
Calcium- and Integrin-Binding Protein 2 (CIB2) in Physiology and Disease: Bright and Dark Sides


Abstract
1. Introduction
2. Structural, Biochemical, and Biophysical Properties of CIB2
2.1. CIB2 Has the Potential to Work as a Ca2+ and Mg2+ Sensor Protein
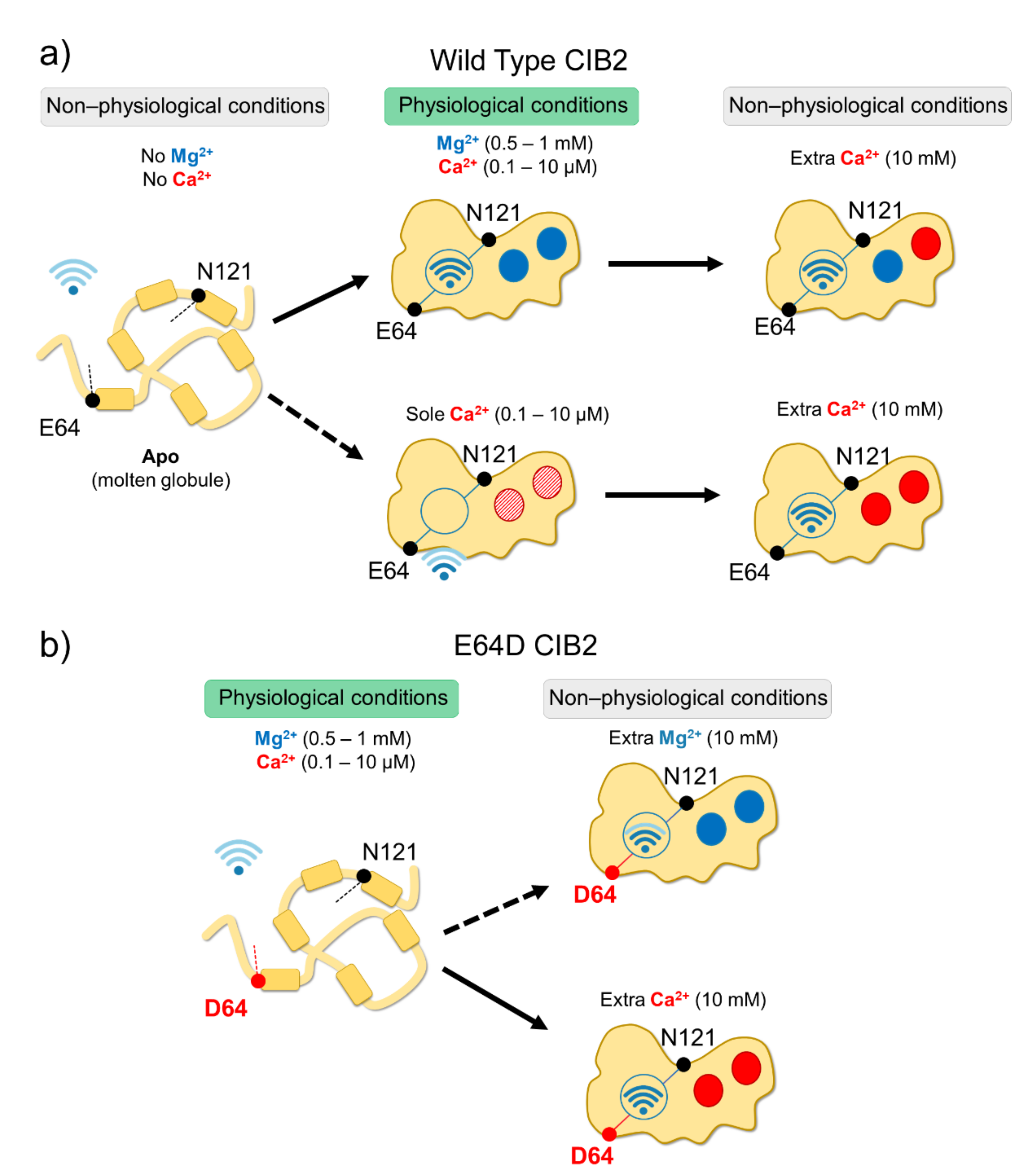
2.2. An Alleged Ca2+ Sensor? CIB2 Constitutively Binds Mg2+ but Has Low Affinity for Ca2+
2.3. An Inter-Domain Allosteric Switch Regulates the Conformational Transitions of CIB2
2.4. CIB2 Is Monomeric, but It Could Dimerize in the Presence of a Target
2.5. CIB2 Myristoylation
3. Broad Expression Pattern and Interaction Promiscuity: Functional Plasticity of CIB2
3.1. Interaction between CIB2 and Integrins
3.2. CIB2 and Ovarian Cancer
3.3. Involvement of CIB2 in HIV Viral Infection
3.4. CIB2, mTORC1 Signaling, and Autophagy
3.5. Putative Roles of CIB2 Investigated by OMICS Techniques
4. Involvement of CIB2 in Hearing and Deafness
4.1. CIB2 as a Member of the Mechanoelectrical Transduction Complex
4.2. CIB2 Interacts with BAIAP2L2
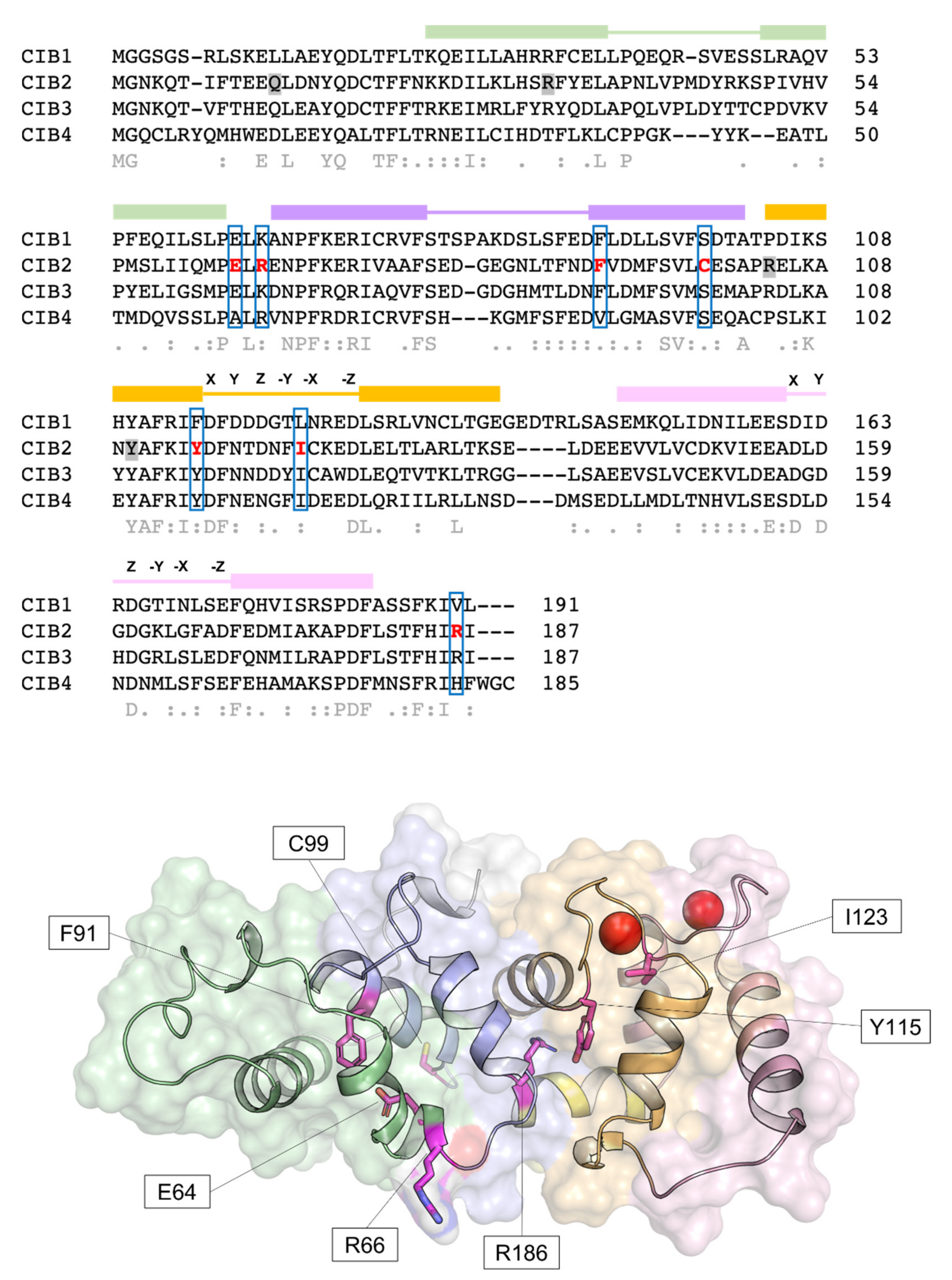
4.3. Mutations in CIB2 Are Associated with Deafness

{kind=link}
{kind=link}
{kind=link}
| cDNA | Protein | Type | Genotype | Ethnicity | Phenotype | Refs. |
|---|---|---|---|---|---|---|
| c.34C > T | p.(Gln12*) | Nonsense | Homozygous | Iranian | NSHI | [58] |
| c.97C > T | p.(Arg33*) | Nonsense | Homozygous | Arabic | HSHI | [76] |
| c.196C > T + c.300_309 | p.(Arg66Trp) + p.(Glu100*) | Missense + null allele | Heterozygous compound | European | HSHI | [75] |
| c.196C > T | p.(Arg66Trp) | Missense | Homozygous | Dutch | NSHI | [78] |
| c.192G > C | p.(Glu64Asp) | Missense | Homozygous | Pakistani | USH | [2] |
| c.97C > T + c.196C > T | p.(Arg33*) + p.(Arg66Trp) | Null allele + missense | Heterozygous compound | Dutch | NSHI | [78] |
| c.223G > A | p.(Val75Met) | Missense | Homozygous | European | NSHI | [75] |
| c.272T > C | p.(Phe91Ser) | Missense | Homozygous | Pakistani | HSHI | [2] |
| c.297C > G | p.(Cys99Trp) | Missense | Homozygous | Pakistani | HSHI | [2] |
| c.310C > T | p.(Arg104*) | Nonsense | Homozygous | Tunisian | NSHI | [77] |
| c.311G > A | p.(Arg104Gln) | Missense | Heterozygous | Not reported | PP | [82] |
| c.330T > A | p.(Tyr110*) | Nonsense | Homozygous | Turkish | HSHI | [58,75] |
| c.344A > G | p.(Tyr115Cys) | Missense | Homozygous | Iranian | HSHI | [75] |
| c.368T > C | p.(Ile123Thr) | Missense | Homozygous | Pakistani | HSHI | [2] |
| c.556C > T | p.(Arg186Trp) | Missense | Homozygous | Hispanic Caribbean Nigeria | NSHL PP | [80,81] |
4.4. CIB2 and Usher Interacome
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seki, N.; Hattori, A.; Hayashi, A.; Kozuma, S.; Ohira, M.; Hori, T.; Saito, T. Structure, expression profile and chromosomal location of an isolog of DNA-PKcs interacting protein (KIP) gene. Biochim. Biophys. Acta 1999, 1444, 143–147. [Google Scholar] [PubMed]
- Riazuddin, S.; Belyantseva, I.A.; Giese, A.P.; Lee, K.; Indzhykulian, A.A.; Nandamuri, S.P.; Yousaf, R.; Sinha, G.P.; Lee, S.; Terrell, D.; et al. Alterations of the CIB2 calcium- and integrin-binding protein cause Usher syndrome type 1J and nonsyndromic deafness DFNB48. Nat. Genet. 2012, 44, 1265–1271. [Google Scholar] [CrossRef] [PubMed]
