar] [CrossRef]
- Yuan, Z.; Lu, Y.; Wei, J.; Wu, J.; Yang, J.; Cai, Z. Abdominal Aortic Aneurysm: Roles of Inflammatory Cells. Front. Immunol. 2021, 11, 609161. [Google Scholar] [CrossRef]
- Shimizu, K.; Mitchell, R.N.; Libby, P. Inflammation and cellular immune responses in abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 987–994. [Google Scholar] [CrossRef]
- Whiteford, J.R.; De Rossi, G.; Woodfin, A. Mutually Supportive Mechanisms of Inflammation and Vascular Remodeling. Int. Rev. Cell Mol. Biol. 2016, 326, 201–278. [Google Scholar]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.D.; Clish, C.B.; Schmidt, B.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat. Immunol. 2001, 2, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.S.; Desai, T.A.; Wu, B.; Schaller, M.; Werlin, E. Pro-resolving lipid mediators in vascular disease. J. Clin. Investig. 2018, 128, 3727–3735. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, G.H.; Dzau, V.J. The emerging concept of vascular remodeling. N. Engl. J. Med. 1994, 330, 1431–1438. [Google Scholar] [CrossRef]
- Gurung, R.; Choong, A.M.; Woo, C.C.; Foo, R.; Sorokin, V. Genetic and Epigenetic Mechanisms Underlying Vascular Smooth Muscle Cell Phenotypic Modulation in Abdominal Aortic Aneurysm. Int. J. Mol. Sci. 2020, 21, 6334. [Google Scholar] [CrossRef]
- Mulvany, M.J. Small artery remodeling in hypertension. Curr. Hypertens. Rep. 2002, 4, 49–55. [Google Scholar] [CrossRef]
- Van Varik, B.J.; Rennenberg, R.J.; Reutelingsperger, C.P.; Kroon, A.A.; de Leeuw, P.W.; Schurgers, L.J. Mechanisms of arterial remodeling: Lessons from genetic diseases. Front. Genet. 2012, 3, 290. [Google Scholar] [CrossRef]
- Schiffrin, E.L. Vascular remodeling in hypertension: Mechanisms and treatment. Hypertension 2012, 59, 367–374. [Google Scholar] [CrossRef]
- Briones, A.M.; Aras-López, R.; Alonso, M.J.; Salaices, M. Small artery remodeling in obesity and insulin resistance. Curr. Vasc. Pharmacol. 2014, 12, 427–437. [Google Scholar] [CrossRef]
- Mulvany, M.J. Small artery remodeling in hypertension: Causes, consequences and therapeutic implications. Med. Biol. Eng. Comput. 2008, 46, 461–467. [Google Scholar] [CrossRef]
- Nørrelund, H.; Christensen, K.L.; Samani, N.J.; Kimber, P.; Mulvany, M.J.; Korsgaard, N. Early narrowed afferent arteriole is a contributor to the development of hypertension. Hypertension 1994, 24, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Briones, A.M.; Xavier, F.E.; Arribas, S.M.; González, M.C.; Rossoni, L.V.; Alonso, M.J.; Salaices, M. Alterations in structure and mechanics of resistance arteries from ouabain-induced hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H193–H201. [Google Scholar] [CrossRef] [PubMed]
- Intengan, H.D.; Schiffrin, E.L. Vascular remodeling in hypertension: Roles of apoptosis, inflammation, and fibrosis. Hypertension 2001, 38 (3 Pt 2), 581–587. [Google Scholar] [CrossRef]
- Dorrance, A.M.; Matin, N.; Pires, P.W. The effects of obesity on the cerebral vasculature. Curr. Vasc. Pharmacol. 2014, 12, 462–472. [Google Scholar] [CrossRef]
- Cahill, P.A.; Redmond, E.M. Vascular endothelium—Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, V.; Vickneson, K.; Kofidis, T.; Woo, C.C.; Lin, X.Y.; Foo, R.; Shanahan, C.M. Role of Vascular Smooth Muscle Cell Plasticity and Interactions in Vessel Wall Inflammation. Front. Immunol. 2020, 11, 599415. [Google Scholar] [CrossRef] [PubMed]
- Nosalski, R.; Guzik, T.J. Perivascular adipose tissue inflammation in vascular disease. Br. J. Pharmacol. 2017, 174, 3496–3513. [Google Scholar] [CrossRef] [PubMed]
- Villacorta, L.; Chang, L. The role of perivascular adipose tissue in vasoconstriction, arterial stiffness, and aneurysm. Horm. Mol. Biol. Clin. Investig. 2015, 21, 137–147. [Google Scholar] [CrossRef]
- Hu, H.; Garcia-Barrio, M.; Jiang, Z.-S.; Chen, Y.E.; Chang, L. Roles of Perivascular Adipose Tissue in Hypertension and Atherosclerosis. Antioxid. Redox Signal. 2021, 34, 736–749. [Google Scholar] [CrossRef]
