
A Dual Anti-Inflammatory and Anti-Proliferative 3-Styrylchromone Derivative Synergistically Enhances the Anti-Cancer Effects of DNA-Damaging Agents on Colon Cancer Cells by Targeting HMGB1-RAGE-ERK1/2 Signaling




Abstract
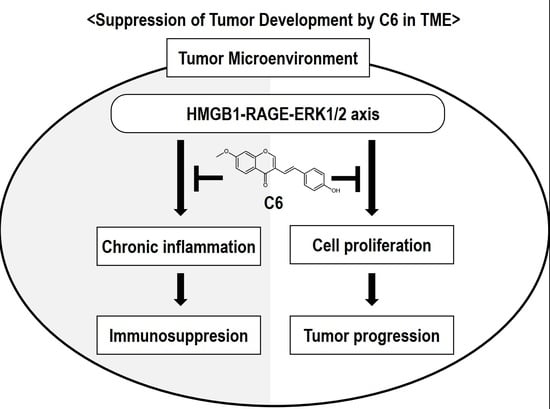
1. Introduction
2. Results
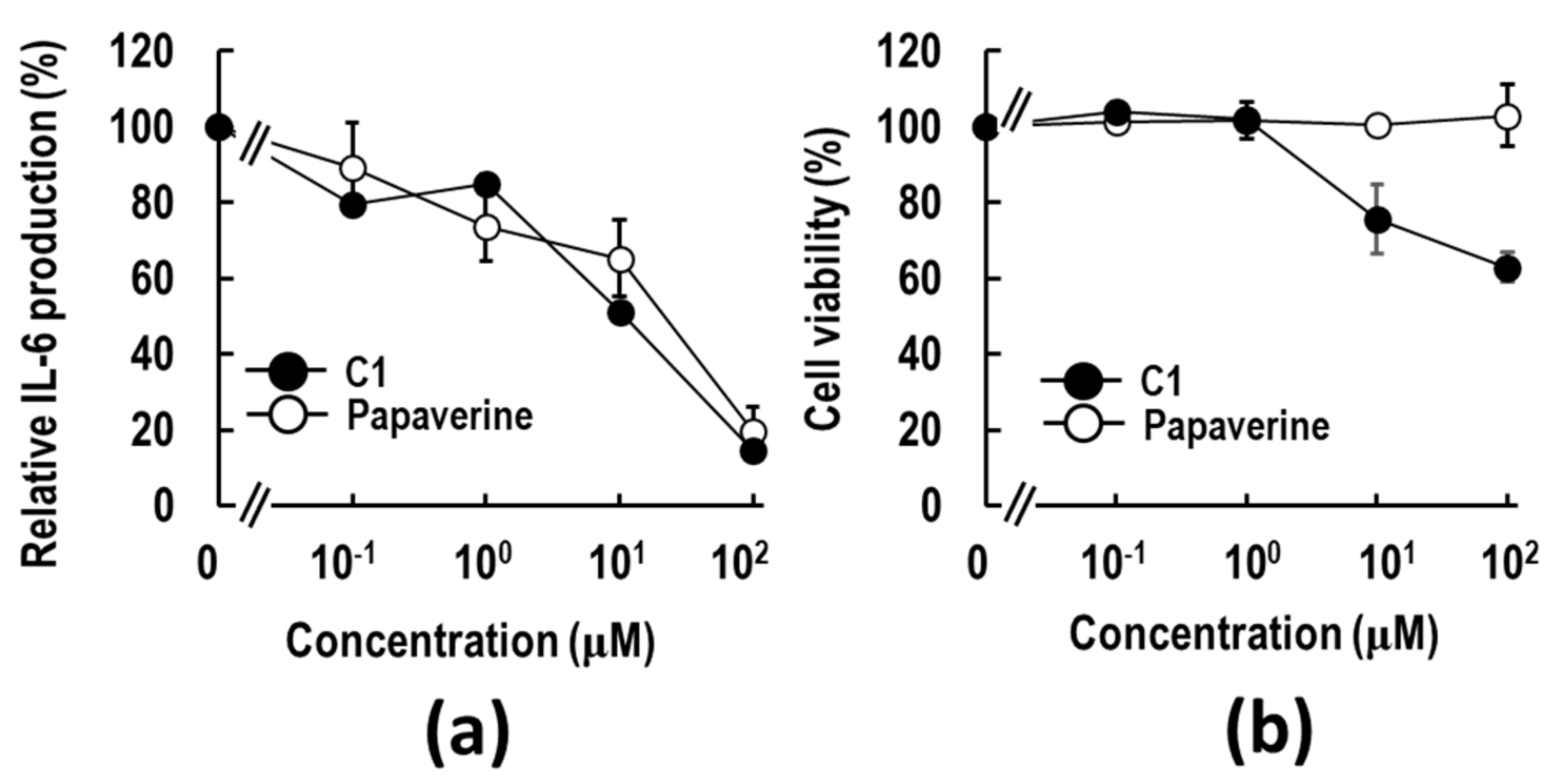
2.1. Discovery of a Papaverine-Mimetic 3-Styrylchromone Derivative
2.2. Three-Dimensional Pharmacophore Similarity Search for C1 Mimetics
2.3. C6 Exerts an Anti-Inflammatory Effect via Suppression of HMGB1-RAGE-ERK1/2 Signaling Pathway in RAW264.7 Cells
2.4. C6 Inhibits Proliferation and Induces Apoptosis in HCT116 Cells
2.5. C6 Suppresses HMGB1-RAGE Signaling by Inhibiting the Activation of ERK 1/2 in HMGB1-Stimulated HCT116 Cells
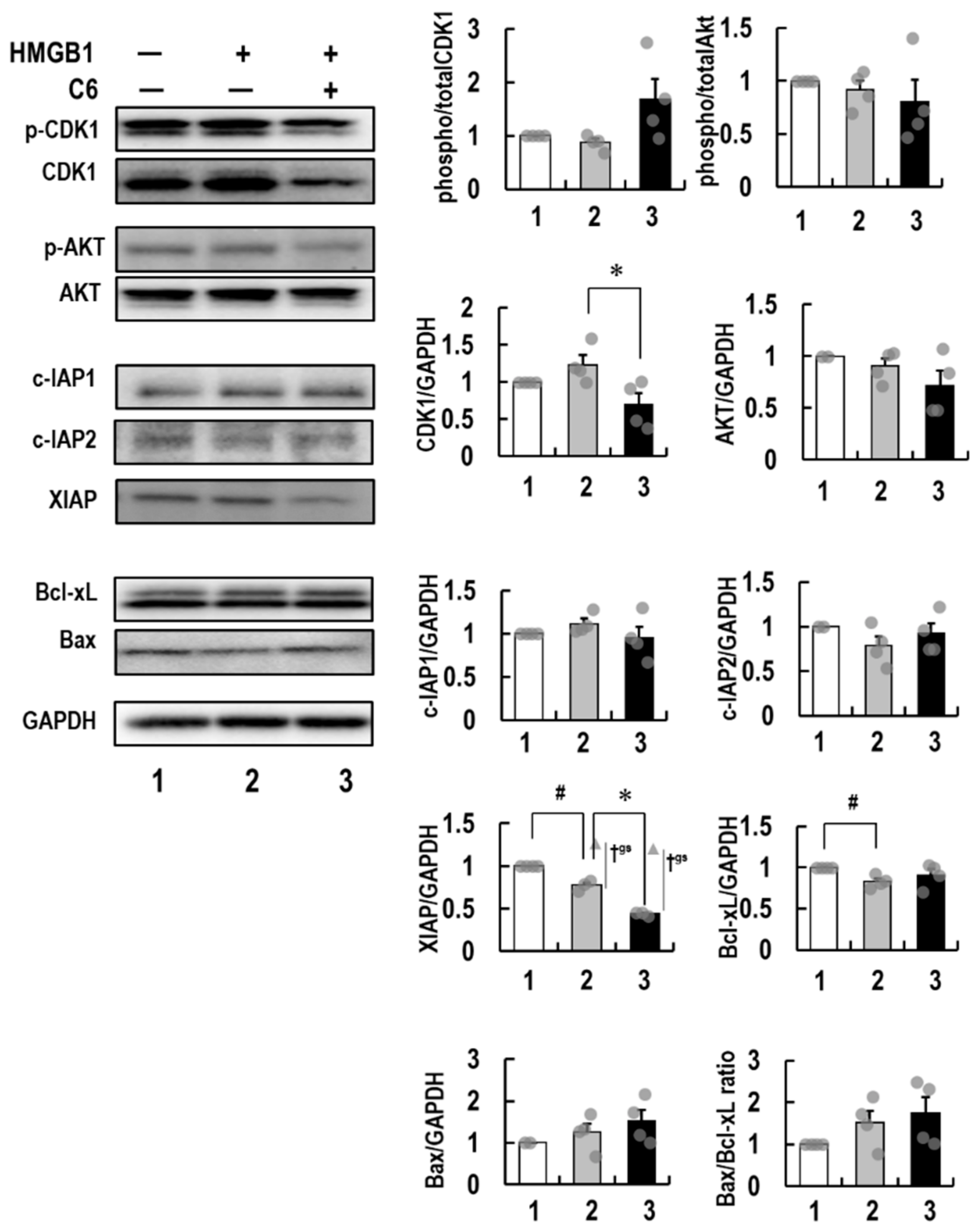
2.6. Molecular Mechanism of Action of C6 in HMGB1-Stimulated HCT116 Cells
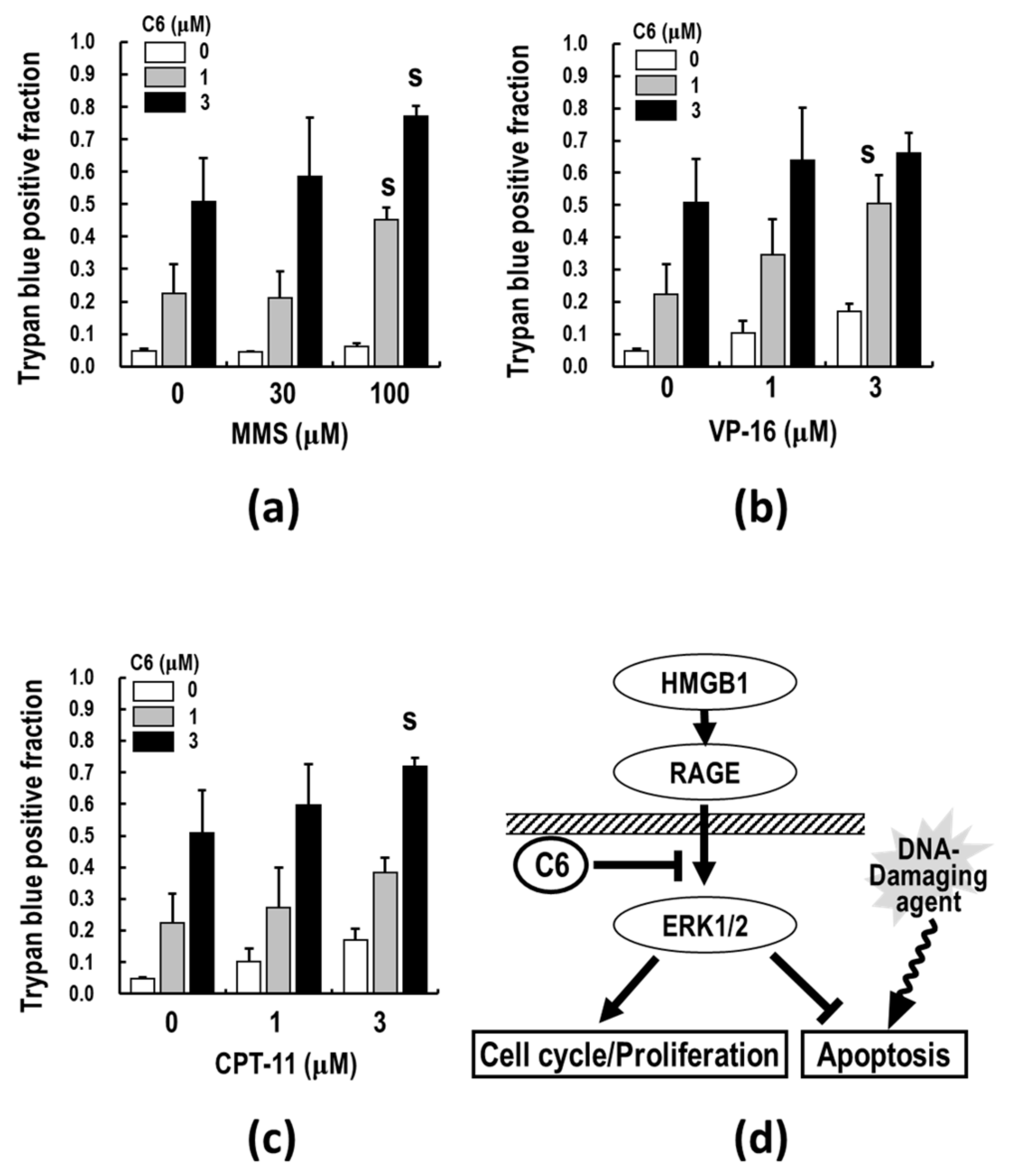
2.7. C6, along with DNA-Damaging Agents, Synergistically Induces Apoptosis
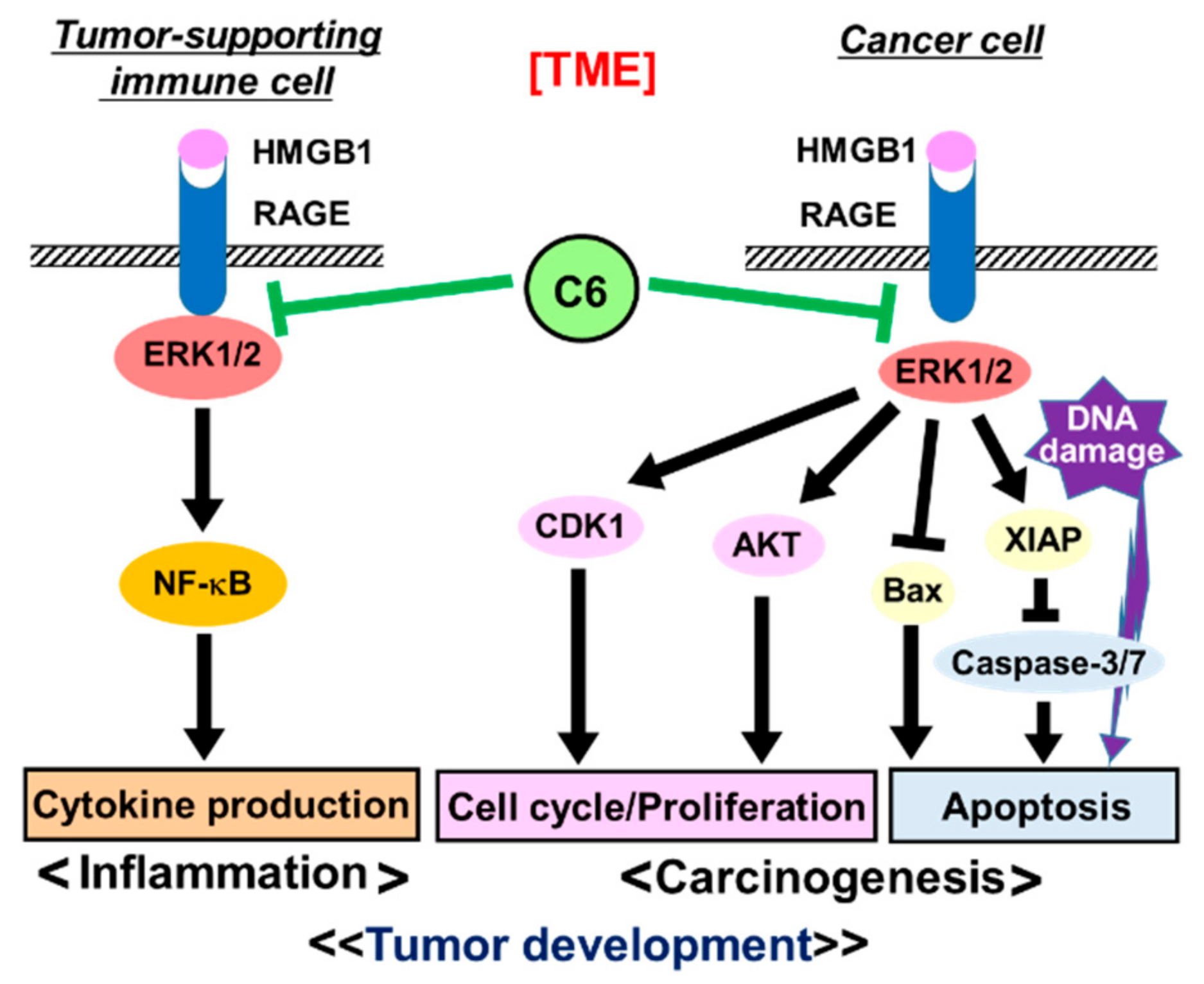
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Synthesis of Test Compounds
4.3. In Silico 3D Pharmacophore Analysis
4.4. Cells and Cell Culture
4.5. Assay for Cell Viability
4.6. Assay for Cell Proliferation
4.7. ELISA for IL-6
4.8. Cell Cycle Analyses
4.9. Caspase Activation Analyses
4.10. Trypan Blue Dye Exclusion Assay
4.11. Western Blotting
4.12. EOBA
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fritz, G. RAGE: A single receptor fits multiple ligands. Trends Biochem. Sci. 2011, 36, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Nisticò, P.; Ciliberto, G. Biological mechanisms linked to inflammation in cancer: Discovery of tumor microenvironment-related biomarkers and their clinical application in solid tumors. Int. J. Biol. Markers 2020, 35 (Suppl. 1), 8–11. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, C.D.; Fuentes, M.K.; Huang, E.H.; Arumugam, T. RAGE and RAGE ligands in cancer. Curr. Mol. Med. 2007, 7, 777–789. [Google Scholar] [CrossRef]
- Palanissami, G.; Paul, S.F.D. RAGE and Its Ligands: Molecular Interplay Between Glycation, Inflammation, and Hallmarks of Cancer—A Review. Horm. Cancer 2018, 9, 295–325. [Google Scholar] [CrossRef]
- Gebhardt, C.; Riehl, A.; Durchdewald, M.; Németh, J.; Fürstenberger, G.; Müller-Decker, K.; Enk, A.; Arnold, B.; Bierhaus, A.; Nawroth, P.P.; et al. RAGE signaling sustains inflammation and promotes tumor development. J. Exp. Med. 2008, 205, 275–285. [Google Scholar] [CrossRef]