- Hager, M.; Bigotti, M.G.; Meszaros, R.; Carmignac, V.; Holmberg, J.; Allamand, V.; Akerlund, M.; Kalamajski, S.; Brancaccio, A.; Mayer, U.; et al. Cib2 binds integrin alpha7Bbeta1D and is reduced in laminin alpha2 chain-deficient muscular dystrophy. J. Biol. Chem. 2008, 283, 24760–24769. [Google Scholar] [CrossRef] [PubMed]
- Blazejczyk, M.; Sobczak, A.; Debowska, K.; Wisniewska, M.B.; Kirilenko, A.; Pikula, S.; Jaworski, J.; Kuznicki, J.; Wojda, U. Biochemical characterization and expression analysis of a novel EF-hand Ca2+ binding protein calmyrin2 (Cib2) in brain indicates its function in NMDA receptor mediated Ca2+ signaling. Arch. Biochem. Biophys. 2009, 487, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Song, X.; Du, L.; Wang, C. Molecular characterization of the sheep CIB1 gene. Mol. Biol. Rep. 2009, 36, 1799–1809. [Google Scholar] [CrossRef] [PubMed]
- Yamniuk, A.P.; Vogel, H.J. Insights into the structure and function of calcium- and integrin-binding proteins. Calcium Bind. Proteins 2006, 1, 150–155. [Google Scholar]
- Dell’Orco, D.; Koch, K.W.; Kreutz, M.R.; Naranjo, J.R.; Schwaller, B. Editorial: Neuronal Calcium Sensors in Health and Disease. Front. Mol. Neurosci. 2019, 12, 278. [Google Scholar] [CrossRef]
- Denofrio, J.C.; Yuan, W.; Temple, B.R.; Gentry, H.R.; Parise, L.V. Characterization of calcium- and integrin-binding protein 1 (CIB1) knockout platelets: Potential compensation by CIB family members. Thromb. Haemost. 2008, 100, 847–856. [Google Scholar]
- Jacoszek, A.; Pollak, A.; Ploski, R.; Oldak, M. Advances in genetic hearing loss: CIB2 gene. Eur. Arch. Otorhinolaryngol. 2017, 274, 1791–1795. [Google Scholar] [CrossRef]
- Corey, D.P.; Akyuz, N.; Holt, J.R. Function and Dysfunction of TMC Channels in Inner Ear Hair Cells. Cold Spring Harb. Perspect. Med. 2019, 9, a033506. [Google Scholar] [CrossRef]
- Holt, J.R.; Tobin, M.; Elferich, J.; Gouaux, E.; Ballesteros, A.; Yan, Z.; Ahmed, Z.M.; Nicolson, T. Putting the Pieces Together: The Hair Cell Transduction Complex. J. Assoc. Res. Otolaryngol. 2021, 22, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Dal Cortivo, G.; Marino, V.; Iacobucci, C.; Vallone, R.; Arlt, C.; Rehkamp, A.; Sinz, A.; Dell’Orco, D. Oligomeric state, hydrodynamic properties and target recognition of human Calcium and Integrin Binding protein 2 (CIB2). Sci. Rep. 2019, 9, 15058. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Bogstie, J.N.; Vogel, H.J. Biophysical and structural studies of the human calcium- and integrin-binding protein family: Understanding their functional similarities and differences. Biochem. Cell Biol. 2012, 90, 646–656. [Google Scholar] [CrossRef] [PubMed]
- Vallone, R.; Dal Cortivo, G.; D’Onofrio, M.; Dell’Orco, D. Preferential Binding of Mg(2+) Over Ca(2+) to CIB2 Triggers an Allosteric Switch Impaired in Usher Syndrome Type 1J. Front. Mol. Neurosci. 2018, 11, 274. [Google Scholar] [CrossRef]
- Sethna, S.; Scott, P.A.; Giese, A.P.J.; Duncan, T.; Jian, X.; Riazuddin, S.; Randazzo, P.A.; Redmond, T.M.; Bernstein, S.L.; Riazuddin, S.; et al. CIB2 regulates mTORC1 signaling and is essential for autophagy and visual function. Nat. Commun. 2021, 12, 3906. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, Y.; Cai, W.Q.; Lu, Y. The Relationship of Sphingosine Kinase 1 With Pyroptosis Provides a New Strategy for Tumor Therapy. Front. Immunol. 2020, 11, 574990. [Google Scholar] [CrossRef] [PubMed]