- Canetti, C.; Leung, B.P.; Culshaw, S.; McInnes, I.; Cunha, F.; Liew, F. IL-18 enhances collagen-induced arthritis by recruiting neutrophils via TNF-alpha and leukotriene B4. J. Immunol. 2003, 171, 1009–1015. [Google Scholar] [CrossRef]
- Chou, R.; Kim, N.D.; Sadik, C.D.; Seung, E.; Lan, Y.; Byme, M.H.; Haribabu, B.; Iwakura, Y.; Luster, A.D. Lipid-Cytokine-Chemokine Cascade Drives Neutrophil Recruitment in a Murine Model of Inflammatory Arthritis. Immunity 2011, 33, 266–278. [Google Scholar] [CrossRef]
- Bäck, M.; Bu, D.X.; Bränström, R.; Sheikine, Y.; Yan, Z.Q.; Hansson, G.K. Leukotriene B4 signaling through NF-kappaB-dependent BLT1 receptors on vascular smooth muscle cells in atherosclerosis and intimal hyperplasia. Proc. Natl. Acad. Sci. USA 2005, 102, 17501–17506. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zukas, A.M.; Hui, Y.; Ricciotti, E.; Puré, E.; Fitzgerald, G.A. Deletion of microsomal prostaglandin E synthase-1 augments prostacyclin and retards atherogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 14507–14512. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Lee, E.; Song, W.; Ricciotti, E.; Rader, D.J.; Lawson, J.A.; Puré, E.; FitzGerald, G.A. Microsomal prostaglandin E synthase-1 deletion suppresses oxidative stress and angiotensin II-induced abdominal aortic aneurysm formation. Circulation 2008, 117, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Ihida-Stansbury, K.; Kothapalli, D.; Tamby, M.C.; Yu, Z.; Chen, L.; Grant, G.; Cheng, Y.; Lawson, J.A.; Assoian, R.K.; et al. Microsomal prostaglandin E2 synthase-1 modulates the response to vascular injury. Circulation 2011, 123, 631–639. [Google Scholar] [CrossRef]
- Motwani, M.P.; Colas, R.A.; George, M.J.; Flint, J.D.; Dalli, J.; Richard-Loendt, A.; De Maeyer, R.P.; Serhan, C.N.; Gilroy, D.W. Pro-resolving mediators promote resolution in a human skin model of UV-killed Escherichia coli-driven acute inflammation. JCI Insight 2018, 3, e94463. [Google Scholar] [CrossRef]
- Sansbury, B.E.; Spite, M. Resolution of Acute Inflammation and the Role of Resolvins in Immunity, Thrombosis, and Vascular Biology. Circ. Res. 2016, 119, 113–130. [Google Scholar] [CrossRef]
- Serhan, C.N.; Clish, C.B.; Brannon, J.; Colgan, S.P.; Chiang, N.; Gronert, K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 2000, 192, 1197–1204. [Google Scholar] [CrossRef]
- Spite, M.; Serhan, C.N. Novel lipid mediators promote resolution of acute inflammation: Impact of aspirin and statins. Circ. Res. 2010, 107, 1170–1184. [Google Scholar] [CrossRef]
- Serhan, C.N.; Libreros, S.; Nshimiyimana, R. E-series resolvin metabolome, biosynthesis and critical role of stereochemistry of specialized pro-resolving mediators (SPMs) in inflammation-resolution: Preparing SPMs for long COVID-19, human clinical trials, and targeted precision nutrition. Semin. Immunol. 2022, 101597. [Google Scholar] [CrossRef]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef]
- Markworth, J.F.; Kaur, G.; Miller, E.G.; Larsen, A.E.; Sinclair, A.J.; Maddipati, K.R.; Cameron-Smith, D. Divergent shifts in lipid mediator profile following supplementation with n-3 docosapentaenoic acid and eicosapentaenoic acid. FASEB J. 2016, 30, 3714–3725. [Google Scholar] [CrossRef]
- Dalli, J.; Chiang, N.; Serhan, C.N. Elucidation of novel 13-series resolvins that increase with atorvastatin and clear infections. Nat. Med. 2015, 21, 1071–1075. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Dalli, J. New pro-resolving n-3 mediators bridge resolution of infectious inflammation to tissue regeneration. Mol. Asp. Med. 2018, 64, 1–17. [Google Scholar] [CrossRef]
- Abdulnour, R.E.; Dalli, J.; Colby, J.K.; Krishnamoorthy, N.; Timmons, J.Y.; Tan, S.H.; Colas, R.A.; Petasis, N.A.; Serhan, C.N.; Levy, B.D. Maresin 1 biosynthesis during platelet-neutrophil interactions is organ-protective. Proc. Natl. Acad. Sci. USA 2014, 111, 16526–16531. [Google Scholar] [CrossRef]
