
Repurposing Dipyridamole in Niemann Pick Type C Disease: A Proof of Concept Study

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. The Expression of Proteins Involved in the Regulation of Adenosine Levels Is Deregulated Both in Fibroblasts from Patients and in NPC1 Mice
2.2. Treatment with Dipyridamole Reduces Intracellular Cholesterol Accumulation in NPC1 Fibroblasts and Increases Mitochondrial Membrane Potential
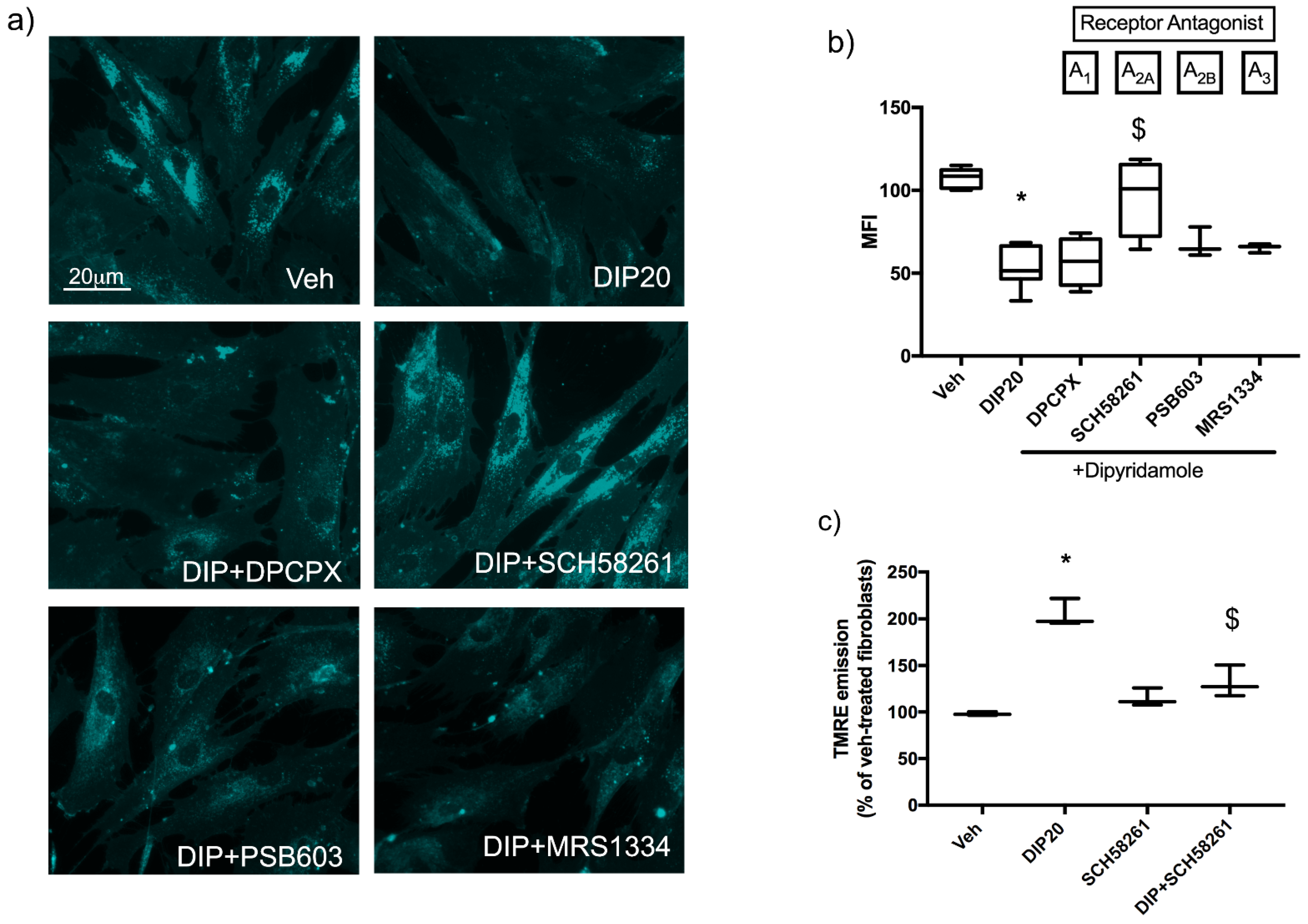
2.3. Dipyridamole Rescues NPC1-Related Phenotypes in Fibroblasts by an A2AR-Dependent Pathway
2.4. Functional Impact of cAMP/PKA and MAPK Pathways on Dipyridamole-Dependent Reduction of Cholesterol Accumulation
3. Discussion
4. Materials and Methods
4.1. Drugs
4.2. Cell Cultures
4.3. Mice
4.4. Real-Time Quantitative Polymerase Chain Reaction (RT-PCR)
4.5. Cell Treatments
4.6. Measurement of Adenosine Levels in Culture Supernatants
4.7. Filipin III Fluorescence and P-ERK1/2 Immunofluorescence
4.8. Mitochondrial Inner Membrane Potential Measurement
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Geberhiwot, T.; Moro, A.; Dardis, A.; Ramaswami, U.; Sirrs, S.; Marfa, M.P.; Vanier, M.T.; Walterfang, M.; Bolton, S.; Dawson, C.; et al. Consensus Clinical Management Guidelines for Niemann-Pick Disease Type C. Orphanet J. Rare Dis 2018, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Chin, S.J.; Fuller, M. Prevalence of Lysosomal Storage Disorders in Australia from 2009 to 2020. Lancet Reg. Health West Pac. 2022, 19, 100344. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T. Complex Lipid Trafficking in Niemann-Pick Disease Type C. J. Inherit. Metab. Dis. 2015, 38, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Hammond, N.; Munkacsi, A.B.; Sturley, S.L. The Complexity of a Monogenic Neurodegenerative Disease: More than Two Decades of Therapeutic Driven Research into Niemann-Pick Type C Disease. Biochim. Biophys. Acta—Mol. Cell Biol. Lipids 2019, 1864, 1109–1123. [Google Scholar] [CrossRef]
- Walterfang, M.; Patenaude, B.; Abel, L.A.; Kluenemann, H.; Bowman, E.A.; Fahey, M.C.; Desmond, P.; Kelso, W.; Velakoulis, D. Subcortical Volumetric Reductions in Adult Niemann-Pick Disease Type C: A Cross-Sectional Study. AJNR Am. J. Neuroradiol. 2013, 34, 1334–1340. [Google Scholar] [CrossRef]
- Vanier, M.T. Niemann-Pick Disease Type C. Orphanet J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef]
- Pineda, M.; Walterfang, M.; Patterson, M.C. Miglustat in Niemann-Pick Disease Type C Patients: A Review. Orphanet J. Rare Dis. 2018, 13, 140. [Google Scholar] [CrossRef]
- Bräuer, A.U.; Kuhla, A.; Holzmann, C.; Wree, A.; Witt, M. Current Challenges in Understanding the Cellular and Molecular Mechanisms in Niemann-Pick Disease Type C1. Int. J. Mol. Sci. 2019, 20, 4392. [Google Scholar] [CrossRef]