- Naik, U.P.; Patel, P.M.; Parise, L.V. Identification of a novel calcium-binding protein that interacts with the integrin alphaIIb cytoplasmic domain. J. Biol. Chem. 1997, 272, 4651–4654. [Google Scholar] [CrossRef]
- Wu, X.; Lieber, M.R. Interaction between DNA-dependent protein kinase and a novel protein, KIP. Mutat. Res. 1997, 385, 13–20. [Google Scholar] [CrossRef]
- Gentry, H.R.; Singer, A.U.; Betts, L.; Yang, C.; Ferrara, J.D.; Sondek, J.; Parise, L.V. Structural and biochemical characterization of CIB1 delineates a new family of EF-hand-containing proteins. J. Biol. Chem. 2005, 280, 8407–8415. [Google Scholar] [CrossRef]
- Liang, X.; Qiu, X.; Dionne, G.; Cunningham, C.L.; Pucak, M.L.; Peng, G.; Kim, Y.H.; Lauer, A.; Shapiro, L.; Muller, U. CIB2 and CIB3 are auxiliary subunits of the mechanotransduction channel of hair cells. Neuron 2021, 109, 2131–2149. [Google Scholar] [CrossRef]
- Kawasaki, H.; Nakayama, S.; Kretsinger, R.H. Classification and evolution of EF-hand proteins. Biometals 1998, 11, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Yamniuk, A.P.; Nguyen, L.T.; Hoang, T.T.; Vogel, H.J. Metal ion binding properties and conformational states of calcium- and integrin-binding protein. Biochemistry 2004, 43, 2558–2568. [Google Scholar] [CrossRef] [PubMed]
- Yamniuk, A.P.; Silver, D.M.; Anderson, K.L.; Martin, S.R.; Vogel, H.J. Domain stability and metal-induced folding of calcium- and integrin-binding protein 1. Biochemistry 2007, 46, 7088–7098. [Google Scholar] [CrossRef] [PubMed]
- Linse, S.; Helmersson, A.; Forsen, S. Calcium binding to calmodulin and its globular domains. J. Biol. Chem. 1991, 266, 8050–8054. [Google Scholar]
- Berridge, M.J.; Bootman, M.D.; Lipp, P. Calcium—A life and death signal. Nature 1998, 395, 645–648. [Google Scholar] [CrossRef]
- Koch, K.W.; Dell’Orco, D. A calcium-relay mechanism in vertebrate phototransduction. ACS Chem. Neurosci. 2013, 4, 909–917. [Google Scholar] [CrossRef]
- Koch, K.W.; Dell’Orco, D. Protein and Signaling Networks in Vertebrate Photoreceptor Cells. Front. Mol. Neurosci. 2015, 8, 67. [Google Scholar] [CrossRef]
- Romani, A.; Scarpa, A. Regulation of cell magnesium. Arch. Biochem. Biophys. 1992, 298, 1–12. [Google Scholar]
- Romani, A.M.; Scarpa, A. Regulation of cellular magnesium. Front. Biosci. 2000, 5, D720–D734. [Google Scholar]
- Gagne, S.M.; Li, M.X.; Sykes, B.D. Mechanism of direct coupling between binding and induced structural change in regulatory calcium binding proteins. Biochemistry 1997, 36, 4386–4392. [Google Scholar] [CrossRef]
- Grabarek, Z. Insights into modulation of calcium signaling by magnesium in calmodulin, troponin C and related EF-hand proteins. Biochim. Biophys. Acta 2011, 1813, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Drake, S.K.; Falke, J.J. Kinetic tuning of the EF-hand calcium binding motif: The gateway residue independently adjusts (i) barrier height and (ii) equilibrium. Biochemistry 1996, 35, 1753–1760. [Google Scholar] [CrossRef] [PubMed]
- Marino, V.; Riva, M.; Zamboni, D.; Koch, K.W.; Dell’Orco, D. Bringing the Ca(2+) sensitivity of myristoylated recoverin into the physiological range. Open Biol. 2021, 11, 200346. [Google Scholar] [CrossRef] [PubMed]