- Bongarzone, S.; Savickas, V.; Luzi, F.; Gee, A.D. Targeting the Receptor for Advanced Glycation Endproducts (RAGE): A Medicinal Chemistry Perspective. J. Med. Chem. 2017, 60, 7213–7232. [Google Scholar] [CrossRef]
- Khambu, B.; Hong, H.; Liu, S.; Liu, G.; Chen, X.; Dong, Z.; Wan, J.; Yin, X.M. The HMGB1-RAGE axis modulates the growth of autophagy-deficient hepatic tumors. Cell Death Dis. 2020, 11, 333. [Google Scholar] [CrossRef]
- Zeng, S.; Feirt, N.; Goldstein, M.; Guarrera, J.; Ippagunta, N.; Ekong, U.; Dun, H.; Lu, Y.; Qu, W.; Schmidt, A.M.; et al. Blockade of receptor for advanced glycation end product (RAGE) attenuates ischemia and reperfusion injury to the liver in mice. Hepatology 2004, 39, 422–432. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Pellegrini, L.; Napolitano, A.; Giorgi, C.; Jube, S.; Preti, A.; Jennings, C.J.; De Marchis, F.; Flores, E.G.; Larson, D.; et al. Aspirin delays mesothelioma growth by inhibiting HMGB1-mediated tumor progression. Cell Death Dis. 2015, 6, e1786. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Yang, X.; Lin, Y.; Shi, Y.; Li, B.; Liu, W.; Yin, W.; Dang, Y.; Chu, Y.; Fan, J.; He, R. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3-CCL2 Signaling. Cancer Res. 2016, 76, 4124–4135. [Google Scholar] [CrossRef]
- McCarthy, J.B.; El-Ashry, D.; Turley, E.A. Hyaluronan, Cancer-Associated Fibroblasts and the Tumor Microenvironment in Malignant Progression. Front. Cell Dev. Biol. 2018, 6, 48. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef]
- Tesi, R.J. MDSC; The Most Important Cell You Have Never Heard of. Trends Pharmacol. Sci. 2019, 40, 4–7. [Google Scholar]
- Gorgulho, C.M.; Romagnoli, G.G.; Bharthi, R.; Lotze, M.T. Johnny on the Spot-Chronic Inflammation Is Driven by HMGB1. Front. Immunol. 2019, 10, 1561. [Google Scholar] [CrossRef]
- Fiuza, C.; Bustin, M.; Talwar, S.; Tropea, M.; Gerstenberger, E.; Shelhamer, J.H.; Suffredini, A.F. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood 2003, 101, 2652–2660. [Google Scholar] [CrossRef]
- Li, H.; Fan, X.; Houghton, J. Tumor microenvironment: The role of the tumor stroma in cancer. J. Cell. Biochem. 2007, 101, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Taylor, M.J.; Postovit, L.M. Microenvironmental regulation of cancer stem cell phenotypes. Curr. Stem Cell Res. Ther. 2012, 7, 197–216. [Google Scholar] [CrossRef] [PubMed]