- Gomes, C.V.; Kaster, M.P.; Tomé, A.R.; Agostinho, P.M.; Cunha, R.A. Adenosine Receptors and Brain Diseases: Neuroprotection and Neurodegeneration. Biochim. Biophys. Acta—Biomembr. 2011, 1808, 1380–1399. [Google Scholar] [CrossRef]
- Latini, S.; Pedata, F. Adenosine in the Central Nervous System: Release Mechanisms and Extracellular Concentrations. J. Neurochem. 2001, 79, 463–484. [Google Scholar] [CrossRef]
- Parkinson, F.E.; Damaraju, V.L.; Graham, K.; Yao, S.Y.M.; Baldwin, S.A.; Cass, C.E.; Young, J.D. Molecular Biology of Nucleoside Transporters and Their Distributions and Functions in the Brain. CTMC 2011, 11, 948–972. [Google Scholar] [CrossRef]
- Burnstock, G. Physiology and Pathophysiology of Purinergic Neurotransmission. Physiol. Rev. 2007, 87, 659–797. [Google Scholar] [CrossRef]
- Zimmermann, H.; Zebisch, M.; Sträter, N. Cellular Function and Molecular Structure of Ecto-Nucleotidases. Purinergic Signal. 2012, 8, 437–502. [Google Scholar] [CrossRef]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and Classification of Adenosine Receptors—An Update. Pharm. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef]
- Lee, C.-C.; Chang, C.-P.; Lin, C.-J.; Lai, H.-L.; Kao, Y.-H.; Cheng, S.-J.; Chen, H.-M.; Liao, Y.-P.; Faivre, E.; Buée, L.; et al. Adenosine Augmentation Evoked by an ENT1 Inhibitor Improves Memory Impairment and Neuronal Plasticity in the APP/PS1 Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 8936–8952. [Google Scholar] [CrossRef]
- Kao, Y.-H.; Lin, M.-S.; Chen, C.-M.; Wu, Y.-R.; Chen, H.-M.; Lai, H.-L.; Chern, Y.; Lin, C.-J. Targeting ENT1 and Adenosine Tone for the Treatment of Huntington’s Disease. Hum. Mol. Genet. 2017, 26, 467–478. [Google Scholar] [CrossRef]
- Zhou, S.; Xu, S.; Yan, Y.; Yu, H.; Ling, S.; Luo, J. Decreased Purinergic Inhibition of Synaptic Activity in a Mouse Model of Niemann-Pick Disease Type C. Hippocampus 2011, 21, 212–219. [Google Scholar] [CrossRef]
- Visentin, S.; De Nuccio, C.; Bernardo, A.; Pepponi, R.; Ferrante, A.; Minghetti, L.; Popoli, P. The Stimulation of Adenosine A2A Receptors Ameliorates the Pathological Phenotype of Fibroblasts from Niemann-Pick Type C Patients. J. Neurosci. 2013, 33, 15388–15393. [Google Scholar] [CrossRef][Green Version]
- Ferrante, A.; De Nuccio, C.; Pepponi, R.; Visentin, S.; Martire, A.; Bernardo, A.; Minghetti, L.; Popoli, P. Stimulation of Adenosine A2A Receptors Reduces Intracellular Cholesterol Accumulation and Rescues Mitochondrial Abnormalities in Human Neural Cell Models of Niemann-Pick C1. Neuropharmacology 2016, 103, 155–162. [Google Scholar] [CrossRef]