- La Verde, V.; Dominici, P.; Astegno, A. Determination of Hydrodynamic Radius of Proteins by Size Exclusion Chromatography. Bio-Protocol 2017, 7, e2230. [Google Scholar]
- Giese, A.P.J.; Tang, Y.Q.; Sinha, G.P.; Bowl, M.R.; Goldring, A.C.; Parker, A.; Freeman, M.J.; Brown, S.D.M.; Riazuddin, S.; Fettiplace, R.; et al. CIB2 interacts with TMC1 and TMC2 and is essential for mechanotransduction in auditory hair cells. Nat. Commun. 2017, 8, 43. [Google Scholar] [CrossRef]
- Weljie, A.M.; Yamniuk, A.P.; Yoshino, H.; Izumi, Y.; Vogel, H.J. Protein conformational changes studied by diffusion NMR spectroscopy: Application to helix-loop-helix calcium binding proteins. Protein Sci. 2003, 12, 228–236. [Google Scholar] [CrossRef]
- Sobczak, A.; Blazejczyk, M.; Piszczek, G.; Zhao, G.; Kuznicki, J.; Wojda, U. Calcium-binding calmyrin forms stable covalent dimers in vitro, but in vivo is found in monomeric form. Acta Biochim. Pol. 2005, 52, 469–476. [Google Scholar]
- Blamey, C.J.; Ceccarelli, C.; Naik, U.P.; Bahnson, B.J. The crystal structure of calcium- and integrin-binding protein 1: Insights into redox regulated functions. Protein Sci. 2005, 14, 1214–1221. [Google Scholar] [CrossRef]
- Wright, M.H.; Heal, W.P.; Mann, D.J.; Tate, E.W. Protein myristoylation in health and disease. J. Chem. Biol. 2010, 3, 19–35. [Google Scholar] [CrossRef]
- Ames, J.B.; Tanaka, T.; Stryer, L.; Ikura, M. Portrait of a myristoyl switch protein. Curr. Opin. Struct. Biol. 1996, 6, 432–438. [Google Scholar] [CrossRef]
- Jarman, K.E.; Moretti, P.A.; Zebol, J.R.; Pitson, S.M. Translocation of sphingosine kinase 1 to the plasma membrane is mediated by calcium- and integrin-binding protein 1. J. Biol. Chem. 2010, 285, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Blazejczyk, M.; Wojda, U.; Sobczak, A.; Spilker, C.; Bernstein, H.G.; Gundelfinger, E.D.; Kreutz, M.R.; Kuznicki, J. Ca2+-independent binding and cellular expression profiles question a significant role of calmyrin in transduction of Ca2+-signals to Alzheimer’s disease-related presenilin 2 in forebrain. Biochim. Biophys. Acta 2006, 1762, 66–72. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hennigs, J.K.; Burhenne, N.; Stahler, F.; Winnig, M.; Walter, B.; Meyerhof, W.; Schmale, H. Sweet taste receptor interacting protein CIB1 is a general inhibitor of InsP3-dependent Ca2+ release in vivo. J. Neurochem. 2008, 106, 2249–2262. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Stabler, S.M.; Ames, J.B.; Baskakov, I.; Monteiro, M.J. Calcium binding sequences in calmyrin regulates interaction with presenilin-2. Exp. Cell Res. 2004, 300, 440–454. [Google Scholar] [CrossRef] [PubMed]
- Heineke, J.; Auger-Messier, M.; Correll, R.N.; Xu, J.; Benard, M.J.; Yuan, W.; Drexler, H.; Parise, L.V.; Molkentin, J.D. CIB1 is a regulator of pathological cardiac hypertrophy. Nat. Med. 2010, 16, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Freeman, T.C., Jr.; Black, J.L.; Bray, H.G.; Dagliyan, O.; Wu, Y.I.; Tripathy, A.; Dokholyan, N.V.; Leisner, T.M.; Parise, L.V. Identification of novel integrin binding partners for calcium and integrin binding protein 1 (CIB1): Structural and thermodynamic basis of CIB1 promiscuity. Biochemistry 2013, 52, 7082–7090. [Google Scholar] [CrossRef] [PubMed]