- Sounni, N.E.; Noel, A. Targeting the tumor microenvironment for cancer therapy. Clin. Chem. 2013, 59, 85–93. [Google Scholar] [CrossRef]
- Germano, G.; Allavena, P.; Mantovani, A. Cytokines as a key component of cancer-related inflammation. Cytokine 2008, 43, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Rincon, M. Interleukin-6: From an inflammatory marker to a target for inflammatory diseases. Trends Immunol. 2012, 33, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Rinaudo-Gaujous, M.; Roblin, X.; Marotte, H.; Paul, S. The antibody response against human and chimeric anti-TNF therapeutic antibodies primarily targets the TNF binding region. Ann. Rheum. Dis. 2015, 74, e40. [Google Scholar] [CrossRef]
- Sun, Z.; Shi, K.; Yang, S.; Liu, J.; Zhou, Q.; Wang, G.; Song, J.; Li, Z.; Zhang, Z.; Yuan, W. Effect of exosomal miRNA on cancer biology and clinical applications. Mol. Cancer 2018, 17, 147. [Google Scholar] [CrossRef]
- Allavena, P.; Garlanda, C.; Borrello, M.G.; Sica, A.; Mantovani, A. Pathways connecting inflammation and cancer. Curr. Opin. Genet. Dev. 2008, 18, 3–10. [Google Scholar] [CrossRef]
- Skrha, J., Jr.; Kalousová, M.; Svarcová, J.; Muravská, A.; Kvasnička, J.; Landová, L.; Zima, T.; Skrha, J. Relationship of soluble RAGE and RAGE ligands HMGB1 and EN-RAGE to endothelial dysfunction in type 1 and type 2 diabetes mellitus. Exp. Clin. Endocrinol. Diabetes Off. J. Ger. Soc. Endocrinol. Ger. Diabetes Assoc. 2012, 120, 277–281. [Google Scholar]
- González, H.; Elgueta, D.; Montoya, A.; Pacheco, R. Neuroimmune regulation of microglial activity involved in neuroinflammation and neurodegenerative diseases. J. Neuroimmunol. 2014, 274, 1–13. [Google Scholar] [CrossRef]
- Manigrasso, M.B.; Pan, J.; Rai, V.; Zhang, J.; Reverdatto, S.; Quadri, N.; DeVita, R.J.; Ramasamy, R.; Shekhtman, A.; Schmidt, A.M. Small Molecule Inhibition of Ligand-Stimulated RAGE-DIAPH1 Signal Transduction. Sci. Rep. 2016, 6, 22450. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.E.; Beltrame, M. Upwardly mobile proteins. Workshop: The role of HMG proteins in chromatin structure, gene expression and neoplasia. EMBO Rep. 2000, 1, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Investig. 2001, 108, 949–955. [Google Scholar] [CrossRef]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Clynes, R.; Moser, B.; Yan, S.F.; Ramasamy, R.; Herold, K.; Schmidt, A.M. Receptor for AGE (RAGE): Weaving tangled webs within the inflammatory response. Curr. Mol. Med. 2007, 7, 743–751. [Google Scholar]
