
New Insights into the Treatment of Glomerular Diseases: When Mechanisms Become Vivid


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
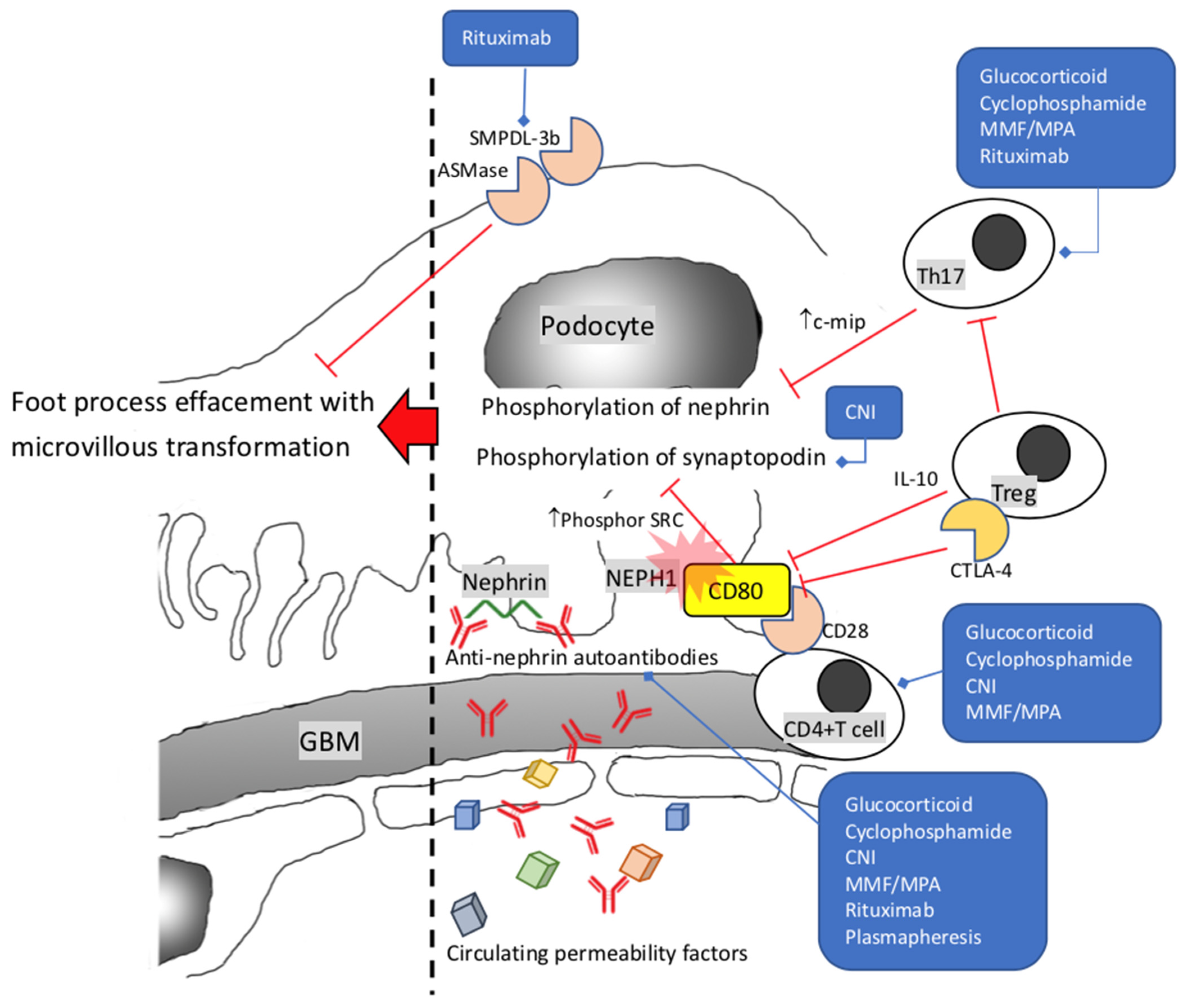
2. Minimal Change Disease and Primary Focal Segmental Glomerulosclerosis
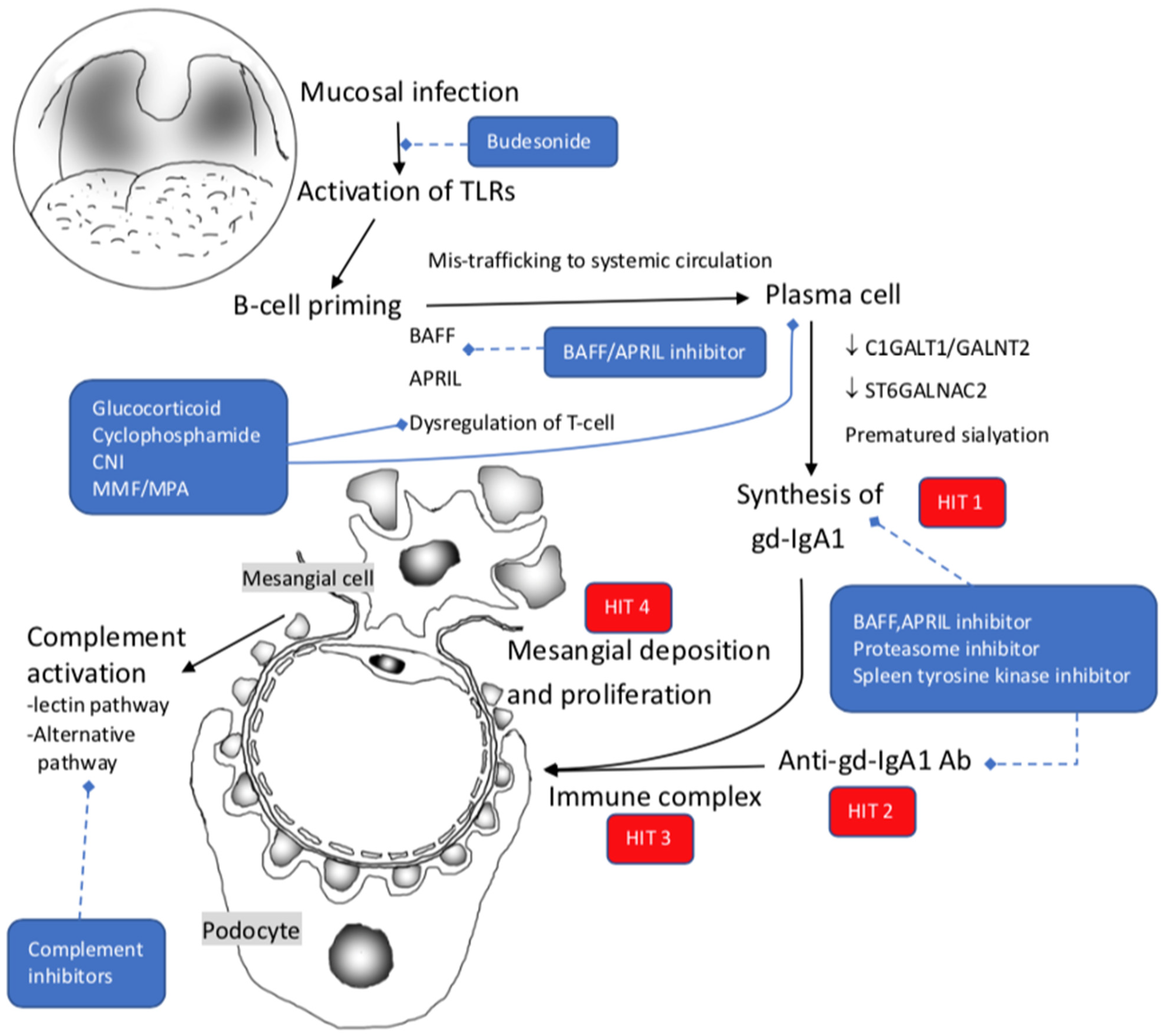
3. IgA Nephropathy
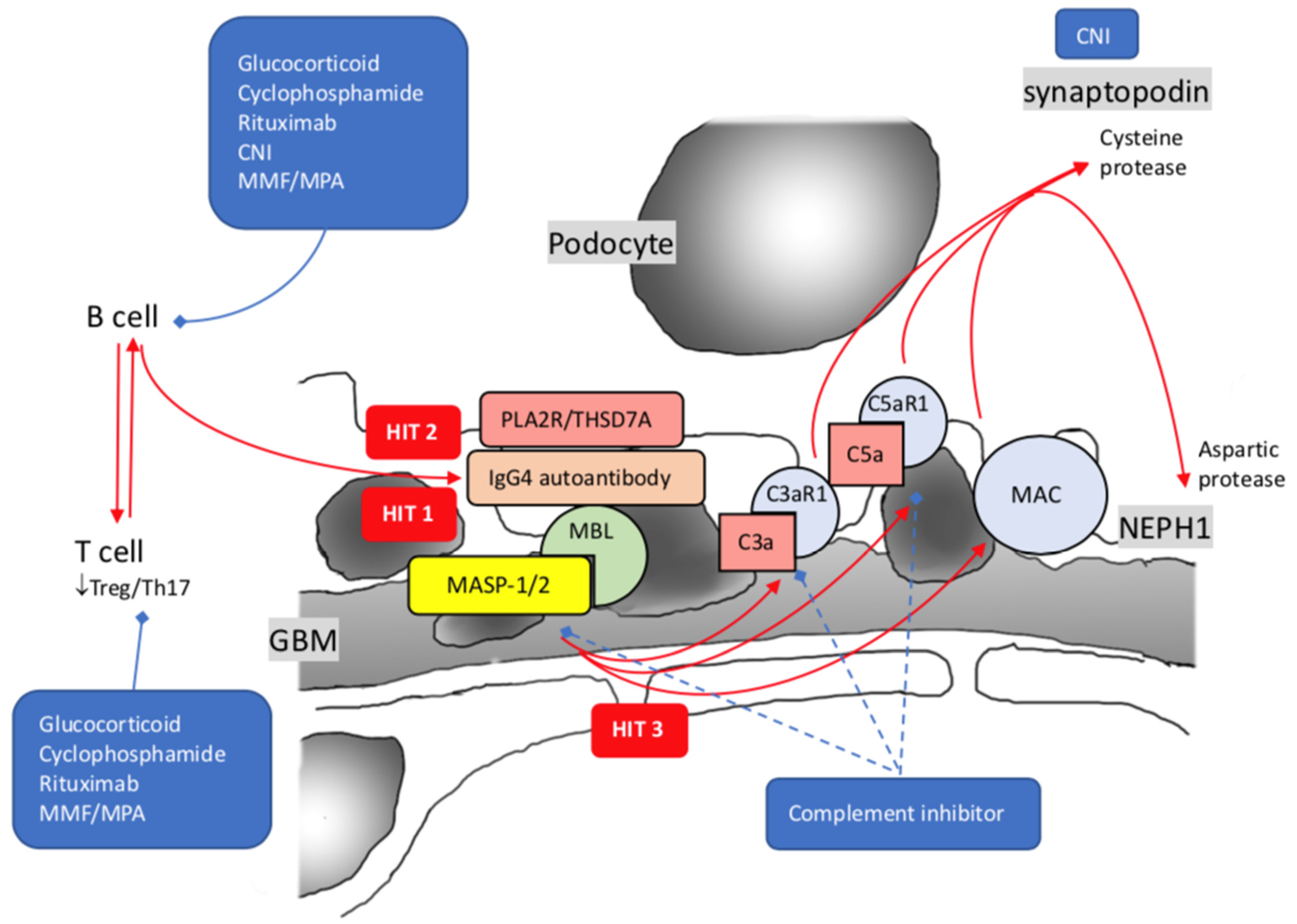
4. Membranous Nephropathy
5. The Glucocorticoid: Still a Drug Full of Uncertainties
6. Cyclophosphamide, Cyclosporine, and Mycophenolate Acid: Indispensable Helpers
6.1. Cyclophosphamide
6.2. Calcineurin Inhibitors
6.3. Mycophenolate Mofetil/Mycophenolic Acid Analogue
7. Rituximab and Complement Inhibitors: The Rising Stars
7.1. Rituximab
7.2. Complement Inhibitors
8. Nrf2 Activator: Accessory Agents for Immunosuppressants
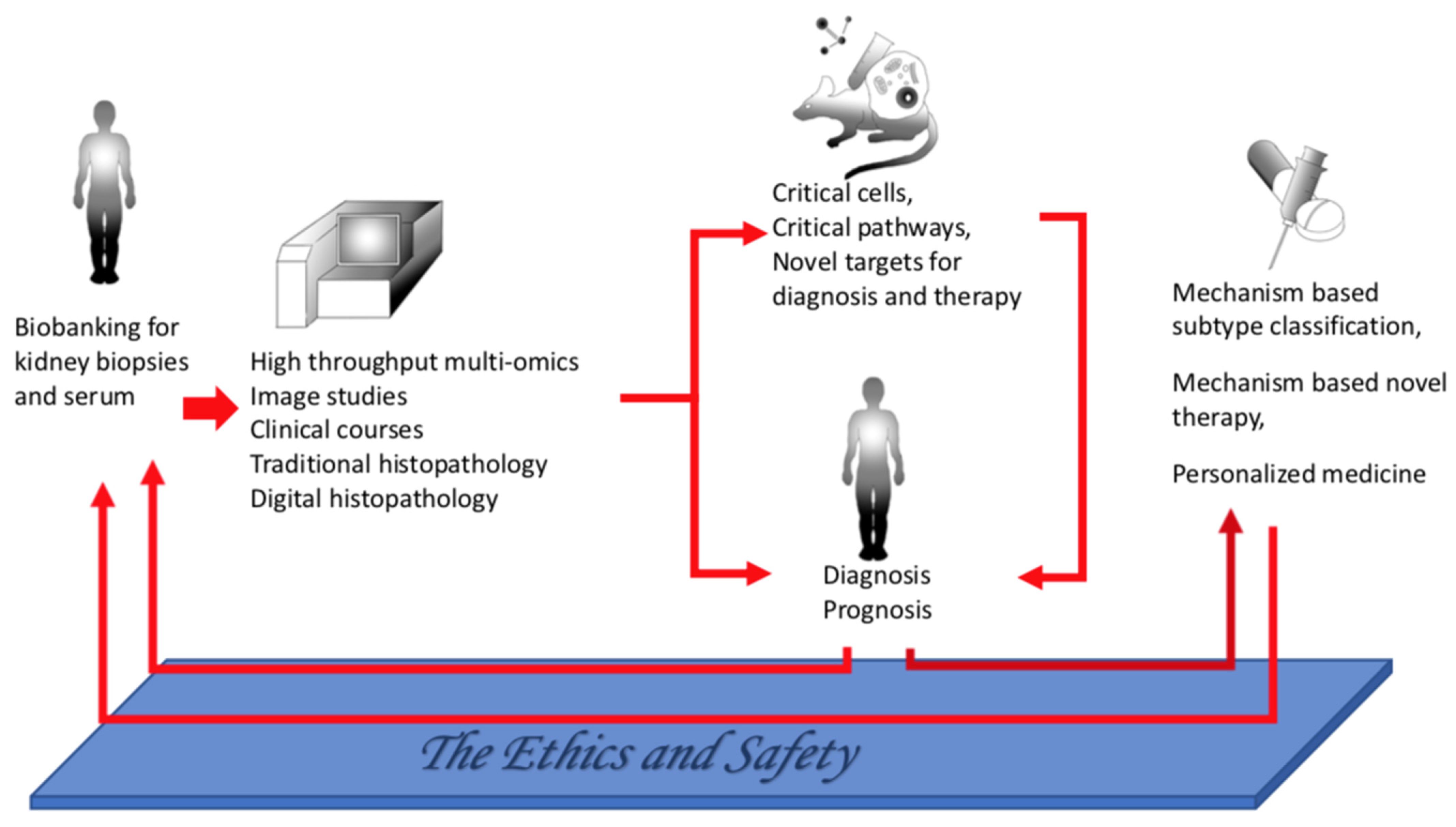
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McGrogan, A.; Franssen, C.F.; de Vries, C.S. The incidence of primary glomerulonephritis worldwide: A systematic review of the literature. Nephrol. Dial. Transplant. 2011, 26, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Trimarchi, H.; Barratt, J.; Cattran, D.C.; Cook, H.T.; Coppo, R.; Haas, M.; Liu, Z.H.; Roberts, I.S.; Yuzawa, Y.; Zhang, H.; et al. Oxford Classification of IgA nephropathy 2016: An update from the IgA Nephropathy Classification Working Group. Kidney Int. 2017, 91, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Miyabe, Y.; Karasawa, K.; Akiyama, K.; Ogura, S.; Takabe, T.; Sugiura, N.; Seki, M.; Iwabuchi, Y.; Hanafusa, N.; Uchida, K.; et al. Grading system utilising the total score of Oxford classification for predicting renal prognosis in IgA nephropathy. Sci. Rep. 2021, 11, 3584. [Google Scholar] [CrossRef]
- Eddy, A.A.; Symons, J.M. Nephrotic syndrome in childhood. Lancet 2003, 362, 629–639. [Google Scholar] [CrossRef]
- Floege, J.; Amann, K. Primary glomerulonephritides. Lancet 2016, 387, 2036–2048. [Google Scholar] [CrossRef]
- Mathieson, P.W. Minimal change nephropathy and focal segmental glomerulosclerosis. Semin. Immunopathol. 2007, 29, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.B.; Anders, H.J.; Susztak, K.; Podestà, M.A.; Remuzzi, G.; Hildebrandt, F.; Romagnani, P. Podocytopathies. Nat. Rev. Dis. Primers 2020, 6, 68. [Google Scholar] [CrossRef]
- D’Agati, V.D.; Kaskel, F.J.; Falk, R.J. Focal segmental glomerulosclerosis. N. Engl. J. Med. 2011, 365, 2398–2411. [Google Scholar] [CrossRef] [PubMed]
- Maas, R.J.; Deegens, J.K.; Smeets, B.; Moeller, M.J.; Wetzels, J.F. Minimal change disease and idiopathic FSGS: Manifestations of the same disease. Nat. Rev. Nephrol. 2016, 12, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Deegens, J.K.; Dijkman, H.B.; Borm, G.F.; Steenbergen, E.J.; van den Berg, J.G.; Weening, J.J.; Wetzels, J.F. Podocyte foot process effacement as a diagnostic tool in focal segmental glomerulosclerosis. Kidney Int. 2008, 74, 1568–1576. [Google Scholar] [CrossRef] [PubMed]
- Wiggins, R.C. The spectrum of podocytopathies: A unifying view of glomerular diseases. Kidney Int. 2007, 71, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Tejani, A. Morphological transition in minimal change nephrotic syndrome. Nephron 1985, 39, 157–159. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.A.; Markowitz, G.S.; Valeri, A.; Appel, G.B. Collapsing glomerulopathy. Semin. Nephrol. 2003, 23, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Shalhoub, R.J. Pathogenesis of lipoid nephrosis: A disorder of T-cell function. Lancet 1974, 2, 556–560. [Google Scholar] [CrossRef]
- MacDonald, N.E.; Wolfish, N.; McLaine, P.; Phipps, P.; Rossier, E. Role of respiratory viruses in exacerbations of primary nephrotic syndrome. J. Pediatr. 1986, 108, 378–382. [Google Scholar] [CrossRef]
- Kim, S.R.; Lee, S.B.; Kim, I.Y.; Lee, D.W.; Rhee, H.; Seong, E.Y.; Song, S.H.; Kwak, I.S. Relapse of minimal change disease following infection with the 2009 pandemic influenza (H1N1) virus. Clin. Exp. Nephrol. 2012, 16, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Hsu, H.C. Histopathological and immunological studies in spontaneous remission of nephrotic syndrome after intercurrent measles infection. Nephron 1986, 42, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, N.; Batwara, R.; McCarthy, E.T.; Sharma, R.; Sharma, M.; Savin, V.J. Serum permeability activity in steroid-resistant minimal change nephrotic syndrome is abolished by treatment of Hodgkin disease. Am. J. Kidney Dis. 2007, 50, 826–829. [Google Scholar] [CrossRef] [PubMed]
- Peces, R.; Sánchez, L.; Gorostidi, M.; Alvarez, J. Minimal change nephrotic syndrome associated with Hodgkin’s lymphoma. Nephrol. Dial. Transplant. 1991, 6, 155–158. [Google Scholar] [CrossRef]
- Rizvi, S.N.; Vaishnava, H. Cyclophosphamide in minimal-change nephrotic syndrome. Lancet 1972, 2, 537–538. [Google Scholar] [CrossRef]
- Black, D.A.; Rose, G.; Brewer, D.B. Controlled trial of prednisone in adult patients with the nephrotic syndrome. Br. Med. J. 1970, 3, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Shimada, M.; Araya, C.; Rivard, C.; Ishimoto, T.; Johnson, R.J.; Garin, E.H. Minimal change disease: A “two-hit” podocyte immune disorder? Pediatr. Nephrol. 2011, 26, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Wakem, P.; Burns, R.P.; Ramirez, F.; Zlotnick, D.; Ferbel, B.; Haidaris, C.G.; Gaspari, A.A. Allergens and irritants transcriptionally upregulate CD80 gene expression in human keratinocytes. J. Investig. Dermatol. 2000, 114, 1085–1092. [Google Scholar] [CrossRef]
- Reiser, J.; von Gersdorff, G.; Loos, M.; Oh, J.; Asanuma, K.; Giardino, L.; Rastaldi, M.P.; Calvaresi, N.; Watanabe, H.; Schwarz, K.; et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J. Clin. Investig. 2004, 113, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Khullar, B.; Balyan, R.; Oswal, N.; Jain, N.; Sharma, A.; Abdin, M.Z.; Bagga, A.; Bhatnagar, S.; Wadhwa, N.; Natchu, U.C.M.; et al. Interaction of CD80 with Neph1: A potential mechanism of podocyte injury. Clin. Exp. Nephrol. 2018, 22, 508–516. [Google Scholar] [CrossRef]
- Shimada, M.; Ishimoto, T.; Lee, P.Y.; Lanaspa, M.A.; Rivard, C.J.; Roncal-Jimenez, C.A.; Wymer, D.T.; Yamabe, H.; Mathieson, P.W.; Saleem, M.A.; et al. Toll-like receptor 3 ligands induce CD80 expression in human podocytes via an NF-kappaB-dependent pathway. Nephrol. Dial. Transplant. 2012, 27, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Shimada, M.; Gabriela, G.; Kosugi, T.; Sato, W.; Lee, P.Y.; Lanaspa, M.A.; Rivard, C.; Maruyama, S.; Garin, E.H.; et al. Toll-like receptor 3 ligand, polyIC, induces proteinuria and glomerular CD80, and increases urinary CD80 in mice. Nephrol. Dial. Transplant. 2013, 28, 1439–1446. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Liu, Y.; He, Y.; Rong, W.; Zhang, M.; Li, L.; Liu, Z.; Zen, K. Podocytes present antigen to activate specific T cell immune responses in inflammatory renal disease. J. Pathol. 2020, 252, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, S.; Kimata, T.; Yamanouchi, S.; Kitao, T.; Kino, J.; Suruda, C.; Kaneko, K. Regulatory T cells and CTLA-4 in idiopathic nephrotic syndrome. Pediatr. Int. 2017, 59, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Kimata, T.; Tsuji, S.; Kino, J.; Kitao, T.; Yamanouchi, S.; Kaneko, K. Close association between proteinuria and regulatory T cells in patients with idiopathic nephrotic syndrome. Pediatr. Nephrol. 2013, 28, 667–669. [Google Scholar] [CrossRef] [PubMed]
- Cara-Fuentes, G.; Wasserfall, C.H.; Wang, H.; Johnson, R.J.; Garin, E.H. Minimal change disease: A dysregulation of the podocyte CD80-CTLA-4 axis? Pediatr. Nephrol. 2014, 29, 2333–2340. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Matsumoto, K. Decreased release of IL-10 by monocytes from patients with lipoid nephrosis. Clin. Exp. Immunol. 1995, 102, 603–607. [Google Scholar] [CrossRef]
- Chen, W.; Wu, Y.; Zhang, G.; Wang, M.; Yang, H.; Li, Q. Gammadeltat Cells Exacerbate Podocyte Injury via the CD28/B7-1-Phosphor-SRC Kinase Pathway. Biomed. Res. Int. 2018, 2018, 5647120. [Google Scholar] [PubMed]
- Zhang, S.-Y.; Kamal, M.; Dahan, K.; Pawlak, A.; Ory, V.; Desvaux, D.; Audard, V.; Candelier, M.; BenMohamed, F.; Matignon, M.; et al. c-mip impairs podocyte proximal signaling and induces heavy proteinuria. Sci. Signal 2010, 3, ra39. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Su, L.; Lin, Q.; Han, Y.; You, P.; Fan, Q. Induction of C-Mip by IL-17 Plays an Important Role in Adriamycin-Induced Podocyte Damage. Cell Physiol. Biochem. 2015, 36, 1274–1290. [Google Scholar] [CrossRef] [PubMed]
- May, C.J.; Welsh, G.I.; Chesor, M.; Lait, P.J.; Schewitz-Bowers, L.P.; Lee, R.W.J.; Saleem, M.A. Human Th17 cells produce a soluble mediator that increases podocyte motility via signaling pathways that mimic PAR-1 activation. Am. J. Physiol. Renal. Physiol. 2019, 317, F913–F921. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; Qin, Y.; Cai, J.F.; Wang, H.Y.; Tao, J.L.; Li, H.; Chen, L.M.; Li, M.X.; Li, X.M.; Li, X.W. Th17/Treg imbalance in adult patients with minimal change nephrotic syndrome. Clin. Immunol. 2011, 139, 314–320. [Google Scholar] [CrossRef]