- Serhan, C.N.; Yacoubian, S.; Yang, R. Anti-inflammatory and proresolving lipid mediators. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 279–312. [Google Scholar] [CrossRef]
- Oh, S.F.; Dona, M.; Fredman, G.; Krishnamoorthy, S.; Irimia, D.; Serhan, C.N. Resolvin E2 formation and impact in inflammation resolution. J. Immunol. 2012, 188, 4527–4534. [Google Scholar] [CrossRef]
- Bang, S.; Xie, Y.K.; Zhang, Z.J.; Wang, Z.; Xu, Z.Z.; Ji, R.R. GPR37 regulates macrophage phagocytosis and resolution of inflammatory pain. J. Clin. Investig. 2018, 128, 3568–3582. [Google Scholar] [CrossRef]
- Chiang, N.; Libreros, S.; Norris, P.C.; De La Rosa, X.; Serhan, C.N. Maresin 1 activates LGR6 receptor promoting phagocyte immunoresolvent functions. J. Clin. Investig. 2019, 129, 5294–5311. [Google Scholar] [CrossRef]
- Flak, M.B.; Koenis, D.S.; Sobrino, A.; Smith, J.; Pistorius, K.; Palmas, F.; Dalli, J. GPR101 mediates the pro-resolving actions of RvD5n-3 DPA in arthritis and infections. J. Clin. Investig. 2020, 30, 359–373. [Google Scholar] [CrossRef]
- Chiang, N.; Fredman, G.; Bäckhed, F.; Oh, S.F.; Vickery, T.; Schmidt, B.A.; Serhan, C.N. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 2012, 484, 524–528. [Google Scholar] [CrossRef]
- Dalli, J.; Winkler, J.W.; Colas, R.A.; Arnardottir, H.; Cheng, C.Y.C.; Chiang, N.; Petasis, N.A.; Serhan, C.N. Resolvin D3 and aspirin-triggered resolvin D3 are potent immunoresolvents. Chem. Biol. 2013, 20, 188–201. [Google Scholar] [CrossRef]
- Arita, M.; Ohira, T.; Sun, Y.-P.; Elangovan, S.; Chiang, N.; Serhan, C.N. Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J. Immunol. 2007, 178, 3912–3917. [Google Scholar] [CrossRef]
- Pirault, J.; Bäck, M. Lipoxin and resolvin receptors transducing the resolution of inflammation in cardiovascular disease. Front. Pharmacol. 2018, 9, 1273. [Google Scholar] [CrossRef]
- Colas, R.A.; Dalli, J.; Chiang, N.; Vlasakov, I.; Sanger, J.M.; Riley, I.R.; Serhan, C.N. Identification and Actions of the Maresin 1 Metabolome in Infectious Inflammation. J. Immunol. 2016, 197, 4444–4452. [Google Scholar] [CrossRef]
- Tulowiecka, N.; Kotlęga, D.; Prowans, P.; Szczuko, M. The role of resolvins: EPA and DHA derivatives can be useful in the prevention and treatment of ischemic stroke. Int. J. Mol. Sci. 2020, 21, 7628. [Google Scholar] [CrossRef]
- Kozłowska, H.; Malinowska, B.; Baranowska-Kuczko, M.; Kusaczuk, M.; Nesterowicz, M.; Kozłowski, M.; Müller, C.E.; Kieć-Kononowicz, K.; Schlicker, E. GPR18-Mediated Relaxation of Human Isolated Pulmonary Arteries. Int. J. Mol. Sci. 2022, 23, 1427. [Google Scholar] [CrossRef]
- Zuo, G.; Zhang, D.; Mu, R.; Shen, H.; Li, X.; Wang, Z.; Li, H.; Chen, G. Resolvin D2 protects against cerebral ischemia/reperfusion injury in rats. Mol. Brain 2018, 11, 9. [Google Scholar] [CrossRef]
- De Palma, G.; Castellano, G.; Del Prete, A.; Sozzani, S.; Fiore, N.; Loverre, A.; Parmentier, M.; Gesualdo, L.; Grandaliano, G.; Schena, F. The possible role of ChemR23/Chemerin axis in the recruitment of dendritic cells in lupus nephritis. Kidney Int. 2011, 79, 1228–1235. [Google Scholar] [CrossRef]
- Rodríguez-Penas, D.; Feijóo-Bandín, S.; García-Rúa, V.; Mosquera-Leal, A.; Durán, D.; Varela, A.; Portolés, M.; Roselló-Lletí, E.; Rivera, M.; Diéguez, C.; et al. The Adipokine Chemerin Induces Apoptosis in Cardiomyocytes. Cell. Physiol. Biochem. 2015, 37, 176–192. [Google Scholar] [CrossRef]
- Hiram, R.; Rizcallah, E.; Marouan, S.; Sirois, C.; Sirois, M.; Morin, C.; Fortin, S.; Rousseau, E. Resolvin E1 normalizes contractility, Ca2+ sensitivity and smooth muscle cell migration rate in TNF-α- and IL-6-pretreated human pulmonary arteries. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L776–L788. [Google Scholar] [CrossRef]
- Ho, K.J.; Spite, M.; Owens, C.D.; Lancero, H.; Kroemer, A.H.K.; Pande, R.; Creager, M.A.; Serhan, C.N.; Conte, M.S. Aspirin-triggered lipoxin and resolvin E1 modulate vascular smooth muscle phenotype and correlate with peripheral atherosclerosis. Am. J. Pathol. 2010, 177, 2116–2123. [Google Scholar] [CrossRef]
