
Cyclin-Dependent Kinase Synthetic Lethality Partners in DNA Damage Response

 , ,
, ,  and
and 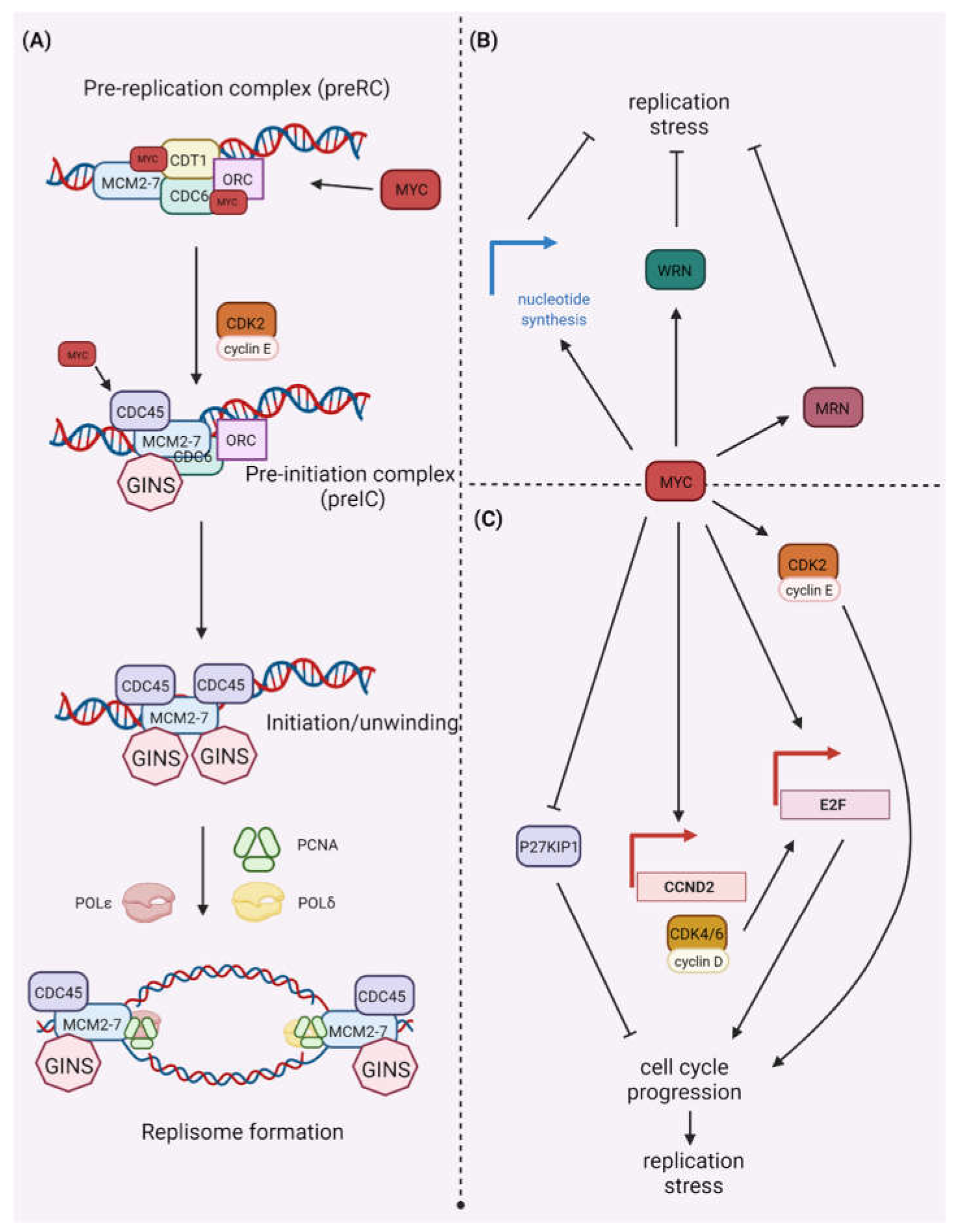{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. CDK/MYC and DDR
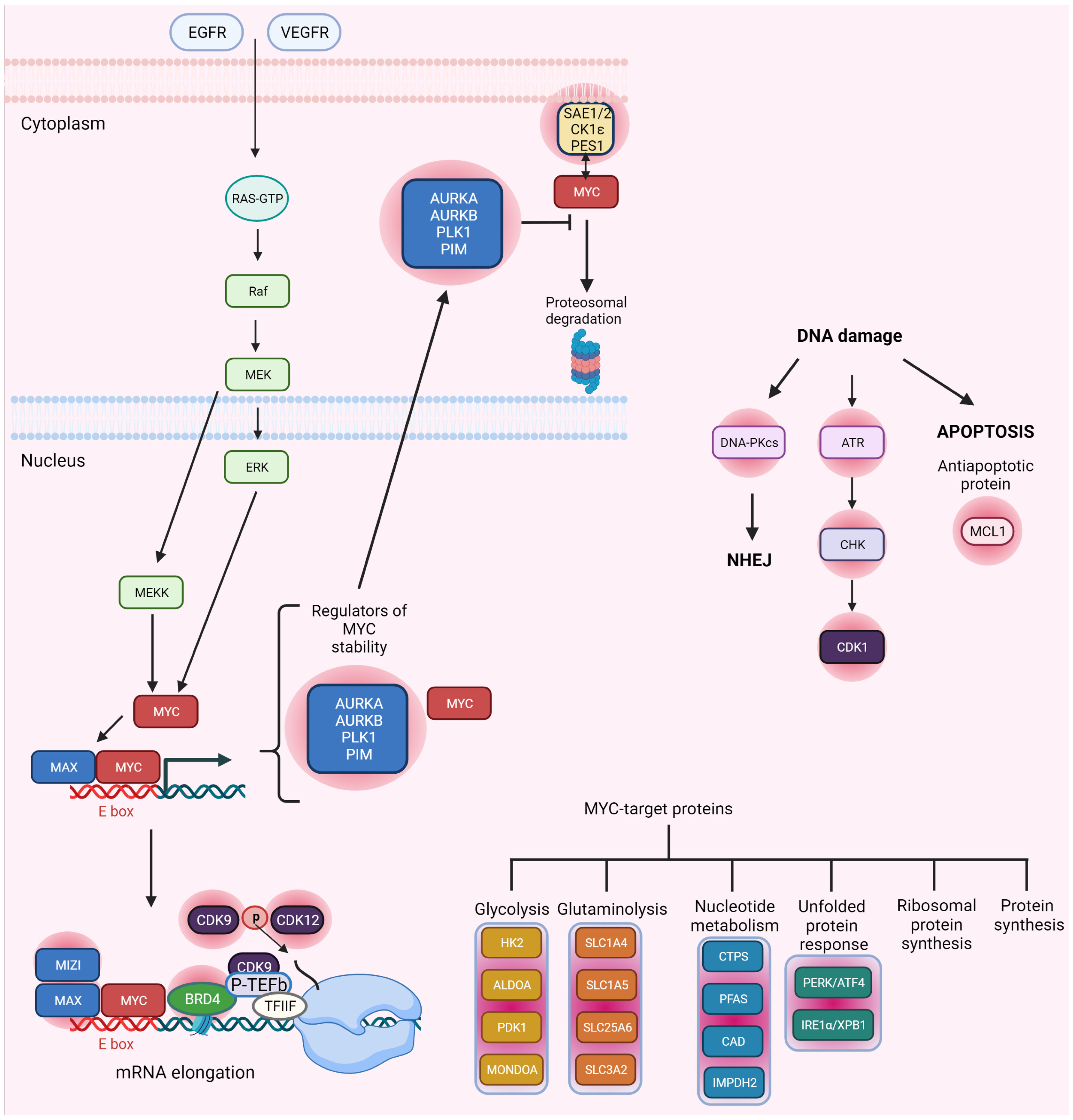
2.1. MYC and DDR
2.2. MYC Synthetic Lethality Partners
2.3. MYC and CDK Synthetic Lethality
3. CDK/TP53 and DDR
4. CDK/PARP and DDR
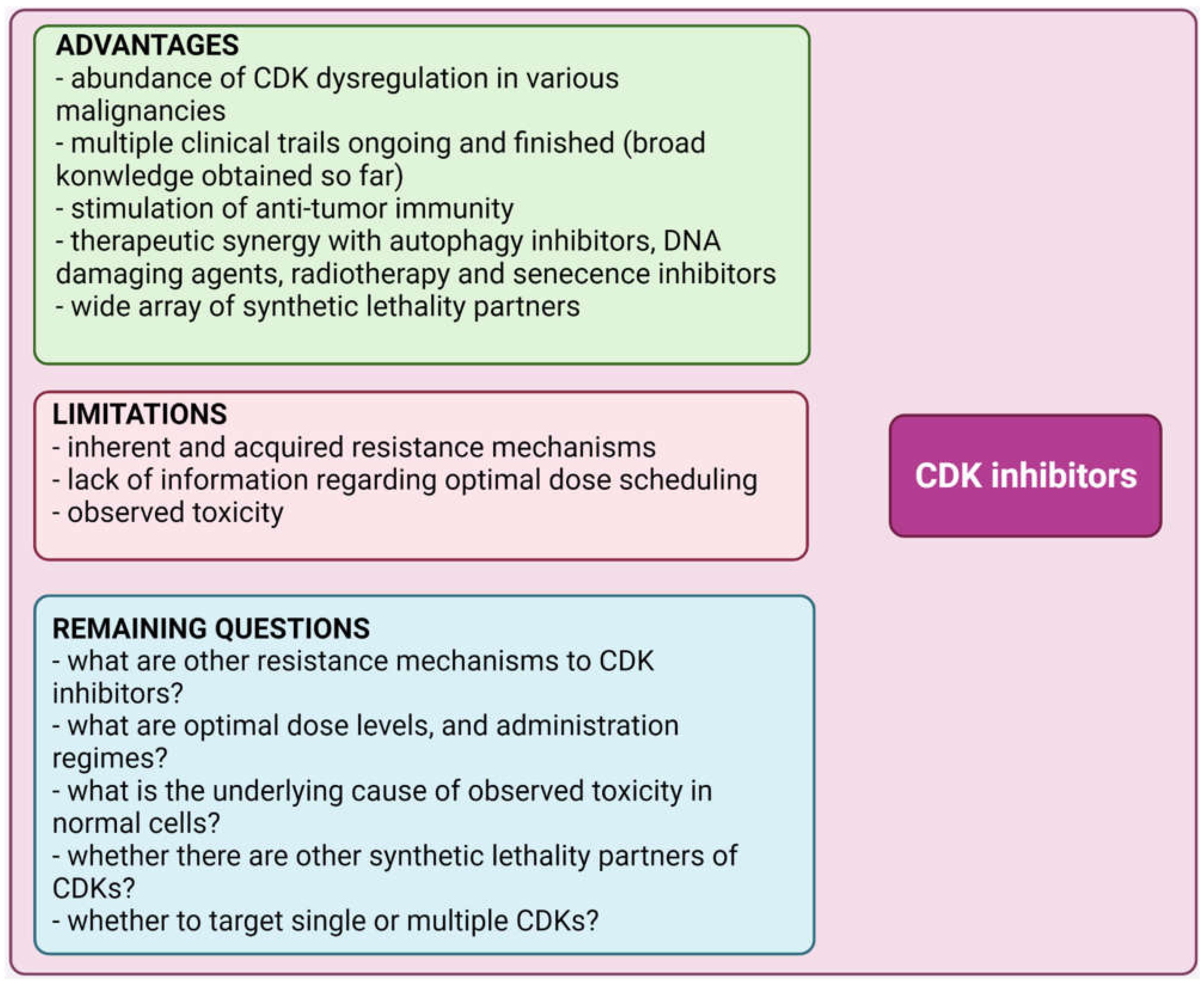
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AKT | RAC-alpha serine/threonine-protein kinase |
| ALDOA | Fructose-bisphosphate aldolase A |
| APAF1 | Apoptotic protease-activating factor 1 |
| APE1 | DNA-(apurinic or apyrimidinic site) endonuclease |
| ARF | Tumor suppressor ARF |
| ARH3 | ADP-ribose glycohydrolase ARH3 |
| ARK5 | AMPK-related protein kinase |
| ATF4 | Cyclic AMP-dependent transcription factor ATF4 |
| ATM | Ataxia telangiectasia mutated |
| ATR | Ataxia telangiectasia and Rad3-related protein |
| AURKA/AURKB | Aurora kinases A and B |
| BAK1 | Bcl-2 homologous antagonist/killer |
| BAX | Pro-apoptotic protein BAX |
| BER | Base excision repair |
| BIM | Bcl-2-like protein 11 |
| BIRC5 | Baculoviral IAP repeat-containing protein 5 |
| BLM | Bloom syndrome protein |
| BRCA1/2 | Breast cancer type 1 susceptibility protein ½ |
| BRD4 | Bromodomain-containing protein 4 |
| CBS | Cystathionine beta synthase |
| CDC45 | Cell division control protein 45 homolog |
| CDC6 | Cell division control protein 6 homolog |
| CDC7 | Cell division cycle 7-related protein kinase |
| CDK | Cyclin-dependent kinase |
| CDKi | Cyclin-dependent kinase inhibitors |
| CDT1 | DNA replication factor Cdt1 |
| CHK1/2 | Checkpoint kinase ½ |
| CK1ε | Casein kinase I isoform epsilon |
| CMG | Replicative helicase CMG |
| CSB | Cockayne syndrome protein B |
| CTPS | CTP synthase 1 |
| DDR | DNA damage response |
| DNA-PKcs | DNA-dependent protein kinase catalytic subunit |
| DSBR | Double-strand break repair |
| DSBs | Double-strand breaks |
| ENO3 | β-enolase |
| FAS/CD95 | Tumor necrosis factor receptor superfamily member 6 |
| FASN | Fatty acid synthase |
| FBXW7 | F-box/WD repeat-containing protein 7 |
| HAT | Histone acetyltransferase |
| HIF-1 | Hypoxia-inducible factor 1 |
| HK2 | Hexokinase 2 |
| HR | Homologous recombination |
| IMPDH2 | Inosine-5-monophosphate dehydrogenase |
| IRE1α | Serine/threonine-protein kinase/endoribonuclease IRE |
| KAT5/TIP60 | Histone acetyltransferase KAT5/TIP60 |
| KU70/80 | X-ray repair cross-complementing protein 5/6 |
| MAPK | Mitogen-activated protein kinases |
| MCL1 | Myeloid leukemia cell differentiation protein |
| MCM2-7 | DNA replication licensing factor MCM2-7 |
| MDM2 | E3 ubiquitin-protein ligase MDM2 |
| MHC | Major histocompatibility complex |
| MIZ1 | Zinc finger and BTB domain-containing protein 17 |
| MONDOA | MLX-interacting protein |
| MRE11 | Double-strand break repair protein MRE11 |
| NAD | Nicotinamide adenine dinucleotide |
| NBS1 | Nibrin |
| NER | Nucleotide excision repair |
| NHEJ | Non-homologous end-joining |
| ORC | Origin recognition complex |
| P27KIP1 | Cyclin-dependent kinase inhibitor 1B |
| PAR | Poly (ADP-ribose) polymers |
| PARG | Poly (ADP-ribose) glycohydrolase |