- De Nuccio, C.; Bernardo, A.; Ferrante, A.; Pepponi, R.; Martire, A.; Falchi, M.; Visentin, S.; Popoli, P.; Minghetti, L. Adenosine A2A Receptor Stimulation Restores Cell Functions and Differentiation in Niemann-Pick Type C-like Oligodendrocytes. Sci. Rep. 2019, 9, 9782. [Google Scholar] [CrossRef]
- Ferrante, A.; Pezzola, A.; Matteucci, A.; Di Biase, A.; Attorri, L.; Armida, M.; Martire, A.; Chern, Y.; Popoli, P. The Adenosine A2A Receptor Agonist T1–11 Ameliorates Neurovisceral Symptoms and Extends the Lifespan of a Mouse Model of Niemann-Pick Type C Disease. Neurobiol. Dis. 2018, 110, 1–11. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Freedman, J.E. Dipyridamole, Cerebrovascular Disease, and the Vasculature. Vasc. Pharmacol. 2008, 48, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Burbulla, L.F.; Krüger, R. The Use of Primary Human Fibroblasts for Monitoring Mitochondrial Phenotypes in the Field of Parkinson’s Disease. JoVE 2012, 68, 4228. [Google Scholar] [CrossRef]
- Ali, R.A.; Gandhi, A.A.; Meng, H.; Yalavarthi, S.; Vreede, A.P.; Estes, S.K.; Palmer, O.R.; Bockenstedt, P.L.; Pinsky, D.J.; Greve, J.M.; et al. Adenosine Receptor Agonism Protects against NETosis and Thrombosis in Antiphospholipid Syndrome. Nat. Commun. 2019, 10, 1916. [Google Scholar] [CrossRef]
- Chang, C.-P.; Wu, K.-C.; Lin, C.-Y.; Chern, Y. Emerging Roles of Dysregulated Adenosine Homeostasis in Brain Disorders with a Specific Focus on Neurodegenerative Diseases. J. Biomed. Sci. 2021, 28, 70. [Google Scholar] [CrossRef]
- Rauniyar, N.; Subramanian, K.; Lavallée-Adam, M.; Martínez-Bartolomé, S.; Balch, W.E.; Yates, J.R. Quantitative Proteomics of Human Fibroblasts with I1061T Mutation in Niemann-Pick C1 (NPC1) Protein Provides Insights into the Disease Pathogenesis. Mol. Cell. Proteom. 2015, 14, 1734–1749. [Google Scholar] [CrossRef]
- Alonso-Andrés, P.; Albasanz, J.L.; Ferrer, I.; Martín, M. Purine-Related Metabolites and Their Converting Enzymes Are Altered in Frontal, Parietal and Temporal Cortex at Early Stages of Alzheimer’s Disease Pathology. Brain Pathol. 2018, 28, 933–946. [Google Scholar] [CrossRef]
- Boison, D.; Aronica, E. Comorbidities in Neurology: Is Adenosine the Common Link? Neuropharmacology 2015, 97, 18–34. [Google Scholar] [CrossRef]
- Gonçalves, F.Q.; Lopes, J.P.; Silva, H.B.; Lemos, C.; Silva, A.C.; Gonçalves, N.; Tomé, Â.R.; Ferreira, S.G.; Canas, P.M.; Rial, D.; et al. Synaptic and Memory Dysfunction in a β-Amyloid Model of Early Alzheimer’s Disease Depends on Increased Formation of ATP-Derived Extracellular Adenosine. Neurobiol. Dis. 2019, 132, 104570. [Google Scholar] [CrossRef]
- Zhou, H.; Wolberg, A.S.; Roubey, R.A.S. Characterization of Monocyte Tissue Factor Activity Induced by IgG Antiphospholipid Antibodies and Inhibition by Dilazep. Blood 2004, 104, 2353–2358. [Google Scholar] [CrossRef]