- Stachowski, J.; Barth, C.; Michałkiewicz, J.; Krynicki, T.; Jarmoliński, T.; Runowski, D.; Lewandowska-Stachowiak, M.; Zaniew, M.; Warzywoda, A.; Bortkiewicz, E.; et al. Th1/Th2 balance and CD45-positive T cell subsets in primary nephrotic syndrome. Pediatr. Nephrol. 2000, 14, 779–785. [Google Scholar] [CrossRef]
- Webendörfer, M.; Reinhard, L.; Stahl, R.A.K.; Wiech, T.; Mittrücker, H.-W.; Harendza, S.; Hoxha, E. Rituximab Induces Complete Remission of Proteinuria in a Patient With Minimal Change Disease and No Detectable B Cells. Front. Immunol. 2020, 11, 586012. [Google Scholar] [CrossRef]
- Fenoglio, R.; Sciascia, S.; Beltrame, G.; Mesiano, P.; Ferro, M.; Quattrocchio, G.; Menegatti, E.; Roccatello, D. Rituximab as a front-line therapy for adult-onset minimal change disease with nephrotic syndrome. Oncotarget 2018, 9, 28799–28804. [Google Scholar] [CrossRef]
- Takemura, S.; Klimiuk, P.; Braun, A.; Goronzy, J.J.; Weyand, C.M. T cell activation in rheumatoid synovium is B cell dependent. J. Immunol. 2001, 167, 4710–4718. [Google Scholar] [CrossRef] [PubMed]
- Silverman, G.J.; Weisman, S. Rituximab therapy and autoimmune disorders: Prospects for anti-B cell therapy. Arthritis Rheum. 2003, 48, 1484–1492. [Google Scholar] [CrossRef]
- Lipsky, P.E. Systemic lupus erythematosus: An autoimmune disease of B cell hyperactivity. Nat. Immunol. 2001, 2, 764–766. [Google Scholar] [CrossRef] [PubMed]
- Silverman, G.J. Anti-CD20 therapy and autoimmune disease: Therapeutic opportunities and evolving insights. Front. Biosci. 2007, 12, 2194–2206. [Google Scholar] [CrossRef] [PubMed]
- MacAulay, A.E.; DeKruyff, R.H.; Goodnow, C.; Umetsu, D.T. Antigen-specific B cells preferentially induce CD4+ T cells to produce IL-4. J. Immunol. 1997, 158, 4171–4179. [Google Scholar] [PubMed]
- Clark, R.; Kupper, T. Old meets new: The interaction between innate and adaptive immunity. J. Investig. Dermatol. 2005, 125, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.; Bagga, A. Rituximab therapy in nephrotic syndrome: Implications for patients’ management. Nat. Rev. Nephrol. 2013, 9, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Hansrivijit, P.; Cheungpasitporn, W.; Thongprayoon, C.; Ghahramani, N. Rituximab therapy for focal segmental glomerulosclerosis and minimal change disease in adults: A systematic review and meta-analysis. BMC Nephrol. 2020, 21, 134. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.C.; Jobson, M.A.; Schober, F.P.; Chang, E.H.; Falk, R.J.; Nachman, P.H.; Pendergraft, W.F. The Evolving Role of Rituximab in Adult Minimal Change Glomerulopathy. Am. J. Nephrol. 2017, 45, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Bai, A.; Robson, S. Beyond ecto-nucleotidase: CD39 defines human Th17 cells with CD161. Purinergic Signal. 2015, 11, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Ravani, P.; Bonanni, A.; Rossi, R.; Caridi, G.; Ghiggeri, G.M. Anti-CD20 Antibodies for Idiopathic Nephrotic Syndrome in Children. Clin. J. Am. Soc. Nephrol. 2016, 11, 710–720. [Google Scholar] [CrossRef] [PubMed]
- van de Veerdonk, F.L.; Lauwerys, B.; Marijnissen, R.J.; Timmermans, K.; Di Padova, F.; Koenders, M.I.; Gutierrez-Roelens, I.; Durez, P.; Netea, M.G.; van der Meer, J.W.M.; et al. The anti-CD20 antibody rituximab reduces the Th17 cell response. Arthritis Rheum. 2011, 63, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Orikasa, M.; Matsui, K.; Oite, T.; Shimizu, F. Massive proteinuria induced in rats by a single intravenous injection of a monoclonal antibody. J. Immunol. 1988, 141, 807–814. [Google Scholar]
- Topham, P.S.; Kawachi, H.; Haydar, S.A.; Chugh, S.; Addona, T.A.; Charron, K.B.; Holzman, L.B.; Shia, M.; Shimizu, F.; Salant, D.J. Nephritogenic mAb 5-1-6 is directed at the extracellular domain of rat nephrin. J. Clin. Investig. 1999, 104, 1559–1566. [Google Scholar] [CrossRef]
- Takeuchi, K.; Naito, S.; Kawashima, N.; Ishigaki, N.; Sano, T.; Kamata, K.; Takeuchi, Y. New Anti-Nephrin Antibody Mediated Podocyte Injury Model. Using a C57BL/6 Mouse Strain. Nephron 2018, 138, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Coward, R.J.; Foster, R.R.; Patton, D.; Ni, L.; Lennon, R.; Bates, D.O.; Harper, S.J.; Mathieson, P.W.; Saleem, M.A. Nephrotic plasma alters slit diaphragm-dependent signaling and translocates nephrin, Podocin, and CD2 associated protein in cultured human podocytes. J. Am. Soc. Nephrol. 2005, 16, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Wernerson, A. Altered ultrastructural distribution of nephrin in minimal change nephrotic syndrome. Nephrol. Dial. Transplant. 2003, 18, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Watts, A.J.; Keller, K.H.; Lerner, G.; Rosales, I.; Collins, A.B.; Sekulic, M.; Waikar, S.S.; Chandraker, A.; Riella, L.V.; Alexander, M.P.; et al. Discovery of Autoantibodies Targeting Nephrin in Minimal Change Disease Supports a Novel Autoimmune Etiology. J. Am. Soc. Nephrol. 2022, 33, 238–252. [Google Scholar] [CrossRef]
- Kienzl-Wagner, K.; Waldegger, S.; Schneeberger, S. Disease Recurrence-The Sword of Damocles in Kidney Transplantation for Primary Focal Segmental Glomerulosclerosis. Front. Immunol. 2019, 10, 1669. [Google Scholar] [CrossRef]
- McCarthy, E.T.; Sharma, M.; Savin, V.J. Circulating permeability factors in idiopathic nephrotic syndrome and focal segmental glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2010, 5, 2115–2121. [Google Scholar] [CrossRef] [PubMed]
- Chebotareva, N.; Vinogradov, A.; Cao, V.; Gindis, A.; Berns, A.; Alentov, I.; Sergeeva, N. Serum levels of plasminogen activator urokinase receptor and cardiotrophin-like cytokine factor 1 in patients with nephrotic syndrome. Clin. Nephrol. 2022, 97, 103–110. [Google Scholar] [CrossRef]
- Doublier, S.; Zennaro, C.; Musante, L.; Spatola, T.; Candiano, G.; Bruschi, M.; Besso, L.; Cedrino, M.; Carraro, M.; Ghiggeri, G.M.; et al. Soluble CD40 ligand directly alters glomerular permeability and may act as a circulating permeability factor in FSGS. PLoS ONE 2017, 12, e0188045. [Google Scholar] [CrossRef]
- Li, Y.; Kang, Y.S.; Dai, C.; Kiss, L.P.; Wen, X.; Liu, Y. Epithelial-to-mesenchymal transition is a potential pathway leading to podocyte dysfunction and proteinuria. Am. J. Pathol. 2008, 172, s299–s308. [Google Scholar] [CrossRef] [PubMed]
- Huber, T.B.; Benzing, T. The slit diaphragm: A signaling platform to regulate podocyte function. Curr. Opin. Nephrol. Hypertens. 2005, 14, 211–216. [Google Scholar] [CrossRef]
- Verma, R.; Wharram, B.; Kovari, I.; Kunkel, R.; Nihalani, D.; Wary, K.; Wiggins, R.; Killen, P.; Holzman, L.B. Fyn binds to and phosphorylates the kidney slit diaphragm component Nephrin. J. Biol. Chem. 2003, 278, 20716–20723. [Google Scholar] [CrossRef]
- Lahdenperä, J.; Kilpeläinen, P.; Liu, X.L.; Pikkarainen, T.; Reponen, P.; Ruotsalainen, V.; Tryggvason, K. Clustering-induced tyrosine phosphorylation of nephrin by Src family kinases. Kidney Int. 2003, 64, 404–413. [Google Scholar] [CrossRef]
- Li, H.; Lemay, S.; Aoudjit, L.; Kawachi, H.; Takano, T. SRC-family kinase Fyn phosphorylates the cytoplasmic domain of nephrin and modulates its interaction with podocin. J. Am. Soc. Nephrol. 2004, 15, 3006–3015. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Sun, N.; Aoudjit, L.; Li, H.; Kawachi, H.; Lemay, S.; Takano, T. Nephrin mediates actin reorganization via phosphoinositide 3-kinase in podocytes. Kidney Int. 2008, 73, 556–566. [Google Scholar] [CrossRef]
- Blasutig, I.M.; New, L.A.; Thanabalasuriar, A.; Dayarathna, T.K.; Goudreault, M.; Quaggin, S.E.; Li, S.S.; Gruenheid, S.; Jones, N.; Pawson, T. Phosphorylated YDXV motifs and Nck SH2/SH3 adaptors act cooperatively to induce actin reorganization. Mol. Cell Biol. 2008, 28, 2035–2046. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Suzuki, K.; Iwamoto, M.; Kawachi, H.; Ohno, M.; Horita, S.; Nitta, K. Decreased tyrosine phosphorylation of nephrin in rat and human nephrosis. Kidney Int. 2008, 73, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, T.; Uchida, K.; Asamiya, Y.; Tsuruta, Y.; Ohno, M.; Horita, S.; Nitta, K. Phosphorylation status of nephrin in human membranous nephropathy. Clin. Exp. Nephrol. 2010, 14, 51–55. [Google Scholar] [CrossRef]
- New, L.A.; Martin, C.E.; Scott, R.; Platt, M.J.; Chahi, A.K.; Stringer, C.D.; Lu, P.; Samborska, B.; Eremina, V.; Takano, T.; et al. Nephrin Tyrosine Phosphorylation Is Required to Stabilize and Restore Podocyte Foot Process Architecture. J. Am. Soc. Nephrol. 2016, 27, 2422–2435. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.C.; Rhodes, G.; Wang, E.; Pruthi, V.; Arif, E.; Saleem, M.A.; Wean, S.E.; Garg, P.; Verma, R.; Holzman, L.B.; et al. Ischemic injury to kidney induces glomerular podocyte effacement and dissociation of slit diaphragm proteins Neph1 and ZO-1. J. Biol. Chem. 2008, 283, 35579–35589. [Google Scholar] [CrossRef]
- Harita, Y.; Kurihara, H.; Kosako, H.; Tezuka, T.; Sekine, T.; Igarashi, T.; Hattori, S. Neph1, a component of the kidney slit diaphragm, is tyrosine-phosphorylated by the Src family tyrosine kinase and modulates intracellular signaling by binding to Grb2. J. Biol. Chem. 2008, 283, 9177–9186. [Google Scholar] [CrossRef]
- Arif, E.; Rathore, Y.S.; Kumari, B.; Ashish, F.; Wong, H.N.; Holzman, L.B.; Nihalani, D. Slit diaphragm protein Neph1 and its signaling: A novel therapeutic target for protection of podocytes against glomerular injury. J. Biol. Chem. 2014, 289, 9502–9518. [Google Scholar] [CrossRef]