- Gaskell, H.; Ge, X.; Nieto, N. High-Mobility Group Box-1 and Liver Disease. Hepatol. Commun. 2018, 2, 1005–1020. [Google Scholar] [CrossRef]
- Yu, L.X.; Yan, L.; Yang, W.; Wu, F.Q.; Ling, Y.; Chen, S.Z.; Tang, L.; Tan, Y.X.; Cao, D.; Wu, M.C.; et al. Platelets promote tumour metastasis via interaction between TLR4 and tumour cell-released high-mobility group box1 protein. Nat. Commun. 2014, 5, 5256. [Google Scholar] [CrossRef]
- Chung, H.W.; Lee, S.G.; Kim, H.; Hong, D.J.; Chung, J.B.; Stroncek, D.; Lim, J.B. Serum high mobility group box-1 (HMGB1) is closely associated with the clinical and pathologic features of gastric cancer. J. Transl. Med. 2009, 7, 38. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Bianchi, M.E.; Crippa, M.P.; Manfredi, A.A.; Mezzapelle, R.; Rovere Querini, P.; Venereau, E. High-mobility group box 1 protein orchestrates responses to tissue damage via inflammation, innate and adaptive immunity, and tissue repair. Immunol. Rev. 2017, 280, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Yang, Z.; Guo, J.; Weng, L.; Tang, W.; Jin, S.; Ma, W. Myeloid-derived suppressor cells-new and exciting players in lung cancer. J. Hematol. Oncol. 2020, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Li, C.; Wang, J.; Xue, L. Myeloid-derived suppressor cells: Roles in the tumor microenvironment and tumor radiotherapy. Int. J. Cancer 2019, 144, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Khambu, B.; Huda, N.; Chen, X.; Antoine, D.J.; Li, Y.; Dai, G.; Köhler, U.A.; Zong, W.X.; Waguri, S.; Werner, S.; et al. HMGB1 promotes ductular reaction and tumorigenesis in autophagy-deficient livers. J. Clin. Investig. 2018, 128, 2419–2435. [Google Scholar] [CrossRef]
- Jube, S.; Rivera, Z.S.; Bianchi, M.E.; Powers, A.; Wang, E.; Pagano, I.; Pass, H.I.; Gaudino, G.; Carbone, M.; Yang, H. Cancer cell secretion of the DAMP protein HMGB1 supports progression in malignant mesothelioma. Cancer Res. 2012, 72, 3290–3301. [Google Scholar] [CrossRef]