| PARPs | Poly [ADP-ribose] polymerases |
| PD-1 | Programmed cell death protein 1 |
| PDK1 | Pyruvate dehydrogenase kinase isoform 1 |
| PERK | PRKR-like endoplasmic reticulum kinase |
| PES1 | Pescadillo homolog |
| PFAS | Phosphoribosylformylglycinamidine synthase |
| PI3K | Phosphatidylinositol 4,5-bisphosphate 3-kinase |
| PIM-1 | Serine/threonine-protein kinase pim-1 |
| PLK1 | Polo-like kinase 1 |
| pRB | Retinoblastoma protein |
| Pre-RC | Pre-replicative complex |
| PTEF-b | Transcription elongation factor |
| RAD51 | DNA repair protein RAD51 homolog |
| RNAP-II | RNA polymerase II |
| ROS | Reactive oxygen species |
| RPA | Replication protein A |
| SAE1/2 | SUMO-activating enzyme subunit ½ |
| SCD | Stearoyl-CoA desaturase |
| SCL7A11 | Cystine/glutamate transporter |
| SLC1A4 | Neutral amino acid transporter A |
| SLC1A5 | Neutral amino acid transporter B(0) |
| SLC25A6 | ADP/ATP translocase 3 |
| SLC3A2 | 4F2 cell-surface antigen heavy chain |
| SSBR | Single-strand break repair |
| SSBs | Single-strand breaks |
| SsDNA | Single-stranded DNA |
| TFAM | Mitochondrial transcription factor A |
| TNBC | Triple-negative breast cancer |
| TP53 | Cellular tumor antigen p53 |
| TRAIL R2/DR540 | Tumor necrosis factor receptor superfamily member 10 |
| UBE2I | SUMO-conjugating enzyme UBC9 |
| UPR | Unfolded protein response |
| WIP1 | Protein phosphatase 1D |
| WRN | Werner syndrome ATP-dependent helicase |
| XBP1 | X-box-binding protein 1 |
| XPB | General transcription and DNA repair factor IIH helicase subunit XPB |
| XPC | DNA repair protein complementing XP-C |
| XPD | General transcription and DNA repair factor IIH helicase subunit XPD |
| XRCC4 | X-ray repair cross-complementing protein 4 |
References
- Panagiotou, E.; Gomatou, G.; Trontzas, I.P.; Syrigos, N.; Kotteas, E. Cyclin-dependent kinase (CDK) inhibitors in solid tumors: A review of clinical trials. Clin. Transl. Oncol. 2021, 24, 161–192. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.H.; Ortega, J.A.; Cavalli, A. Synthetic Lethality through the Lens of Medicinal Chemistry. J. Med. Chem. 2020, 63, 14151–14183. [Google Scholar] [CrossRef] [PubMed]
- Topatana, W.; Juengpanich, S.; Li, S.; Cao, J.; Hu, J.; Lee, J.; Suliyanto, K.; Ma, D.; Zhang, B.; Chen, M.; et al. Advances in synthetic lethality for cancer therapy: Cellular mechanism and clinical translation. J. Hematol. Oncol. 2020, 13, 118. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Stine, Z.E.; Walton, Z.E.; Altman, B.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef]
- Dominguez-Sola, D.; Ying, C.Y.; Grandori, C.; Ruggiero, L.; Chen, B.; Li, M.; Galloway, D.A.; Gu, W.; Gautier, J.; Dalla-Favera, R. Non-transcriptional control of DNA replication by c-Myc. Nature 2007, 448, 445–451. [Google Scholar] [CrossRef]
- Bartkova, J.; Hořejší, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Kotsantis, P.; Petermann, E.; Boulton, S.J. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discov. 2018, 8, 537–555. [Google Scholar] [CrossRef]