- Fukusumi, Y.; Zhang, Y.; Yamagishi, R.; Oda, K.; Watanabe, T.; Matsui, K.; Kawachi, H. Nephrin-Binding Ephrin-B1 at the Slit Diaphragm Controls Podocyte Function through the JNK Pathway. J. Am. Soc. Nephrol. 2018, 29, 1462–1474. [Google Scholar] [CrossRef] [PubMed]
- Yanagida-Asanuma, E.; Asanuma, K.; Kim, K.; Donnelly, M.; Choi, H.Y.; Chang, J.H.; Suetsugu, S.; Tomino, Y.; Takenawa, T.; Faul, C.; et al. Synaptopodin protects against proteinuria by disrupting Cdc42:IRSp53:Mena signaling complexes in kidney podocytes. Am. J. Pathol. 2007, 171, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.M.; Kim, S.H.; North, K.N.; Rennke, H.; A Correia, L.; Tong, H.-Q.; Mathis, B.J.; Rodríguez-Pérez, J.-C.; Allen, P.G.; Beggs, A.; et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet. 2000, 24, 251–256. [Google Scholar] [CrossRef]
- Buvall, L.; Wallentin, H.; Sieber, J.; Andreeva, S.; Choi, H.Y.; Mundel, P.; Greka, A. Synaptopodin Is a Coincidence Detector of Tyrosine versus Serine/Threonine Phosphorylation for the Modulation of Rho Protein Crosstalk in Podocytes. J. Am. Soc. Nephrol. 2017, 28, 837–851. [Google Scholar] [CrossRef]
- Johansen, K.L.; Chertow, G.M.; Foley, R.N.; Gilbertson, D.T.; Herzog, C.A.; Ishani, A.; Israni, A.K.; Ku, E.; Kurella Tamura, M.; Li, S.; et al. 2020 USRDS Annual Data Report: Epidemiology of kidney disease in the United States. Am. J. Kidney Dis. 2021, 77 (Suppl. 1), A7–A8. [Google Scholar] [CrossRef]
- Brown, E.J.; Pollak, M.R.; Barua, M. Genetic testing for nephrotic syndrome and FSGS in the era of next-generation sequencing. Kidney Int. 2014, 85, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Lepori, N.; Zand, L.; Sethi, S.; Fernandez-Juarez, G.; Fervenza, F.C. Clinical and pathological phenotype of genetic causes of focal segmental glomerulosclerosis in adults. Clin. Kidney J. 2018, 11, 179–190. [Google Scholar] [CrossRef]
- Sadowski, C.E.; Lovric, S.; Ashraf, S.; Pabst, W.L.; Gee, H.Y.; Kohl, S.; Engelmann, S.; Vega-Warner, V.; Fang, H.; Halbritter, J.; et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J. Am. Soc. Nephrol. 2015, 26, 1279–1289. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.Z.; Kopp, J.B. Focal Segmental Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2017, 12, 502–517. [Google Scholar] [CrossRef]
- Friedman, D.J.; Pollak, M.R. APOL1 Nephropathy: From Genetics to Clinical Applications. Clin. J. Am. Soc. Nephrol. 2021, 16, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Nourbakhsh, N.; Mak, R.H. Steroid-resistant nephrotic syndrome: Past and current perspectives. Pediatric. Health Med. Ther. 2017, 8, 29–37. [Google Scholar] [CrossRef]
- Rovin, B.H.; Adler, S.G.; Barratt, J.; Bridoux, F.; Burdge, K.A.; Chan, T.M.; Cook, H.T.; Fervenza, F.C.; Gibson, K.L.; Glassock, R.J.; et al. Executive summary of the KDIGO 2021 Guideline for the Management of Glomerular Diseases. Kidney Int. 2021, 100, 753–779. [Google Scholar] [CrossRef]
- Ding, W.Y.; Koziell, A.; McCarthy, H.J.; Bierzynska, A.; Bhagavatula, M.K.; Dudley, J.A.; Inward, C.D.; Coward, R.; Tizard, J.; Reid, C.; et al. Initial steroid sensitivity in children with steroid-resistant nephrotic syndrome predicts post-transplant recurrence. J. Am. Soc. Nephrol. 2014, 25, 1342–1348. [Google Scholar] [CrossRef] [PubMed]
- Julian, B.A.; Woodford, S.Y.; Baehler, R.W.; McMorrow, R.G.; Wyatt, R.J. Familial clustering and immunogenetic aspects of IgA nephropathy. Am. J. Kidney Dis. 1988, 12, 366–370. [Google Scholar] [CrossRef]
- Sato, Y.; Tsukaguchi, H.; Higasa, K.; Kawata, N.; Inui, K.; Linh, T.N.T.; Quynh, T.T.H.; Yoshihiko, I.; Koiwa, F.; Yoshimura, A. Positive renal familial history in IgA nephropathy is associated with worse renal outcomes: A single-center longitudinal study. BMC Nephrol. 2021, 22, 230. [Google Scholar] [CrossRef]
- Rodrigues, J.C.; Haas, M.; Reich, H.N. IgA Nephropathy. Clin. J. Am. Soc. Nephrol. 2017, 12, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Magistroni, R.; D’Agati, V.D.; Appel, G.B.; Kiryluk, K. New developments in the genetics, pathogenesis, and therapy of IgA nephropathy. Kidney Int. 2015, 88, 974–989. [Google Scholar] [CrossRef]
- Baenziger, J.; Kornfeld, S. Structure of the carbohydrate units of IgA1 immunoglobulin. II. Structure of the O-glycosidically linked oligosaccharide units. J. Biol. Chem. 1974, 249, 7270–7281. [Google Scholar] [PubMed]
- Tomana, M.; Niedermeier, W.; Mestecky, J.; Skvaril, F. The differences in carbohydrate composition between the subclasses of IgA immunoglobulins. Immunochemistry 1976, 13, 325–328. [Google Scholar] [CrossRef]
- Tarelli, E.; Smith, A.C.; Hendry, B.M.; Challacombe, S.; Pouria, S. Human serum IgA1 is substituted with up to six O-glycans as shown by matrix assisted laser desorption ionisation time-of-flight mass spectrometry. Carbohydr. Res. 2004, 339, 2329–2335. [Google Scholar] [CrossRef]
- Franc, V.; Řehulka, P.; Raus, M.; Stulík, J.; Novak, J.; Renfrow, M.B.; Šebela, M. Elucidating heterogeneity of IgA1 hinge-region O-glycosylation by use of MALDI-TOF/TOF mass spectrometry: Role of cysteine alkylation during sample processing. J. Proteom. 2013, 92, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Field, M.C.; Dwek, R.A.; Edge, C.J.; Rademacher, T.W. O-linked oligosaccharides from human serum immunoglobulin A1. Biochem. Soc. Trans. 1989, 17, 1034–1035. [Google Scholar] [CrossRef]
- Mattu, T.S.; Pleass, R.J.; Willis, A.C.; Kilian, M.; Wormald, M.R.; Lellouch, A.C.; Rudd, P.M.; Woof, J.M.; Dwek, R.A. The glycosylation and structure of human serum IgA1, Fab, and Fc regions and the role of N-glycosylation on Fcalpha receptor interactions. J. Biol. Chem. 1998, 273, 2260–2272. [Google Scholar] [CrossRef] [PubMed]
- Ohyama, Y.; Renfrow, M.; Novak, J.; Takahashi, K. Aberrantly Glycosylated IgA1 in IgA Nephropathy: What We Know and What We Don’t Know. J. Clin. Med. 2021, 10, 3467. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.C.; Bailey, E.M.; Brenchley, P.; Buck, K.S.; Barratt, J.; Feehally, J. Mesangial IgA1 in IgA nephropathy exhibits aberrant O-glycosylation: Observations in three patients. Kidney Int. 2001, 60, 969–973. [Google Scholar] [CrossRef]
- Takahashi, K.; Raska, M.; Horynová, M.S.; Hall, S.D.; Poulsen, K.; Kilian, M.; Hiki, Y.; Yuzawa, Y.; Moldoveanu, Z.; Julian, B.A.; et al. Enzymatic sialylation of IgA1 O-glycans: Implications for studies of IgA nephropathy. PLoS ONE 2014, 9, e99026. [Google Scholar] [CrossRef]
- Wang, L.; Wang, H.; Li, X.; Fan, J. Bacterial IgA protease-mediated degradation of agIgA1 and agIgA1 immune complexes as a potential therapy for IgA Nephropathy. Sci. Rep. 2016, 6, 30964. [Google Scholar] [CrossRef] [PubMed]
- Blaas, S.H.; Stieber-Gunckel, M.; Falk, W.; Obermeier, F.; Rogler, G. CpG-oligodeoxynucleotides stimulate immunoglobulin A secretion in intestinal mucosal B cells. Clin. Exp. Immunol. 2009, 155, 534–540. [Google Scholar] [CrossRef]
- Cognasse, F.; Acquart, S.; Béniguel, L.; Sabido, O.; Chavarin, P.; Genin, C.; Garraud, O. Differential production of immunoglobulin classes and subclasses by mucosal-type human B-lymphocytes exposed in vitro to CpG oligodeoxynucleotides. Clin. Chem. Lab. Med. 2005, 43, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Takahara, M.; Nagato, T.; Nozaki, Y.; Kumai, T.; Katada, A.; Hayashi, T.; Harabuchi, Y. A proliferation-inducing ligand (APRIL) induced hyper-production of IgA from tonsillar mononuclear cells in patients with IgA nephropathy. Cell Immunol. 2019, 341, 103925. [Google Scholar] [CrossRef]
- Martín-Penagos, L.; Benito-Hernández, A.; Segundo, D.S.; Sango, C.; Azueta, A.; Gómez-Román, J.; Fernández-Fresnedo, G.; López-Hoyos, M.; Ruiz, J.C.; Rodrigo, E. A proliferation-inducing ligand increase precedes IgA nephropathy recurrence in kidney transplant recipients. Clin. Transplant. 2019, 33, e13502. [Google Scholar] [CrossRef] [PubMed]
- Xin, G.; Shi, W.; Xu, L.-X.; Su, Y.; Yan, L.-J.; Li, K.-S. Serum BAFF is elevated in patients with IgA nephropathy and associated with clinical and histopathological features. J. Nephrol. 2013, 26, 683–690. [Google Scholar] [CrossRef]
- Im, J.; Baik, J.E.; Lee, D.; Park, O.J.; Park, D.H.; Yun, C.H.; Han, S.H. Bacterial Lipoproteins Induce BAFF Production via TLR2/MyD88/JNK Signaling Pathways in Dendritic Cells. Front. Immunol. 2020, 11, 564699. [Google Scholar] [CrossRef]
- Hardenberg, G.; Planelles, L.; Schwarte, C.M.; van Bostelen, L.; Le Huong, T.; Hahne, M.; Medema, J.P. Specific TLR ligands regulate APRIL secretion by dendritic cells in a PKR-dependent manner. Eur. J. Immunol. 2007, 37, 2900–2911. [Google Scholar] [CrossRef]
- Batra, A.; Smith, A.C.; Feehally, J.; Barratt, J. T-cell homing receptor expression in IgA nephropathy. Nephrol. Dial. Transplant. 2007, 22, 2540–2548. [Google Scholar] [CrossRef]
- March, A.K.-D.; Bene, M.C.; Renoult, E.; Kessler, M.; Faure, G.C.; Kolopp-Sarda, M.N. Enhanced expression of L-selectin on peripheral blood lymphocytes from patients with IgA nephropathy. Clin. Exp. Immunol. 1999, 115, 542–546. [Google Scholar] [CrossRef]