- Pellegrini, L.; Xue, J.; Larson, D.; Pastorino, S.; Jube, S.; Forest, K.H.; Saad-Jube, Z.S.; Napolitano, A.; Pagano, I.; Negi, V.S.; et al. HMGB1 targeting by ethyl pyruvate suppresses malignant phenotype of human mesothelioma. Oncotarget 2017, 8, 22649–22661. [Google Scholar] [CrossRef]
- Hong, B.; Muili, K.; Bolyard, C.; Russell, L.; Lee, T.J.; Banasavadi-Siddegowda, Y.; Yoo, J.Y.; Yan, Y.; Ballester, L.Y.; Bockhorst, K.H.; et al. Suppression of HMGB1 Released in the Glioblastoma Tumor Microenvironment Reduces Tumoral Edema. Mol. Ther. Oncolytics 2019, 12, 93–102. [Google Scholar] [CrossRef]
- Turovskaya, O.; Foell, D.; Sinha, P.; Vogl, T.; Newlin, R.; Nayak, J.; Nguyen, M.; Olsson, A.; Nawroth, P.P.; Bierhaus, A.; et al. RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis-associated carcinogenesis. Carcinogenesis 2008, 29, 2035–2043. [Google Scholar] [CrossRef]
- Rojas, A.; Figueroa, H.; Morales, E. Fueling inflammation at tumor microenvironment: The role of multiligand/RAGE axis. Carcinogenesis 2010, 31, 334–341. [Google Scholar] [CrossRef]
- Jarosz-Biej, M.; Smolarczyk, R.; Cichoń, T.; Kułach, N. Tumor Microenvironment as A “Game Changer” in Cancer Radiotherapy. Int. J. Mol. Sci. 2019, 20, 3212. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Thompson, P.; Beutler, B. Dexamethasone and pentoxifylline inhibit endotoxin-induced cachectin/tumor necrosis factor synthesis at separate points in the signaling pathway. J. Exp. Med. 1990, 172, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Ching, L.M.; Browne, W.L.; Tchernegovski, R.; Gregory, T.; Baguley, B.C.; Palmer, B.D. Interaction of thalidomide, phthalimide analogues of thalidomide and pentoxifylline with the anti-tumour agent 5,6-dimethylxanthenone-4-acetic acid: Concomitant reduction of serum tumour necrosis factor-alpha and enhancement of anti-tumour activity. Br. J. Cancer 1998, 78, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Tamada, K.; Nakajima, S.; Ogawa, N.; Inada, M.; Shibasaki, H.; Sato, A.; Takasawa, R.; Yoshimori, A.; Suzuki, Y.; Watanabe, N.; et al. Papaverine identified as an inhibitor of high mobility group box 1/receptor for advanced glycation end-products interaction suppresses high mobility group box 1-mediated inflammatory responses. Biochem. Biophys. Res. Commun. 2019, 511, 665–670. [Google Scholar] [CrossRef]