- Primo, L.M.F.; Teixeira, L.K. DNA replication stress: Oncogenes in the spotlight. Genet. Mol. Biol. 2020, 43, e20190138. [Google Scholar] [CrossRef]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc Can Induce DNA Damage, Increase Reactive Oxygen Species, and Mitigate p53 Function: A Mechanism for Oncogene-Induced Genetic Instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Karlsson, A.; Deb-Basu, D.; Cherry, A.; Turner, S.; Ford, J.; Felsher, D.W. Defective double-strand DNA break repair and chromosomal translocations by MYC overexpression. Proc. Natl. Acad. Sci. USA 2003, 100, 9974–9979. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Atkuri, K.R.; Deb-Basu, D.; Adler, A.; Chang, H.Y.; Herzenberg, L.A.; Felsher, D.W. MYC Can Induce DNA Breaks in vivo and in vitro Independent of Reactive Oxygen Species. Cancer Res. 2006, 66, 6598–6605. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.V.; Dominguez-Sola, D.; Wang, L.C.; Hyrien, O.; Gautier, J. Cdc45 Is a Critical Effector of Myc-Dependent DNA Replication Stress. Cell Rep. 2013, 3, 1629–1639. [Google Scholar] [CrossRef] [PubMed]
- Bretones, G.; Delgado, M.D.; León, J. Myc and cell cycle control. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2015, 1849, 506–516. [Google Scholar] [CrossRef]
- Amani, J.; Gorjizadeh, N.; Younesi, S.; Najafi, M.; Ashrafi, A.M.; Irian, S.; Gorjizadeh, N.; Azizian, K. Cyclin-dependent kinase inhibitors (CDKIs) and the DNA damage response: The link between signaling pathways and cancer. DNA Repair 2021, 102, 103103. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-C.; Li, F.; Handler, J.; Huang, C.R.L.; Xiang, Y.; Neretti, N.; Sedivy, J.M.; Zeller, K.I.; Dang, C.V. Global Regulation of Nucleotide Biosynthetic Genes by c-Myc. PLoS ONE 2008, 3, e2722. [Google Scholar] [CrossRef]
- Mannava, S.; Grachtchouk, V.; Wheeler, L.J.; Im, M.; Zhuang, D.; Slavina, E.G.; Mathews, C.K.; Shewach, D.S.; Nikiforov, M.A. Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle 2008, 7, 2392–2400. [Google Scholar] [CrossRef]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide Deficiency Promotes Genomic Instability in Early Stages of Cancer Development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef]
- Grandori, C.; Wu, K.-J.; Fernandez, P.; Ngouenet, C.; Grim, J.; Clurman, B.E.; Moser, M.J.; Oshima, J.; Russell, D.W.; Swisshelm, K.; et al. Werner syndrome protein limits MYC-induced cellular senescence. Genes Dev. 2003, 17, 1569–1574. [Google Scholar] [CrossRef]
- Robinson, K.; Asawachaicharn, N.; Galloway, D.A.; Grandori, C. c-Myc Accelerates S-Phase and Requires WRN to Avoid Replication Stress. PLoS ONE 2009, 4, e5951. [Google Scholar] [CrossRef]
- Petroni, M.; Sardina, F.; Heil, C.; Sahún-Roncero, M.; Colicchia, V.; Veschi, V.; Albini, S.; Fruci, D.; Ricci, B.; Soriani, A.; et al. The MRN complex is transcriptionally regulated by MYCN during neural cell proliferation to control replication stress. Cell Death Differ. 2015, 23, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Pichierri, P.; Ammazzalorso, F.; Bignami, M.; Franchitto, A. The Werner syndrome protein: Linking the replication checkpoint response to genome stability. Aging 2011, 3, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Franchitto, A.; Pirzio, L.M.; Prosperi, E.; Sapora, O.; Bignami, M.; Pichierri, P. Replication fork stalling in WRN-deficient cells is overcome by prompt activation of a MUS81-dependent pathway. J. Cell Biol. 2008, 183, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Luoto, K.R.; Meng, A.X.; Wasylishen, A.; Zhao, H.; Coackley, C.L.; Penn, L.; Bristow, R. Tumor Cell Kill by c-MYC Depletion: Role of MYC-Regulated Genes that Control DNA Double-Strand Break Repair. Cancer Res. 2010, 70, 8748–8759. [Google Scholar] [CrossRef] [PubMed]