- Buren, M.; Yamashita, M.; Suzuki, Y.; Tomino, Y.; Emancipator, S.N. Altered expression of lymphocyte homing chemokines in the pathogenesis of IgA nephropathy. Contrib. Nephrol. 2007, 157, 50–55. [Google Scholar] [PubMed]
- Zheng, N.; Xie, K.; Ye, H.; Dong, Y.; Wang, B.; Luo, N.; Fan, J.; Tan, J.; Chen, W.; Yu, X. TLR7 in B cells promotes renal inflammation and Gd-IgA1 synthesis in IgA nephropathy. JCI Insight 2020, 5, e136965. [Google Scholar] [CrossRef]
- Qin, W.; Zhong, X.; Fan, J.M.; Zhang, Y.J.; Liu, X.R.; Ma, X.Y. External suppression causes the low expression of the Cosmc gene in IgA nephropathy. Nephrol. Dial. Transplant. 2008, 23, 1608–1614. [Google Scholar] [CrossRef]
- Muto, M.; Manfroi, B.; Suzuki, H.; Joh, K.; Nagai, M.; Wakai, S.; Righini, C.; Maiguma, M.; Izui, S.; Tomino, Y.; et al. Toll-Like Receptor 9 Stimulation Induces Aberrant Expression of a Proliferation-Inducing Ligand by Tonsillar Germinal Center B Cells in IgA Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 1227–1238. [Google Scholar] [CrossRef]
- Suzuki, H.; Fan, R.; Zhang, Z.; Brown, R.; Hall, S.; Julian, B.A.; Chatham, W.W.; Suzuki, Y.; Wyatt, R.J.; Moldoveanu, Z.; et al. Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J. Clin. Investig. 2009, 119, 1668–1677. [Google Scholar] [CrossRef] [PubMed]
- Tomana, M.; Novak, J.; Julian, B.A.; Matousovic, K.; Konecny, K.; Mestecky, J. Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J. Clin. Investig. 1999, 104, 73–81. [Google Scholar] [CrossRef]
- Roggenbuck, D.; Mytilinaiou, M.G.; Lapin, S.V.; Reinhold, D.; Conrad, K. Asialoglycoprotein receptor (ASGPR): A peculiar target of liver-specific autoimmunity. Autoimmun. Highlights 2012, 3, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Launay, P.; Grossetête, B.; Arcos-Fajardo, M.; Gaudin, E.; Torres, S.P.; Beaudoin, L.; Patey-Mariaud de Serre, N.; Lehuen, A.; Monteiro, R.C. Fcalpha receptor (CD89) mediates the development of immunoglobulin A (IgA) nephropathy (Berger’s disease). Evidence for pathogenic soluble receptor-Iga complexes in patients and CD89 transgenic mice. J. Exp. Med. 2000, 191, 1999–2009. [Google Scholar] [CrossRef]
- Ebefors, K.; Liu, P.; Lassén, E.; Elvin, J.; Candemark, E.; Levan, K.; Haraldsson, B.; Nyström, J. Mesangial cells from patients with IgA nephropathy have increased susceptibility to galactose-deficient IgA1. BMC Nephrol. 2016, 17, 40. [Google Scholar] [CrossRef]
- Zhao, Y.-F.; Zhu, L.; Liu, L.-J.; Shi, S.-F.; Lv, J.-C.; Zhang, H. Pathogenic role of glycan-specific IgG antibodies in IgA nephropathy. BMC Nephrol. 2017, 18, 301. [Google Scholar] [CrossRef]
- Wu, M.-Y.; Chen, C.-S.; Yiang, G.-T.; Cheng, P.-W.; Chen, Y.-L.; Chiu, H.-C.; Liu, K.-H.; Lee, W.-C.; Li, C.-J. The Emerging Role of Pathogenesis of IgA Nephropathy. J. Clin. Med. 2018, 7, 225. [Google Scholar] [CrossRef]
- Roos, A.; Rastaldi, M.P.; Calvaresi, N.; Oortwijn, B.D.; Schlagwein, N.; Van Gijlswijk-Janssen, D.J.; Stahl, G.; Matsushita, M.; Fujita, T.; van Kooten, C.; et al. Glomerular activation of the lectin pathway of complement in IgA nephropathy is associated with more severe renal disease. J. Am. Soc. Nephrol. 2006, 17, 1724–1734. [Google Scholar] [CrossRef]
- Medjeral-Thomas, N.R.; Lomax-Browne, H.J.; Beckwith, H.; Willicombe, M.; McLean, A.G.; Brookes, P.; Pusey, C.D.; Falchi, M.; Cook, H.T.; Pickering, M.C. Circulating complement factor H-related proteins 1 and 5 correlate with disease activity in IgA nephropathy. Kidney Int. 2017, 92, 942–952. [Google Scholar] [CrossRef]
- Medjeral-Thomas, N.R.; Troldborg, A.; Constantinou, N.; Lomax-Browne, H.J.; Hansen, A.G.; Willicombe, M.; Pusey, C.D.; Cook, H.T.; Thiel, S.; Pickering, M.C. Progressive IgA Nephropathy Is Associated With Low Circulating Mannan-Binding Lectin-Associated Serine Protease-3 (MASP-3) and Increased Glomerular Factor H-Related Protein-5 (FHR5) Deposition. Kidney Int. Rep. 2018, 3, 426–438. [Google Scholar] [CrossRef]
- Cox, S.N.; Sallustio, F.; Serino, G.; Loverre, A.; Pesce, F.; Gigante, M.; Zaza, G.; Stifanelli, P.F.; Ancona, N.; Schena, F.P. Activated innate immunity and the involvement of CX3CR1-fractalkine in promoting hematuria in patients with IgA nephropathy. Kidney Int. 2012, 82, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Kiryluk, K.; Li, Y.; Scolari, F.; Sanna-Cherchi, S.; Choi, M.; Verbitsky, M.; Fasel, D.; Lata, S.; Prakash, S.; Shapiro, S.; et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat. Genet. 2014, 46, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Gharavi, A.G.; Kiryluk, K.; Choi, M.; Li, Y.; Hou, P.; Xie, J.; Sanna-Cherchi, S.; Men, C.J.; A Julian, B.; Wyatt, R.; et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat. Genet. 2011, 43, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Feenstra, B.; Bager, P.; Liu, X.; Hjalgrim, H.; A Nohr, E.; Hougaard, D.M.; Geller, F.; Melbye, M. Genome-wide association study identifies variants in HORMAD2 associated with tonsillectomy. J. Med. Genet. 2017, 54, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-Q.; Li, M.; Zhang, H.; Low, H.-Q.; Wei, X.; Wang, J.-Q.; Sun, L.-D.; Sim, K.S.; Li, Y.; Foo, J.N.; et al. A genome-wide association study in Han Chinese identifies multiple susceptibility loci for IgA nephropathy. Nat. Genet. 2011, 44, 178–182. [Google Scholar] [CrossRef]
- Ai, Z.; Li, M.; Liu, W.; Foo, J.-N.; Mansouri, O.; Yin, P.; Zhou, Q.; Tang, X.; Dong, X.; Feng, S.; et al. Low alpha-defensin gene copy number increases the risk for IgA nephropathy and renal dysfunction. Sci. Transl. Med. 2016, 8, 345ra88. [Google Scholar] [CrossRef] [PubMed]
- Kiryluk, K.; Li, Y.; Moldoveanu, Z.; Suzuki, H.; Reily, C.; Hou, P.; Xie, J.; Mladkova, N.; Prakash, S.; Fischman, C.; et al. GWAS for serum galactose-deficient IgA1 implicates critical genes of the O-glycosylation pathway. PLoS Genet. 2017, 13, e1006609. [Google Scholar] [CrossRef]
- Rauen, T.; Fitzner, C.; Eitner, F.; Sommerer, C.; Zeier, M.; Otte, B.; Panzer, U.; Peters, H.; Benck, U.; Mertens, P.R.; et al. Effects of Two Immunosuppressive Treatment Protocols for IgA Nephropathy. J. Am. Soc. Nephrol. 2018, 29, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Tesar, V.; Troyanov, S.; Bellur, S.; Verhave, J.C.; Cook, H.T.; Feehally, J.; Roberts, I.S.; Cattran, D.; Coppo, R.; On behalf of the VALIGA study of the ERA-EDTA Immunonephrology Working Group. Corticosteroids in IgA Nephropathy: A Retrospective Analysis from the VALIGA Study. J. Am. Soc. Nephrol. 2015, 26, 2248–2258. [Google Scholar] [CrossRef]
- Lv, J.; Zhang, H.; Wong, M.G.; Jardine, M.J.; Hladunewich, M.; Jha, V.; Monaghan, H.; Zhao, M.; Barbour, S.; Reich, H.; et al. Effect of Oral Methylprednisolone on Clinical Outcomes in Patients With IgA Nephropathy: The TESTING Randomized Clinical Trial. JAMA 2017, 318, 432–442. [Google Scholar] [CrossRef]
- Yang, Y.-Z.; Chen, P.; Liu, L.-J.; Cai, Q.-Q.; Shi, S.-F.; Chen, Y.-Q.; Lv, J.-C.; Zhang, H. Comparison of the effects of hydroxychloroquine and corticosteroid treatment on proteinuria in IgA nephropathy: A case-control study. BMC Nephrol. 2019, 20, 297. [Google Scholar] [CrossRef] [PubMed]
- Maes, B.D.; Oyen, R.; Claes, K.; Evenepoel, P.; Kuypers, D.R.; Vanwalleghem, J.; Van Damme, B.; Vanrenterghem, Y.F.C. Mycophenolate mofetil in IgA nephropathy: Results of a 3-year prospective placebo-controlled randomized study. Kidney Int. 2004, 65, 1842–1849. [Google Scholar] [CrossRef]
- Zheng, J.; Bi, T.; Zhu, L.; Liu, L. Efficacy and safety of mycophenolate mofetil for IgA nephropathy: An updated meta-analysis of randomized controlled trials. Exp. Ther. Med. 2018, 16, 1882–1890. [Google Scholar] [CrossRef]
- Lafayette, R.A.; Canetta, P.; Rovin, B.H.; Appel, G.B.; Novak, J.; Nath, K.A.; Sethi, S.; Tumlin, J.A.; Mehta, K.; Hogan, M.; et al. A Randomized, Controlled Trial of Rituximab in IgA Nephropathy with Proteinuria and Renal Dysfunction. J. Am. Soc. Nephrol. 2017, 28, 1306–1313. [Google Scholar] [CrossRef] [PubMed]
- McHeyzer-Williams, M.G.; Ahmed, R. B cell memory and the long-lived plasma cell. Curr. Opin. Immunol. 1999, 11, 172–179. [Google Scholar] [CrossRef]
- O’Connor, B.P.; Cascalho, M.; Noelle, R.J. Short-lived and long-lived bone marrow plasma cells are derived from a novel precursor population. J. Exp. Med. 2002, 195, 737–745. [Google Scholar] [CrossRef]
- Fellström, B.C.; Barratt, J.; Cook, H.; Coppo, R.; Feehally, J.; de Fijter, J.W.; Floege, J.; Hetzel, G.; Jardine, A.G.; Locatelli, F.; et al. Targeted-release budesonide versus placebo in patients with IgA nephropathy (NEFIGAN): A double-blind, randomised, placebo-controlled phase 2b trial. Lancet 2017, 389, 2117–2127. [Google Scholar] [CrossRef]
- Zand, L.; Canetta, P.; Lafayette, R.; Aslam, N.; Jan, N.; Sethi, S.; Fervenza, F.C. An Open-Label Pilot Study of Adrenocorticotrophic Hormone in the Treatment of IgA Nephropathy at High Risk of Progression. Kidney Int. Rep. 2020, 5, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Hartono, C.; Chung, M.; Perlman, A.S.; Chevalier, J.M.; Serur, D.; Seshan, S.V.; Muthukumar, T. Bortezomib for Reduction of Proteinuria in IgA Nephropathy. Kidney Int. Rep. 2018, 3, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Rosenblad, T.; Rebetz, J.; Johansson, M.; Békássy, Z.; Sartz, L.; Karpman, D. Eculizumab treatment for rescue of renal function in IgA nephropathy. Pediatr. Nephrol. 2014, 29, 2225–2228. [Google Scholar] [CrossRef]