- Shock, D.D.; Naik, U.P.; Brittain, J.E.; Alahari, S.K.; Sondek, J.; Parise, L.V. Calcium-dependent properties of CIB binding to the integrin alphaIIb cytoplasmic domain and translocation to the platelet cytoskeleton. Biochem. J. 1999, 342 Pt 3, 729–735. [Google Scholar]
- Spilker, C.; Dresbach, T.; Braunewell, K.H. Reversible translocation and activity-dependent localization of the calcium-myristoyl switch protein VILIP-1 to different membrane compartments in living hippocampal neurons. J. Neurosci. 2002, 22, 7331–7339. [Google Scholar]
- Zhu, W.; Jarman, K.E.; Lokman, N.A.; Neubauer, H.A.; Davies, L.T.; Gliddon, B.L.; Taing, H.; Moretti, P.A.B.; Oehler, M.K.; Pitman, M.R.; et al. CIB2 Negatively Regulates Oncogenic Signaling in Ovarian Cancer via Sphingosine Kinase 1. Cancer Res. 2017, 77, 4823–4834. [Google Scholar] [CrossRef]
- Yuan, W.; Leisner, T.M.; McFadden, A.W.; Wang, Z.; Larson, M.K.; Clark, S.; Boudignon-Proudhon, C.; Lam, S.C.; Parise, L.V. CIB1 is an endogenous inhibitor of agonist-induced integrin alphaIIbbeta3 activation. J. Cell Biol. 2006, 172, 169–175. [Google Scholar] [CrossRef]
- Naik, U.P.; Naik, M.U. Association of CIB with GPIIb/IIIa during outside-in signaling is required for platelet spreading on fibrinogen. Blood 2003, 102, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Ozga, M.; Aghajanian, C.; Myers-Virtue, S.; McDonnell, G.; Jhanwar, S.; Hichenberg, S.; Sulimanoff, I. A systematic review of ovarian cancer and fear of recurrence. Palliat. Support. Care 2015, 13, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Yeung, T.L.; Leung, C.S.; Yip, K.P.; Au Yeung, C.L.; Wong, S.T.; Mok, S.C. Cellular and molecular processes in ovarian cancer metastasis. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. Am. J. Physiol.-Cell Physiol. 2015, 309, C444–C456. [Google Scholar] [CrossRef] [PubMed]
- Godinho-Santos, A.; Hance, A.J.; Goncalves, J.; Mammano, F. CIB1 and CIB2 are HIV-1 helper factors involved in viral entry. Sci. Rep. 2016, 6, 30927. [Google Scholar] [CrossRef]
- Rato, S.; Maia, S.; Brito, P.M.; Resende, L.; Pereira, C.F.; Moita, C.; Freitas, R.P.; Moniz-Pereira, J.; Hacohen, N.; Moita, L.F.; et al. Novel HIV-1 knockdown targets identified by an enriched kinases/phosphatases shRNA library using a long-term iterative screen in Jurkat T-cells. PLoS ONE 2010, 5, e9276. [Google Scholar] [CrossRef]
- Zhou, H.; Xu, M.; Huang, Q.; Gates, A.T.; Zhang, X.D.; Castle, J.C.; Stec, E.; Ferrer, M.; Strulovici, B.; Hazuda, D.J.; et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 2008, 4, 495–504. [Google Scholar] [CrossRef]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef]
- Michel, V.; Booth, K.T.; Patni, P.; Cortese, M.; Azaiez, H.; Bahloul, A.; Kahrizi, K.; Labbe, M.; Emptoz, A.; Lelli, A.; et al. CIB2, defective in isolated deafness, is key for auditory hair cell mechanotransduction and survival. EMBO Mol. Med. 2017, 9, 1711–1731. [Google Scholar] [CrossRef]
- Yang, H.; Jiang, X.; Li, B.; Yang, H.J.; Miller, M.; Yang, A.; Dhar, A.; Pavletich, N.P. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 2017, 552, 368–373. [Google Scholar] [CrossRef]