- Nakajima, S.; Ogawa, N.; Yokoue, N.; Tachibana, H.; Tamada, K.; Okazawa, M.; Sato, A.; Oyama, T.; Abe, H.; Kamiya, T.; et al. Trimebutine attenuates high mobility group box 1-receptor for advanced glycation end-products inflammatory signaling pathways. Biochem. Biophys. Res. Commun. 2020, 533, 1155–1161. [Google Scholar] [CrossRef]
- Inada, M.; Shindo, M.; Kobayashi, K.; Sato, A.; Yamamoto, Y.; Akasaki, Y.; Ichimura, K.; Tanuma, S.I. Anticancer effects of a non-narcotic opium alkaloid medicine, papaverine, in human glioblastoma cells. PLoS ONE 2019, 14, e0216358. [Google Scholar]
- Inada, M.; Sato, A.; Shindo, M.; Yamamoto, Y.; Akasaki, Y.; Ichimura, K.; Tanuma, S.I. Anticancer Non-narcotic Opium Alkaloid Papaverine Suppresses Human Glioblastoma Cell Growth. Anticancer. Res. 2019, 39, 6743–6750. [Google Scholar] [CrossRef]
- Sakagami, H.; Shimada, C.; Kanda, Y.; Amano, O.; Sugimoto, M.; Ota, S.; Soga, T.; Tomita, M.; Sato, A.; Tanuma, S.I.; et al. Effects of 3-styrylchromones on metabolic profiles and cell death in oral squamous cell carcinoma cells. Toxicol. Rep. 2015, 2, 1281–1290. [Google Scholar] [CrossRef]
- Abe, H.; Okazawa, M.; Oyama, T.; Yamazaki, H.; Yoshimori, A.; Kamiya, T.; Tsukimoto, M.; Takao, K.; Sugita, Y.; Sakagami, H.; et al. A Unique Anti-Cancer 3-Styrylchromone Suppresses Inflammatory Response via HMGB1-RAGE Signaling. Medicines 2021, 8, 17. [Google Scholar] [CrossRef]
- Okazawa, M.; Oyama, T.; Abe, H.; Yamazaki, H.; Yoshimori, A.; Tsukimoto, M.; Yoshizawa, K.; Takao, K.; Sugita, Y.; Kamiya, T.; et al. A 3-styrylchromone converted from trimebutine 3D pharmacophore possesses dual suppressive effects on RAGE and TLR4 signaling pathways. Biochem. Biophys. Res. Commun. 2021, 566, 1–8. [Google Scholar] [CrossRef]
- Takao, K.; Ishikawa, R.; Sugita, Y. Synthesis and biological evaluation of 3-styrylchromone derivatives as free radical scavengers and α-glucosidase inhibitors. Chem. Pharm. Bull. 2014, 62, 810–815. [Google Scholar] [CrossRef] [PubMed]
- Shimada, C.; Uesawa, Y.; Ishii-Nozawa, R.; Ishihara, M.; Kagaya, H.; Kanamoto, T.; Terakubo, S.; Nakashima, H.; Takao, K.; Sugita, Y.; et al. Quantitative structure-cytotoxicity relationship of 3-styrylchromones. Anticancer Res. 2014, 34, 5405–5411. [Google Scholar] [PubMed]