- Eischen, C.M.; Weber, J.; Roussel, M.F.; Sherr, C.J.; Cleveland, J.L. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999, 13, 2658–2669. [Google Scholar] [CrossRef]
- Zindy, F.; Eischen, C.M.; Randle, D.H.; Kamijo, T.; Cleveland, J.L.; Sherr, C.J.; Roussel, M.F. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998, 12, 2424–2433. [Google Scholar] [CrossRef]
- Lowe, S.W.; Cepero, E.; Evan, G. Intrinsic tumour suppression. Nature 2004, 432, 307–315. [Google Scholar] [CrossRef]
- Hemann, M.T.; Bric, A.; Teruya-Feldstein, J.; Herbst, A.; Nilsson, J.A.; Cordon-Cardo, C.; Cleveland, J.L.; Tansey, W.P.; Lowe, S.W. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature 2005, 436, 807–811. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. Dissecting p53 tumor suppressor functions in vivo. Cancer Cell 2002, 1, 289–298. [Google Scholar] [CrossRef]
- Felsher, D.W.; Bishop, J.M. Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc. Natl. Acad. Sci. USA 1999, 96, 3940–3944. [Google Scholar] [CrossRef]
- MacLean, K.H.; Kastan, M.B.; Cleveland, J.L. Atm Deficiency Affects Both Apoptosis and Proliferation to Augment Myc-Induced Lymphomagenesis. Mol. Cancer Res. 2007, 5, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Pusapati, R.V.; Rounbehler, R.J.; Hong, S.; Powers, J.T.; Yan, M.; Kiguchi, K.; McArthur, M.J.; Wong, P.K.; Johnson, D.G. ATM promotes apoptosis and suppresses tumorigenesis in response to Myc. Proc. Natl. Acad. Sci. USA 2006, 103, 1446–1451. [Google Scholar] [CrossRef] [PubMed]
- Reimann, M.; Loddenkemper, C.; Rudolph, C.; Schildhauer, I.; Teichmann, B.; Stein, H.; Schlegelberger, B.; Dörken, B.; Schmitt, C.A. The Myc-evoked DNA damage response accounts for treatment resistance in primary lymphomas in vivo. Blood 2007, 110, 2996–3004. [Google Scholar] [CrossRef] [PubMed]
- Shreeram, S.; Hee, W.K.; Demidov, O.N.; Kek, C.; Yamaguchi, H.; Fornace, A.J.; Anderson, C.W.; Appella, E.; Bulavin, D.V. Regulation of ATM/p53-dependent suppression of myc-induced lymphomas by Wip1 phosphatase. J. Exp. Med. 2006, 203, 2793–2799. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Squatrito, M.; Luise, C.; Syed, N.; Perna, D.; Wark, L.; Martinato, F.; Sardella, D.; Verrecchia, A.; Bennett, S.; et al. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature 2007, 448, 1063–1067. [Google Scholar] [CrossRef]
- Korz, C.; Pscherer, A.; Benner, A.; Mertens, D.; Schaffner, C.; Leupolt, E.; Döhner, H.; Stilgenbauer, S.; Lichter, P. Evidence for distinct pathomechanisms in B-cell chronic lymphocytic leukemia and mantle cell lymphoma by quantitative expression analysis of cell cycle and apoptosis-associated genes. Blood 2002, 99, 4554–4561. [Google Scholar] [CrossRef]
- van Attikum, H.; Gasser, S.M. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 2009, 19, 207–217. [Google Scholar] [CrossRef]