- Prasad, A.; Merico, D.; Thiruvahindrapuram, B.; Wei, J.; Lionel, A.C.; Sato, D.; Rickaby, J.; Lu, C.; Szatmari, P.; Roberts, W.; et al. A discovery resource of rare copy number variations in individuals with autism spectrum disorder. G3 Genes Genomes Genet. 2012, 2, 1665–1685. [Google Scholar] [CrossRef]
- Mangino, M.; Cecelja, M.; Menni, C.; Tsai, P.C.; Yuan, W.; Small, K.; Bell, J.; Mitchell, G.F.; AortaGen, C.; Chowienczyk, P.; et al. Integrated multiomics approach identifies calcium and integrin-binding protein-2 as a novel gene for pulse wave velocity. J. Hypertens. 2016, 34, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Vastagh, C.; Solymosi, N.; Farkas, I.; Liposits, Z. Proestrus Differentially Regulates Expression of Ion Channel and Calcium Homeostasis Genes in GnRH Neurons of Mice. Front. Mol. Neurosci. 2019, 12, 137. [Google Scholar] [CrossRef] [PubMed]
- Ajayi, A.F.; Akhigbe, R.E. Staging of the estrous cycle and induction of estrus in experimental rodents: An update. Fertil. Res. Pract. 2020, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Littlewood Evans, A.; Muller, U. Stereocilia defects in the sensory hair cells of the inner ear in mice deficient in integrin alpha8beta1. Nat. Genet. 2000, 24, 424–428. [Google Scholar] [CrossRef]
- Calderwood, D.A.; Shattil, S.J.; Ginsberg, M.H. Integrins and actin filaments: Reciprocal regulation of cell adhesion and signaling. J. Biol. Chem. 2000, 275, 22607–22610. [Google Scholar] [CrossRef]
- Cunningham, C.L.; Muller, U. Molecular Structure of the Hair Cell Mechanoelectrical Transduction Complex. Cold Spring Harb. Perspect. Med. 2019, 9, a033167. [Google Scholar] [CrossRef]
- Pan, B.; Geleoc, G.S.; Asai, Y.; Horwitz, G.C.; Kurima, K.; Ishikawa, K.; Kawashima, Y.; Griffith, A.J.; Holt, J.R. TMC1 and TMC2 are components of the mechanotransduction channel in hair cells of the mammalian inner ear. Neuron 2013, 79, 504–515. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Yao, X.; Li, W.; Du, H.; Tang, M.; Xiong, W.; Chai, R.; Xu, Z. Loss of CIB2 Causes Profound Hearing Loss and Abolishes Mechanoelectrical Transduction in Mice. Front. Mol. Neurosci. 2017, 10, 401. [Google Scholar] [CrossRef]
- Belyantseva, I.A.; Boger, E.T.; Naz, S.; Frolenkov, G.I.; Sellers, J.R.; Ahmed, Z.M.; Griffith, A.J.; Friedman, T.B. Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat. Cell Biol. 2005, 7, 148–156. [Google Scholar] [CrossRef]
- Manor, U.; Disanza, A.; Grati, M.; Andrade, L.; Lin, H.; Di Fiore, P.P.; Scita, G.; Kachar, B. Regulation of stereocilia length by myosin XVa and whirlin depends on the actin-regulatory protein Eps8. Curr. Biol. 2011, 21, 167–172. [Google Scholar] [CrossRef]
- Mauriac, S.A.; Hien, Y.E.; Bird, J.E.; Carvalho, S.D.; Peyroutou, R.; Lee, S.C.; Moreau, M.M.; Blanc, J.M.; Geyser, A.; Medina, C.; et al. Defective Gpsm2/Galphai3 signalling disrupts stereocilia development and growth cone actin dynamics in Chudley-McCullough syndrome. Nat. Commun. 2017, 8, 14907. [Google Scholar] [CrossRef] [PubMed]
- Zampini, V.; Ruttiger, L.; Johnson, S.L.; Franz, C.; Furness, D.N.; Waldhaus, J.; Xiong, H.; Hackney, C.M.; Holley, M.C.; Offenhauser, N.; et al. Eps8 regulates hair bundle length and functional maturation of mammalian auditory hair cells. PLoS Biol. 2011, 9, e1001048. [Google Scholar] [CrossRef]