- Jun, L.; Lin-Lin, S.; Hui, S. Chemerin promotes microangiopathy in diabetic retinopathy via activation of ChemR23 in rat primary microvascular endothelial cells. Mol. Vis. 2021, 27, 575–587. [Google Scholar]
- Yokomizo, T. Two distinct leukotriene B4 receptors, BLT1 and BLT2. J. Biochem. 2015, 157, 65–71. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Dalli, J. The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution. Semin. Immunol. 2015, 27, 200–215. [Google Scholar] [CrossRef]
- Dalli, J.; Serhan, C.N. Identification and structure elucidation of the pro-resolving mediators provides novel leads for resolution pharmacology. Br. J. Pharmacol. 2019, 176, 1024–1037. [Google Scholar] [CrossRef]
- Chiang, N.; Dalli, J.; Colas, R.A.; Serhan, C.N. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J. Exp. Med. 2015, 212, 1203–1217. [Google Scholar] [CrossRef]
- Chiurchiù, V.; Leuti, A.; Dalli, J.; Jacobsson, A.; Battistini, L.; Maccarrone, M.; Serhan, C.N. Proresolving lipid mediators resolvin D1, resolvin D2, and maresin 1 are critical in modulating T cell responses. Sci. Transl. Med. 2016, 8, 353ra111. [Google Scholar] [CrossRef]
- Perez-Hernandez, J.; Chiurchiù, V.; Perruche, S.; You, S. Regulation of T-Cell Immune Responses by Pro-Resolving Lipid Mediators. Front. Immunol. 2021, 12, 768133. [Google Scholar] [CrossRef]
- Lannan, K.L.; Spinelli, S.L.; Blumberg, N.; Phipps, R.P. Maresin 1 induces a novel pro-resolving phenotype in human platelets. J. Thromb. Haemost. 2017, 15, 802–813. [Google Scholar] [CrossRef]
- Elajami, T.K.; Colas, R.A.; Dalli, J.; Chiang, N.; Serhan, C.N.; Welty, F.K. Specialized proresolving lipid mediators in patients with coronary artery disease and their potential for clot remodeling. FASEB J. 2016, 30, 2792–2801. [Google Scholar] [CrossRef]
- Börgeson, E.; Lönn, J.; Bergström, I.; Brodin, V.P.; Ramström, S.; Nayeri, F.; Särndahl, E.; Bengtsson, T. Lipoxin A4 inhibits porphyromonas gingivalis-induced aggregation and reactive oxygen species production by modulating neutrophil-platelet interaction and CD11b expression. Infect. Immun. 2011, 79, 1489–1497. [Google Scholar] [CrossRef]
- Chen, P.; Fenet, B.; Michaud, S.; Tomczyk, N.; Véricel, E.; Lagarde, M.; Guichardant, M. Full characterization of PDX, a neuroprotectin/protectin D1 isomer, which inhibits blood platelet aggregation. FEBS Lett. 2009, 583, 3478–3484. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Filep, J.G.; Zouki, C.; Petasis, N.A.; Hachicha, M.; Serhan, C.N. Anti-inflammatory actions of lipoxin A(4) stable analogs are demonstrable in human whole blood: Modulation of leukocyte adhesion molecules and inhibition of neutrophil-endothelial interactions. Blood 1999, 94, 4132–4142. [Google Scholar] [CrossRef]
- Chatterjee, A.; Sharma, A.; Chen, M.; Toy, R.; Mottola, G.; Conte, M.S. The pro-resolving lipid mediator maresin 1 (MaR1) attenuates inflammatory signaling pathways in vascular smooth muscle and endothelial cells. PLoS ONE 2014, 9, e113480. [Google Scholar] [CrossRef]
- Chattopadhyay, R.; Raghavan, S.; Rao, G.N. Resolvin D1 via prevention of ROS-mediated SHP2 inactivation protects endothelial adherens junction integrity and barrier function. Redox Biol. 2017, 12, 438–455. [Google Scholar] [CrossRef]
- Zhang, M.J.; Sansbury, B.E.; Hellmann, J.; Baker, J.F.; Guo, L.; Parmer, C.M.; Prenner, J.C.; Conklin, D.J.; Bhatnagar, A.; Creager, M.A.; et al. Resolvin D2 enhances postischemic revascularization while resolving inflammation. Circulation 2016, 134, 666–680. [Google Scholar] [CrossRef]
- Spite, M.; Norling, L.V.; Summers, L.; Yang, R.; Cooper, D.; Petasis, N.A.; Flower, R.J.; Perretti, M.; Serhan, C.N. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 2009, 461, 1287–1291. [Google Scholar] [CrossRef]
- Paul-Clark, M.J.; van Cao, T.; Moradi-Bidhendi, N.; Cooper, D.; Gilroy, D.W. 15-epi-lipoxin A4–mediated Induction of Nitric Oxide Explains How Aspirin Inhibits Acute Inflammation. J. Exp. Med. 2004, 200, 69–78. [Google Scholar] [CrossRef]