- Burcoglu-O’Ral, A.; Erkan, D.; Asherson, R. Treatment of Catastrophic Antiphospholipid Syndrome with Defibrotide, a Proposed Vascular Endothelial Cell Modulator. J. Rheumatol. 2002, 29, 2006–2011. [Google Scholar]
- Ferré, S.; Quiroz, C.; Guitart, X.; Rea, W.; Seyedian, A.; Moreno, E.; Casadó-Anguera, V.; Díaz-Ríos, M.; Casadó, V.; Clemens, S.; et al. Pivotal Role of Adenosine Neurotransmission in Restless Legs Syndrome. Front. Neurosci. 2018, 11, 722. [Google Scholar] [CrossRef]
- Wonodi, I.; Gopinath, H.V.; Liu, J.; Adami, H.; Hong, L.E.; Allen-Emerson, R.; McMahon, R.P.; Thaker, G.K. Dipyridamole Monotherapy in Schizophrenia: Pilot of a Novel Treatment Approach by Modulation of Purinergic Signaling. Psychopharmacology 2011, 218, 341–345. [Google Scholar] [CrossRef][Green Version]
- Balakumar, P.; Nyo, Y.H.; Renushia, R.; Raaginey, D.; Oh, A.N.; Varatharajan, R.; Dhanaraj, S.A. Classical and Pleiotropic Actions of Dipyridamole: Not Enough Light to Illuminate the Dark Tunnel? Pharmacol. Res. 2014, 87, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Marcelino, H.; Nogueira, V.C.; Santos, C.R.A.; Quelhas, P.; Carvalho, T.M.A.; Fonseca-Gomes, J.; Tomás, J.; Diógenes, M.J.; Sebastião, A.M.; Cascalheira, J.F. Adenosine Inhibits Human Astrocyte Proliferation Independently of Adenosine Receptor Activation. J. Neurochem. 2020, 153, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Ralevic, V.; Burnstock, G. Receptors for Purines and Pyrimidines. Pharm. Rev. 1998, 50, 413–492. [Google Scholar] [PubMed]
- Fredholm, B.B.; Chern, Y.; Franco, R.; Sitkovsky, M. Aspects of the General Biology of Adenosine A2A Signaling. Prog. Neurobiol. 2007, 83, 263–276. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Warner, D.S.; Warner, M.A. Adenosine: An Old Drug Newly Discovered. Anesthesiology 2009, 111, 904–915. [Google Scholar] [CrossRef]
- Pentchev, P.G.; Kruth, H.S.; Comly, M.E.; Butler, J.D.; Vanier, M.T.; Wenger, D.A.; Patel, S. Type C Niemann-Pick Disease. A Parallel Loss of Regulatory Responses in Both the Uptake and Esterification of Low Density Lipoprotein-Derived Cholesterol in Cultured Fibroblasts. J. Biol. Chem. 1986, 261, 16775–16780. [Google Scholar] [CrossRef]
- Liscum, L.; Faust, J.R. Low Density Lipoprotein (LDL)-Mediated Suppression of Cholesterol Synthesis and LDL Uptake Is Defective in Niemann-Pick Type C Fibroblasts. J. Biol. Chem. 1987, 262, 17002–17008. [Google Scholar] [CrossRef]
- Beltroy, E.P.; Richardson, J.A.; Horton, J.D.; Turley, S.D.; Dietschy, J.M. Cholesterol Accumulation and Liver Cell Death in Mice with Niemann-Pick Type C Disease. Hepatology 2005, 42, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Garver, W.S.; Jelinek, D.; Francis, G.A.; Murphy, B.D. The Niemann-Pick C1 Gene Is Downregulated by Feedback Inhibition of the SREBP Pathway in Human Fibroblasts. J. Lipid Res. 2008, 49, 1090–1102. [Google Scholar] [CrossRef] [PubMed]