- Hirano, K.; Matsuzaki, K.; Yasuda, T.; Nishikawa, M.; Yasuda, Y.; Koike, K.; Maruyama, S.; Yokoo, T.; Matsuo, S.; Kawamura, T.; et al. Association Between Tonsillectomy and Outcomes in Patients With Immunoglobulin A Nephropathy. JAMA Netw. Open 2019, 2, e194772. [Google Scholar] [CrossRef] [PubMed]
- Politano, S.A.; Colbert, G.B.; Hamiduzzaman, N. Nephrotic Syndrome. Prim. Care 2020, 47, 597–613. [Google Scholar] [CrossRef] [PubMed]
- Keri, K.C.; Blumenthal, S.; Kulkarni, V.; Beck, L.; Chongkrairatanakul, T. Primary membranous nephropathy: Comprehensive review and historical perspective. Postgrad. Med. J. 2019, 95, 23–31. [Google Scholar] [CrossRef]
- Couser, W.G. Primary Membranous Nephropathy. Clin. J. Am. Soc. Nephrol. 2017, 12, 983–997. [Google Scholar] [CrossRef]
- Cattran, D.C.; Brenchley, P.E. Membranous nephropathy: Integrating basic science into improved clinical management. Kidney Int. 2017, 91, 566–574. [Google Scholar] [CrossRef]
- Debiec, H.; Guigonis, V.; Mougenot, B.; Decobert, F.; Haymann, J.-P.; Bensman, A.; Deschenes, G.; Ronco, P.M. Antenatal membranous glomerulonephritis due to anti-neutral endopeptidase antibodies. N. Engl. J. Med. 2002, 346, 2053–2060. [Google Scholar] [CrossRef]
- Beck, L.; Bonegio, R.; Lambeau, G.; Beck, D.M.; Powell, D.W.; Cummins, T.D.; Klein, J.B.; Salant, D.J. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N. Engl. J. Med. 2009, 361, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wan, J.; Liu, Y.; Yang, Q.; Liang, W.; Singhal, P.C.; Saleem, M.A.; Ding, G. sPLA2 IB induces human podocyte apoptosis via the M-type phospholipase A2 receptor. Sci. Rep. 2014, 4, 6660. [Google Scholar] [CrossRef]
- Salant, D.J. Does Epitope Spreading Influence Responsiveness to Rituximab in PLA2R-Associated Membranous Nephropathy? Clin. J. Am. Soc. Nephrol. 2019, 14, 1122–1124. [Google Scholar] [CrossRef] [PubMed]
- Seitz-Polski, B.; Dolla, G.; Payre, C.; Girard, C.; Polidori, J.; Zorzi, K.; Birgy-Barelli, E.; Jullien, P.; Courivaud, C.; Krummel, T.; et al. Epitope Spreading of Autoantibody Response to PLA2R Associates with Poor Prognosis in Membranous Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 1517–1533. [Google Scholar] [CrossRef] [PubMed]
- Tomas, N.M.; Beck, L.H.; Meyer-Schwesinger, C.; Seitz-Polski, B.; Ma, H.; Zahner, G.; Dolla, G.; Hoxha, E.; Helmchen, U.; Dabert-Gay, A.-S.; et al. Thrombospondin Type-1 Domain-Containing 7A in Idiopathic Membranous Nephropathy. N. Engl. J. Med. 2014, 371, 2277–2287. [Google Scholar] [CrossRef]
- Ravindran, A.; Moura, M.C.; Fervenza, F.C.; Nasr, S.H.; Alexander, M.P.; Fidler, M.E.; Hernandez, L.P.H.; Zhang, P.; Grande, J.P.; Cornell, L.D.; et al. In Patients with Membranous Lupus Nephritis, Exostosin-Positivity and Exostosin-Negativity Represent Two Different Phenotypes. J. Am. Soc. Nephrol. 2021, 32, 695–706. [Google Scholar] [CrossRef]
- Saïdi, M.; Brochériou, I.; Estève, E.; Tuffet, S.; Amoura, Z.; Miyara, M.; Belenfant, X.; Ulinski, T.; Rouvier, P.; Debiec, H.; et al. The Exostosin Immunohistochemical Status Differentiates Lupus Membranous Nephropathy Subsets with Different Outcomes. Kidney Int. Rep. 2021, 6, 1977–1980. [Google Scholar] [CrossRef] [PubMed]
- Caza, T.N.; Hassen, S.I.; Dvanajscak, Z.; Kuperman, M.; Edmondson, R.; Herzog, C.; Storey, A.; Arthur, J.; Cossey, L.N.; Sharma, S.G.; et al. NELL1 is a target antigen in malignancy-associated membranous nephropathy. Kidney Int. 2020, 99, 967–976. [Google Scholar] [CrossRef]
- Sethi, S.; Debiec, H.; Madden, B.; Vivarelli, M.; Charlesworth, M.C.; Ravindran, A.; Gross, L.; Ulinski, T.; Buob, D.; Tran, C.L.; et al. Semaphorin 3B–associated membranous nephropathy is a distinct type of disease predominantly present in pediatric patients. Kidney Int. 2020, 98, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Caza, T.N.; Hassen, S.I.; Kuperman, M.; Sharma, S.G.; Dvanajscak, Z.; Arthur, J.; Edmondson, R.; Storey, A.; Herzog, C.; Kenan, D.J.; et al. Neural cell adhesion molecule 1 is a novel autoantigen in membranous lupus nephritis. Kidney Int. 2020, 100, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Madden, B.; Debiec, H.; Morelle, J.; Charlesworth, M.C.; Gross, L.; Negron, V.; Buob, D.; Chaudhry, S.; Jadoul, M.; et al. Protocadherin 7–Associated Membranous Nephropathy. J. Am. Soc. Nephrol. 2021, 32, 1249–1261. [Google Scholar] [CrossRef] [PubMed]
- Al-Rabadi, L.F.; Caza, T.; Trivin-Avillach, C.; Rodan, A.R.; Andeen, N.; Hayashi, N.; Williams, B.; Revelo, M.P.; Clayton, F.; Abraham, J.; et al. Serine Protease HTRA1 as a Novel Target Antigen in Primary Membranous Nephropathy. J. Am. Soc. Nephrol. 2021, 32, 1666–1681. [Google Scholar] [CrossRef] [PubMed]
- Rosenzwajg, M.; Languille, E.; Debiec, H.; Hygino, J.; Dahan, K.; Simon, T.; Klatzmann, D.; Ronco, P. B- and T-cell subpopulations in patients with severe idiopathic membranous nephropathy may predict an early response to rituximab. Kidney Int. 2017, 92, 227–237. [Google Scholar] [CrossRef]
- Le, W.-B.; Shi, J.; Zhang, T.; Liu, L.; Qin, H.-Z.; Liang, S.; Zhang, Y.; Zheng, C.-X.; Jiang, S.; Qin, W.-S.; et al. HLA-DRB1*15:01 and HLA-DRB3*02:02 in PLA2R-Related Membranous Nephropathy. J. Am. Soc. Nephrol. 2016, 28, 1642–1650. [Google Scholar] [CrossRef]
- Lv, J.; Hou, W.; Zhou, X.-J.; Liu, G.; Zhou, F.; Zhao, N.; Hou, P.; Zhao, M.; Zhang, H. Interaction between PLA2R1 and HLA-DQA1 Variants Associates with Anti-PLA2R Antibodies and Membranous Nephropathy. J. Am. Soc. Nephrol. 2013, 24, 1323–1329. [Google Scholar] [CrossRef]
- Cui, Z.; Xie, L.-J.; Chen, F.-J.; Pei, Z.-Y.; Zhang, L.-J.; Qu, Z.; Huang, J.; Gu, Q.-H.; Zhang, Y.-M.; Wang, X.; et al. MHC Class II Risk Alleles and Amino Acid Residues in Idiopathic Membranous Nephropathy. J. Am. Soc. Nephrol. 2016, 28, 1651–1664. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Liu, L.; Mladkova, N.; Li, Y.; Ren, H.; Wang, W.; Cui, Z.; Lin, L.; Hu, X.; Yu, X.; et al. The genetic architecture of membranous nephropathy and its potential to improve non-invasive diagnosis. Nat. Commun. 2020, 11, 1600. [Google Scholar] [CrossRef]
- Xu, X.; Wang, G.; Chen, N.; Lu, T.; Nie, S.; Xu, G.; Zhang, P.; Luo, Y.; Wang, Y.; Wang, X.; et al. Long-Term Exposure to Air Pollution and Increased Risk of Membranous Nephropathy in China. J. Am. Soc. Nephrol. 2016, 27, 3739–3746. [Google Scholar] [CrossRef]
- Debiec, H.; Valayannopoulos, V.; Boyer, O.; Nöel, L.-H.; Callard, P.; Sarda, H.; De Lonlay, P.; Niaudet, P.; Ronco, P. Allo-Immune Membranous Nephropathy and Recombinant Aryl Sulfatase Replacement Therapy: A Need for Tolerance Induction Therapy. J. Am. Soc. Nephrol. 2013, 25, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Lehman, A.M.; Albawardi, A.; A Satoskar, A.; Brodsky, S.V.; Nadasdy, G.; A Hebert, L.; Rovin, B.H.; Nadasdy, T. IgG subclass staining in renal biopsies with membranous glomerulonephritis indicates subclass switch during disease progression. Mod. Pathol. 2013, 26, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Olaru, F.; Miner, J.H.; Beck, L.H.J.; Van Der Vlag, J.; Thurman, J.M.; Borza, D.-B. Alternative Pathway Is Essential for Glomerular Complement Activation and Proteinuria in a Mouse Model of Membranous Nephropathy. Front. Immunol. 2018, 9, 1433. [Google Scholar] [CrossRef] [PubMed]
- Tao, M.H.; Canfield, S.M.; Morrison, S.L. The differential ability of human IgG1 and IgG4 to activate complement is determined by the COOH-terminal sequence of the CH2 domain. J. Exp. Med. 1991, 173, 1025–1028. [Google Scholar] [CrossRef]
- Segawa, Y.; Hisano, S.; Matsushita, M.; Fujita, T.; Hirose, S.; Takeshita, M.; Iwasaki, H. IgG subclasses and complement pathway in segmental and global membranous nephropathy. Pediatr. Nephrol. 2010, 25, 1091–1099. [Google Scholar] [CrossRef]
- Brglez, V.; Boyer-Suavet, S.; Seitz-Polski, B. Complement Pathways in Membranous Nephropathy: Complex and Multifactorial. Kidney Int. Rep. 2020, 5, 572–574. [Google Scholar] [CrossRef]
- Haddad, G.; Lorenzen, J.M.; Ma, H.; de Haan, N.; Seeger, H.; Zaghrini, C.; Brandt, S.; Kölling, M.; Wegmann, U.; Kiss, B.; et al. Altered glycosylation of IgG4 promotes lectin complement pathway activation in anti-PLA2R1–associated membranous nephropathy. J. Clin. Investig. 2021, 131, e140453. [Google Scholar] [CrossRef]
- Cattran, D. Management of Membranous Nephropathy: When and What for Treatment. J. Am. Soc. Nephrol. 2005, 16, 1188–1194. [Google Scholar] [CrossRef] [PubMed]
- Donadio, J.V.; Torres, V.E.; Velosa, J.A.; Wagoner, R.D.; Holley, K.E.; Okamura, M.; Ilstrup, D.M.; Chu, C.-P. Idiopathic membranous nephropathy: The natural history of untreated patients. Kidney Int. 1988, 33, 708–715. [Google Scholar] [CrossRef]
- Polanco, N.; Gutiérrez, E.; Covarsí, A.; Ariza, F.; Carreño, A.; Vigil, A.; Baltar, J.; Fernández-Fresnedo, G.; Martín, C.; Pons, S.; et al. Spontaneous Remission of Nephrotic Syndrome in Idiopathic Membranous Nephropathy. J. Am. Soc. Nephrol. 2010, 21, 697–704. [Google Scholar] [CrossRef]
- Jha, V.; Ganguli, A.; Saha, T.K.; Kohli, H.; Sud, K.; Gupta, K.L.; Joshi, K.; Sakhuja, V. A Randomized, Controlled Trial of Steroids and Cyclophosphamide in Adults with Nephrotic Syndrome Caused by Idiopathic Membranous Nephropathy. J. Am. Soc. Nephrol. 2007, 18, 1899–1904. [Google Scholar] [CrossRef] [PubMed]