- Brezinski, M.; Gimbrone, M.A.; Nicolau, K.; Serhan, C.N. Lipoxins stimulate prostacyclin generation by human endothelial cells. FEBS Lett. 1989, 245, 167–172. [Google Scholar] [CrossRef]
- Nascimento-Silva, V.; Arruda, M.A.; Barja-Fidalgo, C.; Fierro, I.M. Aspirin-triggered lipoxin A4 blocks reactive oxygen species generation in endothelial cells: A novel antioxidative mechanism. Thromb. Haemost. 2007, 97, 88–98. [Google Scholar] [PubMed]
- Nascimento-Silva, V.; Arruda, M.A.; Barja-Fidalgo, C.; Villela, C.G.; Fierro, I.M. Novel lipid mediator aspirin-triggered lipoxin A4 induces heme oxygenase-1 in endothelial cells. American journal of physiology. Cell Physiol. 2005, 289, C557–C563. [Google Scholar] [CrossRef]
- Souza, M.C.; Pádua, T.A.; Torres, N.D.; Souza Costa, M.F.; Candéa, A.P.; Maramaldo, T.; Seito, L.N.; Penido, C.; Estato, V.; Antunes, B.; et al. Lipoxin A4 attenuates endothelial dysfunction during experimental cerebral malaria. Int. Immunopharmacol. 2015, 24, 400–407. [Google Scholar] [CrossRef]
- Baker, N.; O’Meara, S.J.; Scannell, M.; Maderna, P.; Godson, C. Lipoxin A4: Anti-inflammatory and anti-angiogenic impact on endothelial cells. J. Immunol. 2009, 182, 3819–3826. [Google Scholar] [CrossRef]
- Cezar-de-Mello, P.F.; Nascimento-Silva, V.; Villela, C.G.; Fierro, I.M. Aspirin-triggered Lipoxin A4 inhibition of VEGF-induced endothelial cell migration involves actin polymerization and focal adhesion assembly. Oncogene 2006, 25, 122–129. [Google Scholar] [CrossRef][Green Version]
- Fierro, I.M.; Kutok, J.L.; Serhan, C.N. Novel lipid mediator regulators of endothelial cell proliferation and migration: Aspirin-triggered-15R-lipoxin A(4) and lipoxin A(4). J. Pharmacol. Exp. Ther. 2002, 300, 385–392. [Google Scholar] [CrossRef]
- Maekawa, T.; Hosur, K.; Abe, T.; Kantarci, A.; Ziogas, A.; Wang, B.; Van Dyke, T.E.; Chavakis, T.; Hajishengallis, G. Antagonistic effects of IL-17 and D-resolvins on endothelial Del-1 expression through a GSK-3β-C/EBPβ pathway. Nat. Commun. 2015, 6, 8272. [Google Scholar] [CrossRef]
- Akagi, D.; Chen, M.; Toy, R.; Chatterjee, A.; Conte, M.S. Systemic delivery of proresolving lipid mediators resolvin D2 and maresin 1 attenuates intimal hyperplasia in mice. FASEB J. 2015, 29, 2504–2513. [Google Scholar] [CrossRef]
- Miyahara, T.; Runge, S.; Chatterjee, A.; Chen, M.; Mottola, G.; Fitzgerald, J.M.; Serhan, C.N.; Conte, M.S. D-series resolvin attenuates vascular smooth muscle cell activation and neointimal hyperplasia following vascular injury. FASEB J. 2013, 27, 2220–2232. [Google Scholar] [CrossRef]
- Petri, M.H.; Laguna-Fernandez, A.; Arnardottir, H.; Wheelock, C.E.; Perretti, M.; Hansson, G.K.; Bäck, M. Aspirin-triggered lipoxin A4 inhibits atherosclerosis progression in apolipoprotein E-/- mice. Br. J. Pharmacol. 2017, 174, 4043–4054. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Mottola, G.; Chatterjee, A.; Lance, K.D.; Chen, M.; Siguenza, I.; Desai, T.A.; Conte, M.S. Perivascular delivery of Resolvin D1 inhibits neointimal hyperplasia in a rat model of arterial injury. J. Vasc. Surg. 2017, 65, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Werlin, E.C.; Chen, M.; Mottola, G.; Chatterjee, A.; Lance, K.D.; Bernards, D.A.; Sansbury, B.E.; Spite, M.; Desai, T.A.; et al. Perivascular delivery of resolvin D1 inhibits neointimal hyperplasia in a rabbit vein graft model. J. Vasc. Surg. 2018, 68, 188S–200S.e4. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Gong, Y.; Zhang, R.; Piao, L.; Li, X.; Liu, Q.; Yan, S.; Shen, Y. Resolvin E1 attenuates injury-induced vascular neointimal formation by inhibition of inflammatory responses and vascular smooth muscle cell migration. FASEB J. 2018, 32, 5413–5425. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.H.; Laguna-Fernández, A.; Gonzalez-Diez, M.; Paulsson-Berne, G.; Hansson, G.K.; Bäck, M. The role of the FPR2/ALX receptor in atherosclerosis development and plaque stability. Cardiovasc. Res. 2015, 105, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Jannaway, M.; Torrens, C.; Warner, J.A.; Sampson, A.P. Resolvin E1, resolvin D1 and resolvin D2 inhibit constriction of rat thoracic aorta and human pulmonary artery induced by the thromboxane mimetic U46619. Br. J. Pharmacol. 2018, 175, 1100–1108. [Google Scholar] [CrossRef]