- Squatrito, M.; Gorrini, C.; Amati, B. Tip60 in DNA damage response and growth control: Many tricks in one HAT. Trends Cell Biol. 2006, 16, 433–442. [Google Scholar] [CrossRef]
- Kusch, T.; Florens, L.; MacDonald, W.H.; Swanson, S.K.; Glaser, R.L.; Yates, J.R., III; Abmayr, S.M.; Washburn, M.P.; Workman, J.L. Acetylation by Tip60 Is Required for Selective Histone Variant Exchange at DNA Lesions. Science 2004, 306, 2084–2087. [Google Scholar] [CrossRef]
- Wang, C.; Fang, H.; Zhang, J.; Gu, Y. Targeting “undruggable” c-Myc protein by synthetic lethality. Front. Med. 2021, 15, 541–550. [Google Scholar] [CrossRef]
- Thng, D.K.H.; Toh, T.B.; Chow, E.K.-H. Capitalizing on Synthetic Lethality of MYC to Treat Cancer in the Digital Age. Trends Pharmacol. Sci. 2021, 42, 166–182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Li, N.; Sun, C.; Jin, Y.; Sheng, X. MYC and the unfolded protein response in cancer: Synthetic lethal partners in crime? EMBO Mol. Med. 2020, 12, e11845. [Google Scholar] [CrossRef] [PubMed]
- Pourdehnad, M.; Truitt, M.L.; Siddiqi, I.N.; Ducker, G.S.; Shokat, K.M.; Ruggero, D. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc. Natl. Acad. Sci. USA 2013, 110, 11988–11993. [Google Scholar] [CrossRef] [PubMed]
- Haikala, H.M.; Anttila, J.; Klefström, J. MYC and AMPK–Save Energy or Die! Front. Cell Dev. Biol. 2017, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.L.; Dang, C.V. MYC, Metabolic Synthetic Lethality, and Cancer. Recent Results Cancer Res. 2016, 207, 73–91. [Google Scholar] [CrossRef]
- Cermelli, S.; Jang, I.S.; Bernard, B.; Grandori, C. Synthetic Lethal Screens as a Means to Understand and Treat MYC-Driven Cancers. Cold Spring Harb. Perspect. Med. 2014, 4, a014209. [Google Scholar] [CrossRef]
- Toyoshima, M.; Howie, H.L.; Imakura, M.; Walsh, R.M.; Annis, J.E.; Chang, A.N.; Frazier, J.; Chau, B.N.; Loboda, A.; Linsley, P.S.; et al. Functional genomics identifies therapeutic targets for MYC-driven cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 9545–9550. [Google Scholar] [CrossRef]
- Goga, A.; Yang, D.; Tward, A.D.; Morgan, D.O.; Bishop, J.M. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat. Med. 2007, 13, 820–827. [Google Scholar] [CrossRef]
- Horiuchi, D.; Kusdra, L.; Huskey, N.E.; Chandriani, S.; Lenburg, M.E.; Gonzalez-Angulo, A.M.; Creasman, K.J.; Bazarov, A.V.; Smyth, J.W.; Davis, S.E.; et al. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J. Exp. Med. 2012, 209, 679–696. [Google Scholar] [CrossRef]
- Kang, J.; Sergio, C.M.; Sutherland, R.L.; Musgrove, E.A. Targeting cyclin-dependent kinase 1 (CDK1) but not CDK4/6 or CDK2 is selectively lethal to MYC-dependent human breast cancer cells. BMC Cancer 2014, 14, 32. [Google Scholar] [CrossRef]
- Santamaría, D.; Barrière, C.; Cerqueira, A.; Hunt, S.; Tardy, C.; Newton, K.; Caceres, J.; Dubus, P.; Malumbres, M.; Barbacid, M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007, 448, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, J.J.; Ebus, M.E.; Geerts, D.; Koster, J.; Lamers, F.; Valentijn, L.J.; Westerhout, E.M.; Versteeg, R.; Caron, H.N. Inactivation of CDK2 is synthetically lethal to MYCN over-expressing cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 12968–12973. [Google Scholar] [CrossRef] [PubMed]