- Cattran, D.C. Mycophenolate mofetil and cyclosporine therapy in membranous nephropathy. Semin. Nephrol. 2003, 23, 272–277. [Google Scholar] [CrossRef]
- Hogan, S.L.; Muller, K.E.; Jennette, J.; Falk, R.J. A review of therapeutic studies of idiopathic membranous glomerulopathy. Am. J. Kidney Dis. 1995, 25, 862–875. [Google Scholar] [CrossRef]
- Fervenza, F.C.; Appel, G.B.; Barbour, S.J.; Rovin, B.H.; Lafayette, R.A.; Aslam, N.; Jefferson, J.A.; Gipson, P.E.; Rizk, D.V.; Sedor, J.R.; et al. Rituximab or Cyclosporine in the Treatment of Membranous Nephropathy. N. Engl. J. Med. 2019, 381, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Cattran, D.C.; Appel, G.B.; Hebert, L.A.; Hunsicker, L.G.; Pohl, M.A.; Hoy, W.E.; Maxwell, D.R.; Kunis, C.L.; For The North American Nephrotic Syndrome. Cyclosporine in patients with steroid-resistant membranous nephropathy: A randomized trial. Kidney Int. 2001, 59, 1484–1490. [Google Scholar] [CrossRef] [PubMed]
- Scolari, F.; Delbarba, E.; Santoro, D.; Gesualdo, L.; Pani, A.; Dallera, N.; Mani, L.-Y.; Santostefano, M.; Feriozzi, S.; Quaglia, M.; et al. Rituximab or Cyclophosphamide in the Treatment of Membranous Nephropathy: The RI-CYCLO Randomized Trial. J. Am. Soc. Nephrol. 2021, 32, 972–982. [Google Scholar] [CrossRef] [PubMed]
- Hofstra, J.M.; Beck, L.; Beck, D.M.; Wetzels, J.F.; Salant, D. Anti-Phospholipase A2 Receptor Antibodies Correlate with Clinical Status in Idiopathic Membranous Nephropathy. Clin. J. Am. Soc. Nephrol. 2011, 6, 1286–1291. [Google Scholar] [CrossRef] [PubMed]
- Hoxha, E.; Thiele, I.; Zahner, G.; Panzer, U.; Harendza, S.; Stahl, R.A. Phospholipase A2 Receptor Autoantibodies and Clinical Outcome in Patients with Primary Membranous Nephropathy. J. Am. Soc. Nephrol. 2014, 25, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Hoxha, E.; Harendza, S.; Pinnschmidt, H.; Panzer, U.; Stahl, R.A.K. PLA2R Antibody Levels and Clinical Outcome in Patients with Membranous Nephropathy and Non-Nephrotic Range Proteinuria under Treatment with Inhibitors of the Renin-Angiotensin System. PLoS ONE 2014, 9, e110681. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Choi, Y.W.; Kim, S.Y.; Moon, J.Y.; Ihm, C.G.; Lee, T.W.; Jeong, K.H.; Yang, S.H.; Kim, Y.S.; Oh, Y.J.; et al. Anti-Phospholipase A2 Receptor Antibody as Prognostic Indicator in Idiopathic Membranous Nephropathy. Am. J. Nephrol. 2015, 42, 250–257. [Google Scholar] [CrossRef]
- Ruggenenti, P.; Debiec, H.; Ruggiero, B.; Chianca, A.; Pellé, T.; Gaspari, F.; Suardi, F.; Gagliardini, E.; Orisio, S.; Benigni, A.; et al. Anti-Phospholipase A2 Receptor Antibody Titer Predicts Post-Rituximab Outcome of Membranous Nephropathy. J. Am. Soc. Nephrol. 2015, 26, 2545–2558. [Google Scholar] [CrossRef]
- Bech, A.P.; Hofstra, J.M.; Brenchley, P.E.; Wetzels, J.F. Association of Anti-PLA2R Antibodies with Outcomes after Immunosuppressive Therapy in Idiopathic Membranous Nephropathy. Clin. J. Am. Soc. Nephrol. 2014, 9, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Lanza, L.; Scudeletti, M.; Puppo, F.; Bosco, O.; Peirano, L.; Filaci, G.; Fecarotta, E.; Vidali, G.; Indiveri, F. Prednisone increases apoptosis in in vitro activated human peripheral blood T lymphocytes. Clin. Exp. Immunol. 1996, 103, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Baris, H.E.; Baris, S.; Karakoc-Aydiner, E.; Gokce, I.; Yildiz, N.; Cicekkoku, D.; Ogulur, I.; Ozen, A.; Alpay, H.; Barlan, I. The effect of systemic corticosteroids on the innate and adaptive immune system in children with steroid responsive nephrotic syndrome. Eur. J. Pediatr. 2016, 175, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Stahn, C.; Buttgereit, F. Genomic and nongenomic effects of glucocorticoids. Nat. Clin. Pr. Rheumatol. 2008, 4, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Kassel, O.; Sancono, A.; Krätzschmar, J.; Kreft, B.; Stassen, M.; Cato, A.C. Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. EMBO J. 2001, 20, 7108–7116. [Google Scholar] [CrossRef]
- Schmid, W.; Strähle, U.; Schütz, G.; Schmitt, J.; Stunnenberg, H. Glucocorticoid receptor binds cooperatively to adjacent recognition sites. EMBO J. 1989, 8, 2257–2263. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. How corticosteroids control inflammation: Quintiles Prize Lecture 2005. J. Cereb. Blood Flow Metab. 2006, 148, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Buttgereit, F.; Scheffold, A. Rapid glucocorticoid effects on immune cells. Steroids 2002, 67, 529–534. [Google Scholar] [CrossRef]
- Buttgereit, F.; Burmester, G.-R.; Brand, M.D. Bioenergetics of immune functions: Fundamental and therapeutic aspects. Immunol. Today 2000, 21, 194–199. [Google Scholar] [CrossRef]
- Bruna, A.; Nicolàs, M.; Munoz, A.; Kyriakis, J.M.; Caelles, C. Glucocorticoid receptor-JNK interaction mediates inhibition of the JNK pathway by glucocorticoids. EMBO J. 2003, 22, 6035–6044. [Google Scholar] [CrossRef]
- Strehl, C.; Gaber, T.; Löwenberg, M.; Hommes, D.W.; Verhaar, A.P.; Schellmann, S.; Hahne, M.; Fangradt, M.; Wagegg, M.; Hoff, P.; et al. Origin and functional activity of the membrane-bound glucocorticoid receptor. Arthritis Care Res. 2011, 63, 3779–3788. [Google Scholar] [CrossRef]
- Achuthan, A.; Aslam, A.S.M.; Nguyen, Q.; Lam, P.-Y.; Fleetwood, A.; Frye, A.; Louis, C.; Lee, K.M.-C.; Smith, J.E.; Cook, A.D.; et al. Glucocorticoids promote apoptosis of proinflammatory monocytes by inhibiting ERK activity. Cell Death Dis. 2018, 9, 267. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Inamdar, N.; Vedeckis, W.V. Transrepression of c-jun gene expression by the glucocorticoid receptor requires both AP-1 sites in the c-jun promoter. Mol. Endocrinol. 1998, 12, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Yang-Yen, H.-F.; Chambard, J.C.; Sun, Y.-L.; Smeal, T.; Schmidt, T.; Drouin, J.; Karin, M. Transcriptional interference between c-Jun and the glucocorticoid receptor: Mutual inhibition of DNA binding due to direct protein-protein interaction. Cell 1990, 62, 1205–1215. [Google Scholar] [CrossRef]
- Auphan, N.; DiDonato, J.A.; Rosette, C.; Helmberg, A.; Karin, M. Immunosuppression by glucocorticoids: Inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science 1995, 270, 286–290. [Google Scholar] [CrossRef]
- McKay, L.I.; Cidlowski, J.A. CBP (CREB binding protein) integrates NF-kappaB (nuclear factor-kappaB) and glucocorticoid receptor physical interactions and antagonism. Mol. Endocrinol. 2000, 14, 1222–1234. [Google Scholar] [PubMed]
- Dumont, A.; Hehner, S.P.; Schmitz, M.L.; Gustafsson, J.A.; Lidén, J.; Okret, S.; van der Saag, P.T.; Wissink, S.; van der Burg, B.; Herrlich, P.; et al. Cross-talk between steroids and NF-kappa B: What language? Trends Biochem. Sci. 1998, 23, 233–235. [Google Scholar] [CrossRef]
- Caldenhoven, E.; Liden, J.; Wissink, S.; Van de Stolpe, A.; Raaijmakers, J.; Koenderman, L.; Okret, S.; Gustafsson, J.A.; Van der Saag, P.T. Negative cross-talk between RelA and the glucocorticoid receptor: A possible mechanism for the antiinflammatory action of glucocorticoids. Mol. Endocrinol. 1995, 9, 401–412. [Google Scholar] [PubMed]
- Shi, J.-X.; Li, J.-S.; Hu, R.; Shi, Y.; Su, X.; Guo, X.-J.; Li, X.-M. Tristetraprolin is involved in the glucocorticoid-mediated interleukin 8 repression. Int. Immunopharmacol. 2014, 22, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Katsuyama, M.; Onodera, T.; Ehama, R.; Hosoi, J.; Tagami, H. Glucocorticoids Enhance Toll-Like Receptor 2 Expression in Human Keratinocytes Stimulated with Propionibacterium acnes or Proinflammatory Cytokines. J. Investig. Dermatol. 2009, 129, 375–382. [Google Scholar] [CrossRef]
- Busillo, J.M.; Azzam, K.M.; Cidlowski, J.A. Glucocorticoids Sensitize the Innate Immune System through Regulation of the NLRP3 Inflammasome. J. Biol. Chem. 2011, 286, 38703–38713. [Google Scholar] [CrossRef] [PubMed]
- Ikezumi, Y.; Suzuki, T.; Karasawa, T.; Hasegawa, H.; Kawachi, H.; Nikolic-Paterson, D.; Uchiyama, M. Contrasting Effects of Steroids and Mizoribine on Macrophage Activation and Glomerular Lesions in Rat Thy-1 Mesangial Proliferative Glomerulonephritis. Am. J. Nephrol. 2010, 31, 273–282. [Google Scholar] [CrossRef] [PubMed]
- van Rossum, E.F.; Lamberts, S.W. Polymorphisms in the glucocorticoid receptor gene and their associations with metabolic parameters and body composition. Recent Prog. Horm. Res. 2004, 59, 333–357. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Su, Y.A.; Chrousos, G.P. Human glucocorticoid receptor isoform beta: Recent understanding of its potential implications in physiology and pathophysiology. Cell Mol. Life Sci. 2009, 66, 3435–3448. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Tuffin, L.J.; Cidlowski, J.A. The physiology of human glucocorticoid receptor beta (hGRbeta) and glucocorticoid resistance. Ann. N. Y. Acad. Sci. 2006, 1069, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Scheschowitsch, K.; Leite, J.A.; Assreuy, J. New Insights in Glucocorticoid Receptor Signaling-More Than Just a Ligand-Binding Receptor. Front. Endocrinol. 2017, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.C.; Oakley, R.H.; Jewell, C.M.; Cidlowski, J.A. Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative beta isoform: A mechanism for the generation of glucocorticoid resistance. Proc. Natl. Acad. Sci. USA 2001, 98, 6865–6870. [Google Scholar] [CrossRef]