- Hiram, R.; Rizcallah, E.; Sirois, C.; Sirois, M.; Morin, C.; Fortin, S.; Rousseau, E. Resolvin D1 reverses reactivity and Ca2+ sensitivity induced by ET-1, TNF-α, and IL-6 in the human pulmonary artery. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1547–H1558. [Google Scholar] [CrossRef][Green Version]
- Kurahara, L.H.; Hiraishi, K.; Yamamura, A.; Zhang, Y.; Abe, K.; Yahiro, E.; Aoki, M.; Koga, K.; Yokomise, H.; Go, T.; et al. Eicosapentaenoic acid ameliorates pulmonary hypertension via inhibition of tyrosine kinase Fyn. J. Mol. Cell. Cardiol. 2020, 148, 50–62. [Google Scholar] [CrossRef]
- von der Weid, P.Y.; Hollenberg, M.D.; Fiorucci, S.; Wallace, J.L. Aspirin-triggered, cyclooxygenase-2-dependent lipoxin synthesis modulates vascular tone. Circulation 2004, 110, 1320–1325. [Google Scholar] [CrossRef]
- Brezinski, D.A.; Nesto, R.W.; Serhan, C.N. Angioplasty triggers intracoronary leukotrienes and lipoxin A4. Impact of aspirin therapy. Circulation 1992, 86, 56–63. [Google Scholar] [CrossRef]
- Wenceslau, C.F.; McCarthy, C.G.; Szasz, T.; Webb, R.C. Lipoxin A4 mediates aortic contraction via RhoA/Rho kinase, endothelial dysfunction and reactive oxygen species. J. Vasc. Res. 2014, 51, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Falk, E. Pathogenesis of atherosclerosis. J. Am. Coll. Cardiol. 2006, 47 (Suppl. S8), C7–C12. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Bornfeldt, K.E. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ. Res. 2016, 118, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 2010, 10, 36–46. [Google Scholar] [CrossRef]
- Merched, A.J.; Ko, K.; Gotlinger, K.H.; Serhan, C.N.; Chan, L. Atherosclerosis: Evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J. 2008, 22, 3595–3606. [Google Scholar] [CrossRef]
- Viola, J.R.; Lemnitzer, P.; Jansen, Y.; Csaba, G.; Winter, C.; Neideck, C.; Silvestre-Roig, C.; Dittmar, G.; Döring, Y.; Drechsler, M.; et al. Resolving Lipid Mediators Maresin 1 and Resolvin D2 Prevent Atheroprogression in Mice. Circ. Res. 2016, 119, 1030–1038. [Google Scholar] [CrossRef]
- Fredman, G.; Hellmann, J.; Proto, J.D.; Kuriakose, G.; Colas, R.A.; Dorweiler, B.; Connolly, E.S.; Solomon, R.; Jones, D.M.; Heyer, E.J.; et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat. Commun. 2016, 7, 12859. [Google Scholar] [CrossRef]
- Bäck, M.; Yurdagul, A., Jr.; Tabas, I.; Öörni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef]
- Hasturk, H.; Abdallah, R.; Kantarci, A.; Nguyen, D.; Giordano, N.; Hamilton, J.; Van Dyke, T.E. Resolvin E1 (RvE1) Attenuates Atherosclerotic Plaque Formation in Diet and Inflammation-Induced atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1123–1133. [Google Scholar] [CrossRef]
- Salic, K.; Morrison, M.C.; Verschuren, L.; Wielinga, P.Y.; Wu, L.; Kleemann, R.; Gjorstrup, P.; Kooistra, T. Resolvin E1 attenuates atherosclerosis in absence of cholesterol-lowering effects and on top of atorvastatin. Atherosclerosis 2016, 250, 158–165. [Google Scholar] [CrossRef]
- Carracedo, M.; Artiach, G.; Witasp, A.; Clària, J.; Carlstrom, M.; Laguna-Fernandez, A.; Stenvinkel, P.; Bäck, M. The G-protein coupled receptor ChemR23 determines smooth muscle cell phenotypic switching to enhance high phosphate-induced vascular calcification. Cardiovasc. Res. 2019, 115, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Zhang, J.; Zhou, X.; Hao, H. Lipoxin A4 and its analog attenuate high fat diet-induced atherosclerosis via Keap1/Nrf2 pathway. Exp. Cell Res. 2022, 412, 113025. [Google Scholar] [CrossRef] [PubMed]