- Garver, W.S.; Jelinek, D.; Oyarzo, J.N.; Flynn, J.; Zuckerman, M.; Krishnan, K.; Chung, B.H.; Heidenreich, R.A. Characterization of Liver Disease and Lipid Metabolism in the Niemann-Pick C1 Mouse. J. Cell. Biochem. 2007, 101, 498–516. [Google Scholar] [CrossRef]
- McGraw, C.; Yang, L.; Levental, I.; Lyman, E.; Robinson, A.S. Membrane Cholesterol Depletion Reduces Downstream Signaling Activity of the Adenosine A2A Receptor. Biochim. Biophys. Acta—Biomembr. 2019, 1861, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Charalambous, C.; Gsandtner, I.; Keuerleber, S.; Milan-Lobo, L.; Kudlacek, O.; Freissmuth, M.; Zezula, J. Restricted Collision Coupling of the A2A Receptor Revisited. J. Biol. Chem. 2008, 283, 9276–9288. [Google Scholar] [CrossRef]
- Esquejo, R.M.; Roqueta-Rivera, M.; Shao, W.; Phelan, P.E.; Seneviratne, U.; am Ende, C.W.; Hershberger, P.M.; Machamer, C.E.; Espenshade, P.J.; Osborne, T.F. Dipyridamole Inhibits Lipogenic Gene Expression by Retaining SCAP-SREBP in the Endoplasmic Reticulum. Cell Chem. Biol. 2021, 28, 169–179.e7. [Google Scholar] [CrossRef]
- Sloka, S.; Metz, L.M.; Hader, W.; Starreveld, Y.; Yong, V.W. Reduction of Microglial Activity in a Model of Multiple Sclerosis by Dipyridamole. J. Neuroinflamm. 2013, 10, 855. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pepponi, R.; De Simone, R.; De Nuccio, C.; Visentin, S.; Matteucci, A.; Bernardo, A.; Popoli, P.; Ferrante, A. Repurposing Dipyridamole in Niemann Pick Type C Disease: A Proof of Concept Study. Int. J. Mol. Sci. 2022, 23, 3456. https://doi.org/10.3390/ijms23073456
Pepponi R, De Simone R, De Nuccio C, Visentin S, Matteucci A, Bernardo A, Popoli P, Ferrante A. Repurposing Dipyridamole in Niemann Pick Type C Disease: A Proof of Concept Study. International Journal of Molecular Sciences. 2022; 23(7):3456. https://doi.org/10.3390/ijms23073456
Chicago/Turabian StylePepponi, Rita, Roberta De Simone, Chiara De Nuccio, Sergio Visentin, Andrea Matteucci, Antonietta Bernardo, Patrizia Popoli, and Antonella Ferrante. 2022. "Repurposing Dipyridamole in Niemann Pick Type C Disease: A Proof of Concept Study" International Journal of Molecular Sciences 23, no. 7: 3456. https://doi.org/10.3390/ijms23073456
APA StylePepponi, R., De Simone, R., De Nuccio, C., Visentin, S., Matteucci, A., Bernardo, A., Popoli, P., & Ferrante, A. (2022). Repurposing Dipyridamole in Niemann Pick Type C Disease: A Proof of Concept Study. International Journal of Molecular Sciences, 23(7), 3456. https://doi.org/10.3390/ijms23073456