- Ito, K.; Yamamura, S.; Essilfie-Quaye, S.; Cosio, B.; Ito, M.; Barnes, P.J.; Adcock, I.M. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J. Exp. Med. 2006, 203, 7–13. [Google Scholar] [CrossRef]
- Oakley, R.H.; Cidlowski, J.A. Cellular Processing of the Glucocorticoid Receptor Gene and Protein: New Mechanisms for Generating Tissue-specific Actions of Glucocorticoids. J. Biol. Chem. 2011, 286, 3177–3184. [Google Scholar] [CrossRef] [PubMed]
- Szatmáry, Z.; Garabedian, M.J.; Vilček, J. Inhibition of Glucocorticoid Receptor-mediated Transcriptional Activation by p38 Mitogen-activated Protein (MAP) Kinase. J. Biol. Chem. 2004, 279, 43708–43715. [Google Scholar] [CrossRef]
- Rogatsky, I.; Logan, S.K.; Garabedian, M.J. Antagonism of glucocorticoid receptor transcriptional activation by the c-Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 2050–2055. [Google Scholar] [CrossRef]
- McKay, L.I.; Cidlowski, J.A. Cross-talk between nuclear factor-kappa B and the steroid hormone receptors: Mechanisms of mutual antagonism. Mol. Endocrinol. 1998, 12, 45–56. [Google Scholar] [CrossRef]
- Petta, I.; Dejager, L.; Ballegeer, M.; Lievens, S.; Tavernier, J.; De Bosscher, K.; Libert, C. The Interactome of the Glucocorticoid Receptor and Its Influence on the Actions of Glucocorticoids in Combatting Inflammatory and Infectious Diseases. Microbiol. Mol. Biol. Rev. 2016, 80, 495–522. [Google Scholar] [CrossRef]
- Van Bogaert, T.; De Bosscher, K.; Libert, C. Crosstalk between TNF and glucocorticoid receptor signaling pathways. Cytokine Growth Factor Rev. 2010, 21, 275–286. [Google Scholar] [CrossRef]
- Ji, N.; Kovalovsky, A.; Fingerle-Rowson, G.; Guentzel, M.N.; Forsthuber, T.G. Macrophage migration inhibitory factor promotes resistance to glucocorticoid treatment in EAE. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e139. [Google Scholar] [CrossRef]
- Wang, F.-F.; Zhu, L.-A.; Zou, Y.-Q.; Zheng, H.; Wilson, A.; Yang, C.-D.; Shen, N.; Wallace, D.J.; Weisman, M.H.; Chen, S.-L.; et al. New insights into the role and mechanism of macrophage migration inhibitory factor in steroid-resistant patients with systemic lupus erythematosus. Arthritis Res. Ther. 2012, 14, R103. [Google Scholar] [CrossRef]
- Tsuruoka, S.; Sugimoto, K.-I.; Fujimura, A.; Imai, M.; Asano, Y.; Muto, S. P-Glycoprotein-Mediated Drug Secretion in Mouse Proximal Tubule Perfused In Vitro. J. Am. Soc. Nephrol. 2001, 12, 177–181. [Google Scholar] [CrossRef]
- Yates, C.R.; Chang, C.; Kearbey, J.D.; Yasuda, K.; Schuetz, E.G.; Miller, D.D.; Dalton, J.T.; Swaan, P. Structural Determinants of P-Glycoprotein-Mediated Transport of Glucocorticoids. Pharm. Res. 2003, 20, 1794–1803. [Google Scholar] [CrossRef]
- Cuppen, B.V.J.; Pardali, K.; Kraan, M.C.; Marijnissen, A.C.A.; Yrlid, L.; Olsson, M.; Bijlsma, J.W.J.; Lafeber, F.P.J.G.; Fritsch-Stork, R.D.E. Polymorphisms in the multidrug-resistance 1 gene related to glucocorticoid response in rheumatoid arthritis treatment. Rheumatol. Int. 2017, 37, 531–536. [Google Scholar] [CrossRef]
- Schmidt, J.; Metselaar, J.M.; Wauben, M.H.M.; Toyka, K.V.; Storm, G.; Gold, R. Drug targeting by long-circulating liposomal glucocorticosteroids increases therapeutic efficacy in a model of multiple sclerosis. Brain 2003, 126 Pt 8, 1895–1904. [Google Scholar] [CrossRef]
- Metselaar, J.M.; Wauben, M.H.M.; Wagenaar-Hilbers, J.P.A.; Boerman, O.C.; Storm, G. Complete remission of experimental arthritis by joint targeting of glucocorticoids with long-circulating liposomes. Arthritis Care Res. 2003, 48, 2059–2066. [Google Scholar] [CrossRef]
- Ozbakir, B.; Crielaard, B.; Metselaar, J.M.; Storm, G.; Lammers, T. Liposomal corticosteroids for the treatment of inflammatory disorders and cancer. J. Control. Release 2014, 190, 624–636. [Google Scholar] [CrossRef]
- Schäcke, H.; Döcke, W.-D.; Asadullah, K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol. Ther. 2002, 96, 23–43. [Google Scholar] [CrossRef]
- Schäcke, H.; Berger, M.; Rehwinkel, H.; Asadullah, K. Selective glucocorticoid receptor agonists (SEGRAs): Novel ligands with an improved therapeutic index. Mol. Cell. Endocrinol. 2007, 275, 109–117. [Google Scholar] [CrossRef]
- Schäcke, H.; Schottelius, A.; Döcke, W.-D.; Strehlke, P.; Jaroch, S.; Schmees, N.; Rehwinkel, H.; Hennekes, H.; Asadullah, K. Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. Proc. Natl. Acad. Sci. USA 2003, 101, 227–232. [Google Scholar] [CrossRef]
- Yu, M.; Ren, Q.; Yu, S.Y. Role of nephrin phosphorylation inducted by dexamethasone and angiotensin II in podocytes. Mol. Biol. Rep. 2014, 41, 3591–3595. [Google Scholar] [CrossRef]
- Dou, C.; Zhang, H.; Ke, G.; Zhang, L.; Lian, Z.; Chen, X.; Zhao, X.; Chen, Y.; Li, R.; Ma, J.; et al. The Krüppel-like factor 15-NFATc1 axis ameliorates podocyte injury: A novel rationale for using glucocorticoids in proteinuria diseases. Clin. Sci. 2020, 134, 1305–1318. [Google Scholar] [CrossRef]
- Wang, Y.; Jarad, G.; Tripathi, P.; Pan, M.; Cunningham, J.; Martin, D.R.; Liapis, H.; Miner, J.H.; Chen, F. Activation of NFAT Signaling in Podocytes Causes Glomerulosclerosis. J. Am. Soc. Nephrol. 2010, 21, 1657–1666. [Google Scholar] [CrossRef]
- Yu, S.; Yu, L. Dexamethasone Resisted Podocyte Injury via Stabilizing TRPC6 Expression and Distribution. Evidence-Based Complement. Altern. Med. 2012, 2012, 652059. [Google Scholar] [CrossRef]
- Mallipattu, S.K.; Guo, Y.; Revelo, M.P.; Roa-Peña, L.; Miller, T.; Ling, J.; Shankland, S.J.; Bialkowska, A.B.; Ly, V.; Estrada, C.; et al. Krüppel–Like Factor 15 Mediates Glucocorticoid-Induced Restoration of Podocyte Differentiation Markers. J. Am. Soc. Nephrol. 2017, 28, 166–184. [Google Scholar] [CrossRef]
- Ransom, R.F.; Lam, N.G.; Hallett, M.A.; Atkinson, S.J.; Smoyer, W.E. Glucocorticoids protect and enhance recovery of cultured murine podocytes via actin filament stabilization. Kidney Int. 2005, 68, 2473–2483. [Google Scholar] [CrossRef] [PubMed]
- McCaffrey, J.C.; Webb, N.J.; Poolman, T.M.; Fresquet, M.; Moxey, C.; Zeef, L.A.H.; Donaldson, I.J.; Ray, D.W.; Lennon, R. Glucocorticoid therapy regulates podocyte motility by inhibition of Rac1. Sci. Rep. 2017, 7, 6725. [Google Scholar] [CrossRef]
- Robins, R.; Baldwin, C.; Aoudjit, L.; Côté, J.-F.; Gupta, I.R.; Takano, T. Rac1 activation in podocytes induces the spectrum of nephrotic syndrome. Kidney Int. 2017, 92, 349–364. [Google Scholar] [CrossRef]
- Yu, S.; Li, Y. Dexamethasone inhibits podocyte apoptosis by stabilizing the PI3K/Akt signal pathway. Biomed. Res. Int. 2013, 2013, 326986. [Google Scholar]
- Wada, T.; Pippin, J.W.; Marshall, C.B.; Griffin, S.V.; Shankland, S.J. Dexamethasone Prevents Podocyte Apoptosis Induced by Puromycin Aminonucleoside: Role of p53 and Bcl-2–Related Family Proteins. J. Am. Soc. Nephrol. 2005, 16, 2615–2625. [Google Scholar] [CrossRef] [PubMed]
- Hales, B.F. Effects of phosphoramide mustard and acrolein, cytotoxic metabolites of cyclophosphamide, on mouse limb development in vitro. Teratology 1989, 40, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, K. DNA-binding products of nornitrogen mustard, a metabolite of cyclophosphamide. Chem. Interactions 1987, 61, 75–88. [Google Scholar] [CrossRef]
- Pass, G.J.; Carrie, D.; Lorimore, S.; Wright, E.; Houston, B.; Henderson, C.J.; Boylan, M.; Wolf, C.R. Role of Hepatic Cytochrome P450s in the Pharmacokinetics and Toxicity of Cyclophosphamide: Studies with the Hepatic Cytochrome P450 Reductase Null Mouse. Cancer Res. 2005, 65, 4211–4217. [Google Scholar] [CrossRef]
- Magni, M.; Shammah, S.; Schiró, R.; Mellado, W.; Dalla-Favera, R.; Gianni, A.M. Induction of cyclophosphamide-resistance by aldehyde-dehydrogenase gene transfer. Blood 1996, 87, 1097–1103. [Google Scholar] [CrossRef]
- Agarwal, D.P.; Eitzen, U.V.; Meier-Tackmann, D.; Goedde, H.W. Metabolism of Cyclophosphamide by Aldehyde Dehydrogenases. In Enzymology and Molecular Biology of Carbonyl Metabolism 5; Springer: Boston, MA, USA, 1995; Volume 372, pp. 115–122. [Google Scholar]
- Hurd, E.R.; Giuliano, V.J. The effect of cyclophosphamide on b and t lymphocytes in patients with connective tissue diseases. Arthritis Care Res. 1975, 18, 67–75. [Google Scholar] [CrossRef]
- Kanakry, C.G.; Ganguly, S.; Zahurak, M.; Bolaños-Meade, J.; Thoburn, C.; Perkins, B.; Fuchs, E.J.; Jones, R.J.; Hess, A.D.; Luznik, L. Aldehyde Dehydrogenase Expression Drives Human Regulatory T Cell Resistance to Posttransplantation Cyclophosphamide. Sci. Transl. Med. 2013, 5, 211ra157. [Google Scholar] [CrossRef]
- Bao, L.; Hao, C.; Wang, J.; Wang, D.; Zhao, Y.; Li, Y.; Yao, W. High-Dose Cyclophosphamide Administration Orchestrates Phenotypic and Functional Alterations of Immature Dendritic Cells and Regulates Th Cell Polarization. Front. Pharmacol. 2020, 11, 775. [Google Scholar] [CrossRef]
- Williams, C.R.; Gooch, J.L. Calcineurin inhibitors and immunosuppression – a tale of two isoforms. Expert Rev. Mol. Med. 2012, 14, e14. [Google Scholar] [CrossRef]