- Laguna-Fernandez, A.; Checa, A.; Carracedo, M.; Artiach, G.; Petri, M.H.; Baumgartner, R.; Forteza, M.J.; Jiang, X.; Andonova, T.; Walker, M.E.; et al. ERV1/ChemR23 Signaling Protects against Atherosclerosis by Modifying Oxidized Low-Density Lipoprotein Uptake and Phagocytosis in Macrophages. Circulation 2018, 138, 1693–1705. [Google Scholar] [CrossRef]
- Drechsler, M.; de Jong, R.; Rossaint, J.; Viola, J.R.; Leoni, G.; Wang, J.M.; Grommes, J.; Hinkel, R.; Kupatt, C.; Weber, C.; et al. Annexin A1 counteracts chemokine-induced arterial myeloid cell recruitment. Circ. Res. 2015, 116, 827–835. [Google Scholar] [CrossRef]
- Arnardottir, H.; Thul, S.; Pawelzik, S.-C.; Karadimou, G.; Artiach, G.; Gallina, A.L.; Mysdotter, V.; Carracedo, M.; Tarnawski, L.; Caravaca, A.S.; et al. The resolvin D1 receptor GPR32 transduces inflammation resolution and atheroprotection. J. Clin. Investig. 2021, 131, e142883. [Google Scholar] [CrossRef] [PubMed]
- Johnston, K.; Rutherford, R.B.; Tilson, M.; Shah, D.M.; Hollier, L.; Stanley, J.C. Suggested standards for reporting on arterial aneurysms. Subcommittee on Reporting Standards for Arterial Aneurysms, Ad Hoc Committee on Reporting Standards, Society for Vascular Surgery and North Am Chapter, International Society for Cardiovascular Surgery. J. Vasc. Surg. 1991, 13, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Dua, A.; Kuy, S.; Lee, C.J.; Upchurch, G.R., Jr.; Desai, S.S. Epidemiology of aortic aneurysm repair in the United States from 2000 to 2010. J. Vasc. Surg. 2014, 59, 1512–1517. [Google Scholar] [CrossRef] [PubMed]
- Skotsimara, G.; Antonopoulos, A.; Oikonomou, E.; Papastamos, C.; Siasos, G.; Tousoulis, D. Aortic Wall Inflammation in the Pathogenesis, Diagnosis and Treatment of Aortic Aneurysms. Inflammation 2022. [Google Scholar] [CrossRef]
- Meital, L.T.; Sandow, S.L.; Calder, P.C.; Russell, F.D. Abdominal aortic aneurysm and omega-3 polyunsaturated fatty acids: Mechanisms, animal models, and potential treatment. Prostaglandins Leukot. Essent. Fat. Acids 2017, 118, 1–9. [Google Scholar] [CrossRef]
- Quintana, R.A.; Taylor, W.R. Cellular Mechanisms of Aortic Aneurysm Formation. Circ. Res. 2019, 124, 607–618. [Google Scholar] [CrossRef]
- Reed, D.; Reed, C.; Stemmermann, G.; Hayashi, T. Are aortic aneurysms caused by atherosclerosis? Circulation 1992, 85, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Pillai, P.S.; Leeson, S.; Porter, T.F.; Owens, C.D.; Kim, J.M.; Conte, M.S.; Serhan, C.N.; Gelman, S. Chemical mediators of inflammation and resolution in post-operative abdominal aortic aneurysm patients. Inflammation 2012, 35, 98–113. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.H.; Thul, S.; Andonova, T.; Lindquist-Liljeqvist, M.; Jin, H.; Skenteris, N.T.; Arnardottir, H.; Maegdefessel, L.; Caidahl, K.; Perretti, M.; et al. Resolution of Inflammation Through the Lipoxin and ALX–FPR2 Receptor Pathway Protects against Abdominal Aortic Aneurysms. JACC Basic Transl. Sci. 2018, 3, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Pope, N.H.; Salmon, M.; Davis, J.P.; Chatterjee, A.; Su, G.; Conte, M.S.; Ailawadi, G.; Upchurch, G.R., Jr. D-series resolvins inhibit murine abdominal aortic aneurysm formation and increase M2 macrophage polarization. FASEB J. 2016, 30, 4192–4201. [Google Scholar] [CrossRef]
- Spinosa, M.; Su, G.; Salmon, M.D.; Lu, G.; Cullen, J.M.; Fashandi, A.Z.; Hawkins, R.B.; Montgomery, W.; Meher, A.K.; Conte, M.S.; et al. Resolvin D1 decreases abdominal aortic aneurysm formation by inhibiting NETosis in a mouse model. J. Vasc. Surg. 2018, 68, 93S–103S. [Google Scholar] [CrossRef]
- Elder, C.T.; Filiberto, A.C.; Su, G.; Ladd, Z.; Leroy, V.; Pruitt, E.Y.; Lu, G.; Jiang, Z.; Sharma, A.K.; Upchurch, G.R., Jr. Maresin 1 activates LGR6 signaling to inhibit smooth muscle cell activation and attenuate murine abdominal aortic aneurysm formation. FASEB J. 2021, 35, e21780. [Google Scholar] [CrossRef]
- Satish, M.; Agrawal, D.K. Pro-resolving lipid mediators in the resolution of neointimal hyperplasia pathogenesis in atherosclerotic diseases. Expert Rev. Cardiovasc. Ther. 2019, 17, 177–184. [Google Scholar] [CrossRef]