- Yan, K.; Zong, W.; Du, H.; Zhai, X.; Ren, R.; Liu, S.; Xiong, W.; Wang, Y.; Xu, Z. BAIAP2L2 is required for the maintenance of mechanotransducing stereocilia of cochlear hair cells. J. Cell. Physiol. 2022, 237, 774–788. [Google Scholar] [CrossRef] [PubMed]
- Bowl, M.R.; Simon, M.M.; Ingham, N.J.; Greenaway, S.; Santos, L.; Cater, H.; Taylor, S.; Mason, J.; Kurbatova, N.; Pearson, S.; et al. A large scale hearing loss screen reveals an extensive unexplored genetic landscape for auditory dysfunction. Nat. Commun. 2017, 8, 886. [Google Scholar] [CrossRef]
- Booth, K.T.; Kahrizi, K.; Babanejad, M.; Daghagh, H.; Bademci, G.; Arzhangi, S.; Zareabdollahi, D.; Duman, D.; El-Amraoui, A.; Tekin, M.; et al. Variants in CIB2 cause DFNB48 and not USH1J. Clin. Genet. 2018, 93, 812–821. [Google Scholar] [CrossRef]
- Talbi, S.; Bonnet, C.; Riahi, Z.; Boudjenah, F.; Dahmani, M.; Hardelin, J.P.; Wong Jun Tai, F.; Louha, M.; Ammar-Khodja, F.; Petit, C. Genetic heterogeneity of congenital hearing impairment in Algerians from the Ghardaia province. Int. J. Pediatr. Otorhinolaryngol. 2018, 112, 1–5. [Google Scholar] [CrossRef]
- Souissi, A.; Ben Said, M.; Ben Ayed, I.; Elloumi, I.; Bouzid, A.; Mosrati, M.A.; Hasnaoui, M.; Belcadhi, M.; Idriss, N.; Kamoun, H.; et al. Novel pathogenic mutations and further evidence for clinical relevance of genes and variants causing hearing impairment in Tunisian population. J. Adv. Res. 2021, 31, 13–24. [Google Scholar] [CrossRef]
- Seco, C.Z.; Giese, A.P.; Shafique, S.; Schraders, M.; Oonk, A.M.; Grossheim, M.; Oostrik, J.; Strom, T.; Hegde, R.; van Wijk, E.; et al. Novel and recurrent CIB2 variants, associated with nonsyndromic deafness, do not affect calcium buffering and localization in hair cells. Eur. J. Hum. Genet. 2016, 24, 542–549. [Google Scholar] [CrossRef]
- Richard, E.M.; Santos-Cortez, R.L.P.; Faridi, R.; Rehman, A.U.; Lee, K.; Shahzad, M.; Acharya, A.; Khan, A.A.; Imtiaz, A.; Chakchouk, I.; et al. Global genetic insight contributed by consanguineous Pakistani families segregating hearing loss. Hum. Mutat. 2019, 40, 53–72. [Google Scholar] [CrossRef]
- Adeyemo, A.; Faridi, R.; Chattaraj, P.; Yousaf, R.; Tona, R.; Okorie, S.; Bharadwaj, T.; Nouel-Saied, L.M.; Acharya, A.; Schrauwen, I.; et al. Genomic analysis of childhood hearing loss in the Yoruba population of Nigeria. Eur. J. Hum. Genet. 2022, 30, 42–52. [Google Scholar] [CrossRef]
- Patel, K.; Giese, A.P.; Grossheim, J.M.; Hegde, R.S.; Delio, M.; Samanich, J.; Riazuddin, S.; Frolenkov, G.I.; Cai, J.; Ahmed, Z.M.; et al. A Novel C-Terminal CIB2 (Calcium and Integrin Binding Protein 2) Mutation Associated with Non-Syndromic Hearing Loss in a Hispanic Family. PLoS ONE 2015, 10, e0133082. [Google Scholar] [CrossRef]
- Fuster-Garcia, C.; Garcia-Garcia, G.; Jaijo, T.; Fornes, N.; Ayuso, C.; Fernandez-Burriel, M.; Sanchez-De la Morena, A.; Aller, E.; Millan, J.M. High-throughput sequencing for the molecular diagnosis of Usher syndrome reveals 42 novel mutations and consolidates CEP250 as Usher-like disease causative. Sci. Rep. 2018, 8, 17113. [Google Scholar] [CrossRef] [PubMed]
- Booth, K.T.; Azaiez, H.; Kahrizi, K.; Simpson, A.C.; Tollefson, W.T.; Sloan, C.M.; Meyer, N.C.; Babanejad, M.; Ardalani, F.; Arzhangi, S.; et al. PDZD7 and hearing loss: More than just a modifier. Am. J. Med. Genet. A 2015, 167, 2957–2965. [Google Scholar] [CrossRef]