- Takao, K.; Hoshi, K.; Sakagami, H.; Shi, H.; Bandow, K.; Nagai, J.; Uesawa, Y.; Tomomura, A.; Tomomura, M.; Sugita, Y. Further Quantitative Structure-Cytotoxicity Relationship Analysis of 3-Styrylchromones. Anticancer Res. 2020, 40, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, N.; Nakajima, S.; Tamada, K.; Yokoue, N.; Tachibana, H.; Okazawa, M.; Oyama, T.; Abe, H.; Yamazaki, H.; Yoshimori, A.; et al. Trimebutine suppresses Toll-like receptor 2/4/7/8/9 signaling pathways in macrophages. Arch. Biochem. Biophys. 2021, 711, 109029. [Google Scholar] [CrossRef]
- Ishihara, K.; Tsutsumi, K.; Kawane, S.; Nakajima, M.; Kasaoka, T. The receptor for advanced glycation end-products (RAGE) directly binds to ERK by a D-domain-like docking site. FEBS Lett. 2003, 550, 107–113. [Google Scholar] [CrossRef]
- Rotin, L.E.; Gronda, M.; MacLean, N.; Hurren, R.; Wang, X.; Lin, F.H.; Wrana, J.; Datti, A.; Barber, D.L.; Minden, M.D.; et al. Ibrutinib synergizes with poly(ADP-ribose) glycohydrolase inhibitors to induce cell death in AML cells via a BTK-independent mechanism. Oncotarget 2016, 7, 2765–2779. [Google Scholar] [CrossRef][Green Version]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef]
- Liu, L.; Xu, Y.; Reiter, R.J.; Pan, Y.; Chen, D.; Liu, Y.; Pu, X.; Jiang, L.; Li, Z. Inhibition of ERK1/2 Signaling Pathway is Involved in Melatonin’s Antiproliferative Effect on Human MG-63 Osteosarcoma Cells. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2016, 39, 2297–2307. [Google Scholar] [CrossRef]
- Zhang, P.; Kawakami, H.; Liu, W.; Zeng, X.; Strebhardt, K.; Tao, K.; Huang, S.; Sinicrope, F.A. Targeting CDK1 and MEK/ERK Overcomes Apoptotic Resistance in BRAF-Mutant Human Colorectal Cancer. Mol. Cancer Res. MCR 2018, 16, 378–389. [Google Scholar] [CrossRef]
- Lau, H.W.; Ma, H.T.; Yeung, T.K.; Tam, M.Y.; Zheng, D.; Chu, S.K.; Poon, R.Y.C. Quantitative differences between cyclin-dependent kinases underlie the unique functions of CDK1 in human cells. Cell Rep. 2021, 37, 109808. [Google Scholar] [CrossRef]
- Nitulescu, G.M.; Van De Venter, M.; Nitulescu, G.; Ungurianu, A.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Grădinaru, D.; Tsatsakis, A.; Tsoukalas, D.; et al. The Akt pathway in oncology therapy and beyond (Review). Int. J. Oncol. 2018, 53, 2319–2331. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zhao, W.; Tong, P.; Li, P.; Zhao, Y.; Li, H.; Liang, J. Comprehensive molecular characterization of inhibitors of apoptosis proteins (IAPs) for therapeutic targeting in cancer. BMC Med. Genom. 2020, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Peña-Blanco, A.; García-Sáez, A.J. Bax, Bak and beyond—Mitochondrial performance in apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef] [PubMed]