- Campaner, S.; Doni, M.; Hydbring, P.; Verrecchia, A.; Bianchi, L.; Sardella, D.; Schleker, T.; Perna, D.; Tronnersjö, S.; Murga, M.; et al. Cdk2 suppresses cellular senescence induced by the c-myc oncogene. Nat. Cell Biol. 2009, 12, 54–59. [Google Scholar] [CrossRef]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J., 3rd. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef] [PubMed]
- Puissant, A.; Frumm, S.M.; Alexe, G.; Bassil, C.F.; Qi, J.; Chanthery, Y.H.; Nekritz, E.A.; Zeid, R.; Gustafson, W.C.; Greninger, P.; et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013, 3, 308–323. [Google Scholar] [CrossRef]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 Inhibition Suppresses Super-Enhancer-Linked Oncogenic Transcription in MYCN-Driven Cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef]
- Hashiguchi, T.; Bruss, N.; Best, S.; Lam, V.; Danilova, O.; Paiva, C.J.; Wolf, J.; Gilbert, E.W.; Okada, C.Y.; Kaur, P.; et al. Cyclin-Dependent Kinase-9 Is a Therapeutic Target in MYC-Expressing Diffuse Large B-Cell Lymphoma. Mol. Cancer Ther. 2019, 18, 1520–1532. [Google Scholar] [CrossRef]
- Gregory, G.; Hogg, S.; Kats, L.; Vidacs, E.; Baker, A.J.; Gilan, O.; Lefebure, M.; Martin, B.P.; Dawson, M.A.; Johnstone, R.; et al. CDK9 inhibition by dinaciclib potently suppresses Mcl-1 to induce durable apoptotic responses in aggressive MYC-driven B-cell lymphoma in vivo. Leukemia 2014, 29, 1437–1441. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef]
- Lui, G.; Grandori, C.; Kemp, C.J. CDK12: An emerging therapeutic target for cancer. J. Clin. Pathol. 2018, 71, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Li, B.-B.; Wang, B.; Zhu, C.-M.; Tang, D.; Pang, J.; Zhao, J.; Sun, C.-H.; Qiu, M.-J.; Qian, Z.-R. Cyclin-dependent kinase 7 inhibitor THZ1 in cancer therapy. Chronic Dis. Transl. Med. 2019, 5, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Vodicka, P.; Andera, L.; Opattova, A.; Vodickova, L. The Interactions of DNA Repair, Telomere Homeostasis, and p53 Mutational Status in Solid Cancers: Risk, Prognosis, and Prediction. Cancers 2021, 13, 479. [Google Scholar] [CrossRef]
- Zhang, Y.-X.; Pan, W.-Y.; Chen, J. p53 and its isoforms in DNA double-stranded break repair. J. Zhejiang Univ. Sci. B 2019, 20, 457–466. [Google Scholar] [CrossRef]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef]
- Smith, M.L.; Seo, Y.R. p53 regulation of DNA excision repair pathways. Mutagenesis 2002, 17, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Bacevic, K.; Lossaint, G.; Achour, T.N.; Georget, V.; Fisher, D.; Dulić, V. Cdk2 strengthens the intra-S checkpoint and counteracts cell cycle exit induced by DNA damage. Sci. Rep. 2017, 7, 13429. [Google Scholar] [CrossRef] [PubMed]
- Nekova, T.S.; Kneitz, S.; Einsele, H.; Bargou, R.; Stuhler, G. Silencing of CDK2, but not CDK1, separates mitogenic from anti-apoptotic signaling, sensitizing p53 defective cells for synthetic lethality. Cell Cycle 2016, 15, 3203–3209. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cheng, C.K.; Gustafson, W.C.; Charron, E.; Houseman, B.T.; Zunder, E.; Goga, A.; Gray, N.S.; Pollok, B.; Oakes, S.A.; James, C.D.; et al. Dual blockade of lipid and cyclin-dependent kinases induces synthetic lethality in malignant glioma. Proc. Natl. Acad. Sci. USA 2012, 109, 12722–12727. [Google Scholar] [CrossRef]