- Macian, F. NFAT proteins: Key regulators of T-cell development and function. Nat. Rev. Immunol. 2005, 5, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Hilchey, S.P.; Palshikar, M.G.; Emo, J.A.; Li, D.; Garigen, J.; Wang, J.; Mendelson, E.S.; Cipolla, V.; Thakar, J.; Zand, M.S. Cyclosporine a directly affects human and mouse b cell migration in vitro by disrupting a hIF-1 alphadependent, o2 sensing, molecular switch. BMC Immunol. 2020, 21, 13. [Google Scholar] [CrossRef] [PubMed]
- IIsrani, A.; Brozena, S.; Pankewycz, O.; Grossman, R.; Bloom, R. Conversion to tacrolimus for the treatment of cyclosporine-associated nephrotoxicity in heart transplant recipients. Am. J. Kidney Dis. 2002, 39, e16.1–e16.5. [Google Scholar] [CrossRef]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ellis, M.J.; Gomez, J.A.; Eisner, W.; Fennell, W.; Howell, D.N.; Ruiz, P.; Fields, T.A.; Spurney, R.F. Mechanisms of the proteinuria induced by Rho GTPases. Kidney Int. 2012, 81, 1075–1085. [Google Scholar] [CrossRef]
- Faul, C.; Donnelly, M.; Merscher-Gomez, S.; Chang, Y.H.; Franz, S.; Delfgaauw, J.; Chang, J.-M.; Choi, H.Y.; Campbell, K.N.; Kim, K.; et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat. Med. 2008, 14, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Natsumeda, Y.; Ohno, S.; Kawasaki, H.; Konno, Y.; Weber, G.; Suzuki, K. Two distinct cDNAs for human IMP dehydrogenase. J. Biol. Chem. 1990, 265, 5292–5295. [Google Scholar] [CrossRef]
- Jain, J.; Almquist, S.J.; Ford, P.J.; Shlyakhter, D.; Wang, Y.; Nimmesgern, E.; Germann, U.A. Regulation of inosine monophosphate dehydrogenase type I and type II isoforms in human lymphocytes. Biochem. Pharmacol. 2003, 67, 767–776. [Google Scholar] [CrossRef]
- Nagai, M.; Natsumeda, Y.; Weber, G. Proliferation-linked regulation of type II IMP dehydrogenase gene in human normal lymphocytes and HL-60 leukemic cells. Cancer Res. 1992, 52, 258–261. [Google Scholar] [PubMed]
- Carr, S.F.; Papp, E.; Wu, J.C.; Natsumeda, Y. Characterization of human type I and type II IMP dehydrogenases. J. Biol. Chem. 1993, 268, 27286–27290. [Google Scholar] [CrossRef]
- Nakamura, M.; Ogawa, N.; Shalabi, A.; Maley, W.R.; Longo, D.; Burdick, J.F. Positive effect on T-cell regulatory apoptosis by mycophenolate mofetil. Clin. Transplant. 2001, 15, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Cohn, R.G.; Mirkovich, A.; Dunlap, B.; Burton, P.; Chiu, S.-H.; Eugui, E.; Caulfield, J.P. Mycophenolic Acid Increases Apoptosis, Lysosomes and Lipid Droplets in Human Lymphoid and Monocytic Cell Lines. Transplantation 1999, 68, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Čolić, M.; Stojić-Vukanić, Z.; Pavlović, B.; Jandrić, D.; Stefanoska, I. Mycophenolate mofetil inhibits differentiation, maturation and allostimulatory function of human monocyte-derived dendritic cells. Clin. Exp. Immunol. 2003, 134, 63–69. [Google Scholar] [CrossRef]
- A Blaheta, R.; Leckel, K.; Wittig, B.; Zenker, D.; Oppermann, E.; Harder, S.; Scholz, M.; Weber, S.; Schuldes, H.; Encke, A.; et al. Inhibition of endothelial receptor expression and of T-cell ligand activity by mycophenolate mofetil. Transpl. Immunol. 1998, 6, 251–259. [Google Scholar] [CrossRef]
- Hatakeyama, K.; Harada, T.; Kagamiyama, H. IMP dehydrogenase inhibitors reduce intracellular tetrahydrobiopterin levels through reduction of intracellular GTP levels. Indications of the regulation of GTP cyclohydrolase I activity by restriction of GTP availability in the cells. J. Biol. Chem. 1992, 267, 20734–20739. [Google Scholar] [CrossRef]
- Senda, M.; DeLustro, B.; Eugui, E.; Natsumeda, Y. Mycophenolic acid, an inhibitor of IMP dehydrogenase that is also an immunosuppressive agent, suppresses the cytokine-induced nitric oxide production in mouse and rat vascular endothelial cells. Transplantation 1995, 60, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Hackl, A.; Ehren, R.; Weber, L.T. Effect of mycophenolic acid in experimental, nontransplant glomerular diseases: New mechanisms beyond immune cells. Pediatr. Nephrol. 2016, 32, 1315–1322. [Google Scholar] [CrossRef]
- Nakajo, A.; Khoshnoodi, J.; Takenaka, H.; Hagiwara, E.; Watanabe, T.; Kawakami, H.; Kurayama, R.; Sekine, Y.; Bessho, F.; Takahashi, S.; et al. Mizoribine Corrects Defective Nephrin Biogenesis by Restoring Intracellular Energy Balance. J. Am. Soc. Nephrol. 2007, 18, 2554–2564. [Google Scholar] [CrossRef]
- Wei, C.; Möller, C.C.; Altintas, M.; Li, J.; Schwarz, K.; Zacchigna, S.; Xie, L.; Henger, A.; Schmid, H.; Rastaldi, M.P.; et al. Modification of kidney barrier function by the urokinase receptor. Nat. Med. 2007, 14, 55–63. [Google Scholar] [CrossRef]
- Cheng, C.-C.; Lee, Y.-F.; Lan, J.-L.; Wu, M.-J.; Hsieh, T.-Y.; Lin, N.-N.; Wang, J.-M.; Chiu, Y.-T. Mycophenolate mofetil alleviates lupus nephritis through urokinase receptor signaling in a mice model. Lupus 2013, 22, 554–561. [Google Scholar] [CrossRef]
- Looney, R.J. B cell-targeted therapy for rheumatoid arthritis: An update on the evidence. Drugs 2006, 66, 625–639. [Google Scholar] [CrossRef]
- Fervenza, F.; Cosio, F.; Erickson, S.; Specks, U.; Herzenberg, A.; Dillon, J.; Leung, N.; Cohen, I.; Wochos, D.; Bergstralh, E.; et al. Rituximab treatment of idiopathic membranous nephropathy. Kidney Int. 2008, 73, 117–125. [Google Scholar] [CrossRef]
- Cragg, M.S.; Walshe, C.A.; Ivanov, A.O.; Glennie, M.J. The Biology of CD20 and Its Potential as a Target for mAb Therapy. Curr. Dir. Autoimmun. 2004, 8, 140–174. [Google Scholar]
- Vallerskog, T.; Gunnarsson, I.; Widhe, M.; Risselada, A.; Klareskog, L.; van Vollenhoven, R.; Malmström, V.; Trollmo, C. Treatment with rituximab affects both the cellular and the humoral arm of the immune system in patients with SLE. Clin. Immunol. 2007, 122, 62–74. [Google Scholar] [CrossRef]
- Sfikakis, P.; Souliotis, V.; Fragiadaki, K.; Moutsopoulos, H.; Boletis, J.; Theofilopoulos, A. Increased expression of the FoxP3 functional marker of regulatory T cells following B cell depletion with rituximab in patients with lupus nephritis. Clin. Immunol. 2007, 123, 66–73. [Google Scholar] [CrossRef]
- Fornoni, A.; Sageshima, J.; Wei, C.; Merscher-Gomez, S.; Aguillon-Prada, R.; Jauregui, A.N.; Li, J.; Mattiazzi, A.; Ciancio, G.; Chen, L.; et al. Rituximab Targets Podocytes in Recurrent Focal Segmental Glomerulosclerosis. Sci. Transl. Med. 2011, 3, 85ra46. [Google Scholar] [CrossRef]
- Dick, J.; Gan, P.-Y.; Ford, S.L.; Odobasic, D.; Alikhan, M.A.; Loosen, S.H.; Hall, P.; Westhorpe, C.L.; Li, A.; Ooi, J.D.; et al. C5a receptor 1 promotes autoimmunity, neutrophil dysfunction and injury in experimental anti-myeloperoxidase glomerulonephritis. Kidney Int. 2017, 93, 615–625. [Google Scholar] [CrossRef]
- Menne, J.; Delmas, Y.; Fakhouri, F.; Licht, C.; Lommelé, Å.; Minetti, E.E.; Provôt, F.; Rondeau, E.; Sheerin, N.S.; Wang, J.; et al. Outcomes in patients with atypical hemolytic uremic syndrome treated with eculizumab in a long-term observational study. BMC Nephrol. 2019, 20, 125. [Google Scholar] [CrossRef]
- Jayne, D.R.W.; Merkel, P.A.; Schall, T.J.; Bekker, P.; ADVOCATE Study Group. Avacopan for the Treatment of ANCA-Associated Vasculitis. N. Engl. J. Med. 2021, 384, 599–609. [Google Scholar] [CrossRef]
- Ebihara, S.; Tajima, H.; Ono, M. Nuclear factor erythroid 2-related factor 2 is a critical target for the treatment of glucocorticoid-resistant lupus nephritis. Arthritis Res. Ther. 2016, 18, 139. [Google Scholar] [CrossRef] [PubMed]
- Adcock, I. Glucocorticoid-regulated Transcription Factors. Pulm. Pharmacol. Ther. 2001, 14, 211–219. [Google Scholar] [CrossRef]
- Guan, F.-J.; Peng, Q.-Q.; Wang, L.-L.; Yan, X.-B.; Dong, C.; Jiang, X.-H. Histone deacetylase-2 expression and activity in children with nephrotic syndrome with different glucocorticoid response. Pediatr. Nephrol. 2017, 33, 269–276. [Google Scholar] [CrossRef]
- Adenuga, D.; Caito, S.; Yao, H.; Sundar, I.K.; Hwang, J.-W.; Chung, S.; Rahman, I. Nrf2 deficiency influences susceptibility to steroid resistance via HDAC2 reduction. Biochem. Biophys. Res. Commun. 2010, 403, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Lim, A.Y.H.; Tan, W.S.D.; Abisheganaden, J.; Wong, W.S.F. Restoration of HDAC2 and Nrf2 by andrographolide overcomes corticosteroid resistance in chronic obstructive pulmonary disease. J. Cereb. Blood Flow Metab. 2020, 177, 3662–3673. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, D.-W.; Chang, C.-C.; Hsu, Y.-C.; Lin, C.-L. New Insights into the Treatment of Glomerular Diseases: When Mechanisms Become Vivid. Int. J. Mol. Sci. 2022, 23, 3525. https://doi.org/10.3390/ijms23073525
Lin D-W, Chang C-C, Hsu Y-C, Lin C-L. New Insights into the Treatment of Glomerular Diseases: When Mechanisms Become Vivid. International Journal of Molecular Sciences. 2022; 23(7):3525. https://doi.org/10.3390/ijms23073525
Chicago/Turabian StyleLin, Da-Wei, Cheng-Chih Chang, Yung-Chien Hsu, and Chun-Liang Lin. 2022. "New Insights into the Treatment of Glomerular Diseases: When Mechanisms Become Vivid" International Journal of Molecular Sciences 23, no. 7: 3525. https://doi.org/10.3390/ijms23073525
APA StyleLin, D.-W., Chang, C.-C., Hsu, Y.-C., & Lin, C.-L. (2022). New Insights into the Treatment of Glomerular Diseases: When Mechanisms Become Vivid. International Journal of Molecular Sciences, 23(7), 3525. https://doi.org/10.3390/ijms23073525