- Collins, M.J.; Li, X.; Lv, W.; Yang, C.; Protack, C.D.; Muto, A.; Jadlowiec, C.C.; Shu, C.; Dardik, A. Therapeutic strategies to combat neointimal hyperplasia in vascular grafts. Expert Rev. Cardiovasc. Ther. 2012, 10, 635–647. [Google Scholar] [CrossRef]
- Muto, A.; Fitzgerald, T.N.; Pimiento, J.M.; Maloney, S.P.; Teso, D.; Paszkowiak, J.J.; Westvik, T.S.; Kudo, F.A.; Nishibe, T.; Dardik, A. Smooth muscle cell signal transduction: Implications of vascular biology for vascular surgeons. J. Vasc. Surg. 2007, 45 (Suppl. A(6S)), A15–A24. [Google Scholar] [CrossRef]
- Petri, M.H.; Laguna-Fernandez, A.; Tseng, C.N.; Hedin, U.; Perretti, M.; Bäck, M. Aspirin-triggered 15-epi-lipoxin A4 signals through FPR2/ALX in vascular smooth muscle cells and protects against intimal hyperplasia after carotid ligation. Int. J. Cardiol. 2015, 179, 370–372. [Google Scholar] [CrossRef]
- Artiach, G.; Carracedo, M.; Clària, J.; Laguna-Fernandez, A.; Bäck, M. Opposing Effects on Vascular Smooth Muscle Cell Proliferation and Macrophage-induced Inflammation Reveal a Protective Role for the Proresolving Lipid Mediator Receptor ChemR23 in Intimal Hyperplasia. Front. Pharmacol. 2018, 9, 1327. [Google Scholar] [CrossRef] [PubMed]
- Makino, Y.; Miyahara, T.; Nitta, J.; Miyahara, K.; Seo, A.; Kimura, M.; Suhara, M.; Akai, A.; Akagi, D.; Yamamoto, K.; et al. Proresolving Lipid Mediators Resolvin D1 and Protectin D1 Isomer Attenuate Neointimal Hyperplasia in the Rat Carotid Artery Balloon Injury Model. J. Surg. Res. 2019, 233, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Mottola, G.; Werlin, E.C.; Wu, B.; Chen, M.; Chatterjee, A.; Schaller, M.S.; Conte, M.S. Oral Resolvin D1 attenuates early inflammation but not intimal hyperplasia in a rat carotid angioplasty model. Prostaglandins Other Lipid Mediat. 2020, 146, 106401. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, M.; Origasa, H.; Matsuzaki, M.; Matsuzawa, Y.; Saito, Y.; Ishikawa, Y.; Oikawa, S.; Sasaki, J.; Hishida, H.; Itakura, H.; et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): A randomised open-label, blinded endpoint analysis. Lancet 2007, 369, 1090–1098. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Steg, P.G.; Miller, M.; Brinton, E.A.; Jacobson, T.A.; Ketchum, S.B.; Doyle, R.T., Jr.; Juliano, R.A.; Jiao, L.; Granowitz, C.; et al. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N. Engl. J. Med. 2019, 380, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Lincoff, A.M.; Garcia, M.; Bash, D.; Ballantyne, C.M.; Barter, P.J.; Davidson, M.H.; Kastelein, J.J.P.; Koenig, W.; McGuire, D.K.; et al. Effect of High-Dose Omega-3 Fatty Acids vs Corn Oil on Major Adverse Cardiovascular Events in Patients at High Cardiovascular Risk: The STRENGTH Randomized Clinical Trial. JAMA 2020, 324, 2268–2280. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz del Campo, L.S.; Rodrigues-Díez, R.; Salaices, M.; Briones, A.M.; García-Redondo, A.B. Specialized Pro-Resolving Lipid Mediators: New Therapeutic Approaches for Vascular Remodeling. Int. J. Mol. Sci. 2022, 23, 3592. https://doi.org/10.3390/ijms23073592
Díaz del Campo LS, Rodrigues-Díez R, Salaices M, Briones AM, García-Redondo AB. Specialized Pro-Resolving Lipid Mediators: New Therapeutic Approaches for Vascular Remodeling. International Journal of Molecular Sciences. 2022; 23(7):3592. https://doi.org/10.3390/ijms23073592
Chicago/Turabian StyleDíaz del Campo, Lucía Serrano, Raquel Rodrigues-Díez, Mercedes Salaices, Ana M. Briones, and Ana B. García-Redondo. 2022. "Specialized Pro-Resolving Lipid Mediators: New Therapeutic Approaches for Vascular Remodeling" International Journal of Molecular Sciences 23, no. 7: 3592. https://doi.org/10.3390/ijms23073592
APA StyleDíaz del Campo, L. S., Rodrigues-Díez, R., Salaices, M., Briones, A. M., & García-Redondo, A. B. (2022). Specialized Pro-Resolving Lipid Mediators: New Therapeutic Approaches for Vascular Remodeling. International Journal of Molecular Sciences, 23(7), 3592. https://doi.org/10.3390/ijms23073592