- Jabbour-Leung, N.A.; Chen, X.; Bui, T.; Jiang, Y.; Yang, D.; Vijayaraghavan, S.; McArthur, M.J.; Hunt, K.K.; Keyomarsi, K. Sequential Combination Therapy of CDK Inhibition and Doxorubicin Is Synthetically Lethal in p53-Mutant Triple-Negative Breast Cancer. Mol. Cancer Ther. 2016, 15, 593–607. [Google Scholar] [CrossRef]
- Kalan, S.; Amat, R.; Schachter, M.M.; Kwiatkowski, N.; Abraham, B.; Liang, Y.; Zhang, T.; Olson, C.M.; Larochelle, S.; Young, R.A.; et al. Activation of the p53 Transcriptional Program Sensitizes Cancer Cells to Cdk7 Inhibitors. Cell Rep. 2017, 21, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Herceg, Z.; Wang, Z.-Q. Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutat. Res. Mol. Mech. Mutagen. 2001, 477, 97–110. [Google Scholar] [CrossRef]
- Curtin, N.J.; Szabo, C. Poly(ADP-ribose) polymerase inhibition: Past, present and future. Nat. Rev. Drug Discov. 2020, 19, 711–736. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Zhang, Y.; Chen, S.; Weng, X.; Rao, Y.; Fang, H. Mechanism and current progress of Poly ADP-ribose polymerase (PARP) inhibitors in the treatment of ovarian cancer. Biomed. Pharmacother. 2020, 123, 109661. [Google Scholar] [CrossRef]
- Przybycinski, J.; Nalewajska, M.; Marchelek-Mysliwiec, M.; Dziedziejko, V.; Pawlik, A. Poly-ADP-ribose polymerases (PARPs) as a therapeutic target in the treatment of selected cancers. Expert Opin. Ther. Targets 2019, 23, 773–785. [Google Scholar] [CrossRef]
- Aly, A.; Ganesan, S. BRCA1, PARP, and 53BP1: Conditional synthetic lethality and synthetic viability. J. Mol. Cell Biol. 2011, 3, 66–74. [Google Scholar] [CrossRef]
- Yap, T.A.; Sandhu, S.K.; Carden, C.P.; de Bono, J.S. Poly(ADP-Ribose) polymerase (PARP) inhibitors: Exploiting a synthetic lethal strategy in the clinic. CA Cancer J. Clin. 2011, 61, 31–49. [Google Scholar] [CrossRef]
- Johnson, N.; Li, Y.C.; Walton, Z.E.; Cheng, K.A.; Li, D.; Rodig, S.J.; Moreau, L.A.; Unitt, C.; Bronson, R.T.; Thomas, H.D.; et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat. Med. 2011, 17, 875–882. [Google Scholar] [CrossRef]
- Xia, Q.; Cai, Y.; Peng, R.; Wu, G.; Shi, Y.; Jiang, W. The CDK1 inhibitor RO3306 improves the response of BRCA-proficient breast cancer cells to PARP inhibition. Int. J. Oncol. 2013, 44, 735–744. [Google Scholar] [CrossRef]
- Alagpulinsa, D.A.; Ayyadevara, S.; Yaccoby, S.; Reis, R.J.S. A Cyclin-Dependent Kinase Inhibitor, Dinaciclib, Impairs Homologous Recombination and Sensitizes Multiple Myeloma Cells to PARP Inhibition. Mol. Cancer Ther. 2015, 15, 241–250. [Google Scholar] [CrossRef]
- Johnson, S.F.; Cruz, C.; Greifenberg, A.K.; Dust, S.; Stover, D.; Chi, D.; Primack, B.; Cao, S.; Bernhardy, A.J.; Coulson, R.; et al. CDK12 Inhibition Reverses De Novo and Acquired PARP Inhibitor Resistance in BRCA Wild-Type and Mutated Models of Triple-Negative Breast Cancer. Cell Rep. 2016, 17, 2367–2381. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, M.; Dries, R.; Grassetti, A.V.; Dust, S.; Gao, Y.; Huang, H.; Sharma, B.; Day, D.S.; Kwiatkowski, N.; Pomaville, M.; et al. CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat. Commun. 2019, 10, 1757. [Google Scholar] [CrossRef] [PubMed]