- Nishio, S.Y.; Takumi, Y.; Usami, S.I. Laser-capture micro dissection combined with next-generation sequencing analysis of cell type-specific deafness gene expression in the mouse cochlea. Hear. Res. 2017, 348, 87–97. [Google Scholar] [CrossRef]
- Aparisi, M.J.; Aller, E.; Fuster-Garcia, C.; Garcia-Garcia, G.; Rodrigo, R.; Vazquez-Manrique, R.P.; Blanco-Kelly, F.; Ayuso, C.; Roux, A.F.; Jaijo, T.; et al. Targeted next generation sequencing for molecular diagnosis of Usher syndrome. Orphanet J. Rare Dis. 2014, 9, 168. [Google Scholar] [CrossRef]
- Ahmed, Z.M.; Frolenkov, G.I.; Riazuddin, S. Usher proteins in inner ear structure and function. Physiol. Genom. 2013, 45, 987–989. [Google Scholar] [CrossRef]
- Cosgrove, D.; Zallocchi, M. Usher protein functions in hair cells and photoreceptors. Int. J. Biochem. Cell Biol. 2014, 46, 80–89. [Google Scholar] [CrossRef]
- Jouret, G.; Poirsier, C.; Spodenkiewicz, M.; Jaquin, C.; Gouy, E.; Arndt, C.; Labrousse, M.; Gaillard, D.; Doco-Fenzy, M.; Lebre, A.S. Genetics of Usher Syndrome: New Insights From a Meta-analysis. Otol. Neurotol. 2019, 40, 121–129. [Google Scholar] [CrossRef]
- Shen, X.; Valencia, C.A.; Szostak, J.W.; Dong, B.; Liu, R. Scanning the human proteome for calmodulin-binding proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 5969–5974. [Google Scholar] [CrossRef]
- O’Connell, D.J.; Bauer, M.; Linse, S.; Cahill, D.J. Probing calmodulin protein-protein interactions using high-content protein arrays. Methods Mol. Biol. 2011, 785, 289–303. [Google Scholar] [CrossRef]



| CIB2 | Integrin | Method | Ref. | ||||
|---|---|---|---|---|---|---|---|
| Origin | Tag | Name | Origin | Seq. | Assay | T (°C), Ions | |
| M. | Yes | αIIb | M. | LVLAMWKAGFFKRNR | ELISA | 25 °C | [8] |
| M. | No | α7B | M. | LAADWHPELGPDGHPVPATA | ITF | 20 °C, Ca2+ | [3] |
| H. | Yes | αIIb α7B_Mα7B_C | H. M. M. | LVLAMWKVGFFKRNRPPLEEDDEEGQ LVLLLWKLGFFKRA LAADWHPELGPDGHPVPATA | ITF | 25 °C, apo, Mg2+ and Ca2+ | [13] |
| H. | No | α7B_M | H. | LVLLLWKLGFFKRA | ITF | 37 °C, Mg2+ Ca2+ | [14] |
| H. | Yes | α7B_Mα7B_C | H. | LVLLLWKLGFFKRA LAADWHPELGPDGHPVPATA | SPR | 25 °C, Mg2+ vs. Mg2+ and Ca2+ | [12] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dal Cortivo, G.; Dell’Orco, D. Calcium- and Integrin-Binding Protein 2 (CIB2) in Physiology and Disease: Bright and Dark Sides. Int. J. Mol. Sci. 2022, 23, 3552. https://doi.org/10.3390/ijms23073552
Dal Cortivo G, Dell’Orco D. Calcium- and Integrin-Binding Protein 2 (CIB2) in Physiology and Disease: Bright and Dark Sides. International Journal of Molecular Sciences. 2022; 23(7):3552. https://doi.org/10.3390/ijms23073552
Chicago/Turabian StyleDal Cortivo, Giuditta, and Daniele Dell’Orco. 2022. "Calcium- and Integrin-Binding Protein 2 (CIB2) in Physiology and Disease: Bright and Dark Sides" International Journal of Molecular Sciences 23, no. 7: 3552. https://doi.org/10.3390/ijms23073552
APA StyleDal Cortivo, G., & Dell’Orco, D. (2022). Calcium- and Integrin-Binding Protein 2 (CIB2) in Physiology and Disease: Bright and Dark Sides. International Journal of Molecular Sciences, 23(7), 3552. https://doi.org/10.3390/ijms23073552