- Kciuk, M.; Gielecińska, A.; Mujwar, S.; Mojzych, M.; Kontek, R. Cyclin-dependent kinases in DNA damage response. Biochim. Biophys. Acta 2022, 1877, 188716. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, L.; Hei, R.; Li, X.; Cai, H.; Wu, X.; Zheng, Q.; Cai, C. CDK Inhibitors in Cancer Therapy, an Overview of Recent Development. Am. J. Cancer Res. 2021, 11, 1913–1935. [Google Scholar]
- Reinius, M.A.V.; Smyth, E. Anti-cancer therapy with cyclin-dependent kinase inhibitors: Impact and challenges. Expert Rev. Mol. Med. 2021, 23, e6. [Google Scholar] [CrossRef]
- Johnson, N.; Shapiro, G.I. Cyclin-dependent kinases (cdks) and the DNA damage response: Rationale for cdk inhibitor–chemotherapy combinations as an anticancer strategy for solid tumors. Expert Opin. Ther. Targets 2010, 14, 1199–1212. [Google Scholar] [CrossRef]
- Mandal, R.; Becker, S.; Strebhardt, K. Targeting CDK9 for Anti-Cancer Therapeutics. Cancers 2021, 13, 2181. [Google Scholar] [CrossRef]
- Sava, G.; Fan, H.; Coombes, R.C.; Buluwela, L.; Ali, S. CDK7 inhibitors as anticancer drugs. Cancer Metastasis Rev. 2020, 39, 805–823. [Google Scholar] [CrossRef]
- Tadesse, S.; Duckett, D.R.; Monastyrskyi, A. The promise and current status of CDK12/13 inhibition for the treatment of cancer. Futur. Med. Chem. 2021, 13, 117–141. [Google Scholar] [CrossRef]
- Salmaninejad, A.; Valilou, S.F.; Shabgah, A.G.; Aslani, S.; Alimardani, M.; Pasdar, A.; Sahebkar, A. PD-1/PD-L1 pathway: Basic biology and role in cancer immunotherapy. J. Cell. Physiol. 2019, 234, 16824–16837. [Google Scholar] [CrossRef]
- Thoma, O.-M.; Neurath, M.F.; Waldner, M.J. Cyclin-Dependent Kinase Inhibitors and Their Therapeutic Potential in Colorectal Cancer Treatment. Front. Pharmacol. 2021, 12, 757120. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Topatana, W.; Juengpanich, S.; Cao, J.; Hu, J.; Zhang, B.; Ma, D.; Cai, X.; Chen, M. Development of synthetic lethality in cancer: Molecular and cellular classification. Signal Transduct. Target. Ther. 2020, 5, 241. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kciuk, M.; Gielecińska, A.; Mujwar, S.; Mojzych, M.; Kontek, R. Cyclin-Dependent Kinase Synthetic Lethality Partners in DNA Damage Response. Int. J. Mol. Sci. 2022, 23, 3555. https://doi.org/10.3390/ijms23073555
Kciuk M, Gielecińska A, Mujwar S, Mojzych M, Kontek R. Cyclin-Dependent Kinase Synthetic Lethality Partners in DNA Damage Response. International Journal of Molecular Sciences. 2022; 23(7):3555. https://doi.org/10.3390/ijms23073555
Chicago/Turabian StyleKciuk, Mateusz, Adrianna Gielecińska, Somdutt Mujwar, Mariusz Mojzych, and Renata Kontek. 2022. "Cyclin-Dependent Kinase Synthetic Lethality Partners in DNA Damage Response" International Journal of Molecular Sciences 23, no. 7: 3555. https://doi.org/10.3390/ijms23073555
APA StyleKciuk, M., Gielecińska, A., Mujwar, S., Mojzych, M., & Kontek, R. (2022). Cyclin-Dependent Kinase Synthetic Lethality Partners in DNA Damage Response. International Journal of Molecular Sciences, 23(7), 3555. https://doi.org/10.3390/ijms23073555