- Greaves, G.; Milani, M.; Butterworth, M.; Carter, R.J.; Byrne, D.P.; Eyers, P.A.; Luo, X.; Cohen, G.M.; Varadarajan, S. BH3-only proteins are dispensable for apoptosis induced by pharmacological inhibition of both MCL-1 and BCL-X(L). Cell Death Differ. 2019, 26, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.S.; Mitchell, D.L.; Vasquez, K.M. High mobility group protein B1 enhances DNA repair and chromatin modification after DNA damage. Proc. Natl. Acad. Sci. USA 2008, 105, 10320–10325. [Google Scholar] [CrossRef]
- Olson, M.E.; Guselle, N.J.; O’Handley, R.M.; Swift, M.L.; McAllister, T.A.; Jelinski, M.D.; Morck, D.W. Giardia and Cryptosporidium in dairy calves in British Columbia. Can. Vet. J. La Rev. Vet. Can. 1997, 38, 703–706. [Google Scholar]
- Mandke, P.; Vasquez, K.M. Interactions of high mobility group box protein 1 (HMGB1) with nucleic acids: Implications in DNA repair and immune responses. DNA Repair 2019, 83, 102701. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Modeling 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Shibui, Y.; Oyama, T.; Okazawa, M.; Yoshimori, A.; Abe, H.; Uchiumi, F.; Tanuma, S.I. Structural insights into the active site of poly(ADP-ribose) glycohydrolase using docking modes of 6-hydroxy-3H-xanthen-3-one derivative inhibitors. Bioorg. Med. Chem. 2020, 28, 115249. [Google Scholar] [CrossRef]
- Tanuma, S.I.; Katsuragi, K.; Oyama, T.; Yoshimori, A.; Shibasaki, Y.; Asawa, Y.; Yamazaki, H.; Makino, K.; Okazawa, M.; Ogino, Y.; et al. Structural Basis of Beneficial Design for Effective Nicotinamide Phosphoribosyltransferase Inhibitors. Molecules 2020, 25, 3633. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Compound | R1 | R2 | IL-6 Production Inhibition EC50 (μM) * |
| C1 | –OCH3 | –OCH3 | 11.1 ± 7.63 |
| C2 | –OCH3 | –F | >10 |
| C3 | –OCH3 | –Cl | >10 |
| C4 | –OCH3 | –N(CH3)2 | >10 |
| C5 | –OCH3 | –H | >10 |
| C6 | –OCH3 | –OH | 0.018 ± 0.025 |
| C7 | –H | –OH | 0.020 ± 0.016 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanuma, S.-i.; Oyama, T.; Okazawa, M.; Yamazaki, H.; Takao, K.; Sugita, Y.; Amano, S.; Abe, T.; Sakagami, H. A Dual Anti-Inflammatory and Anti-Proliferative 3-Styrylchromone Derivative Synergistically Enhances the Anti-Cancer Effects of DNA-Damaging Agents on Colon Cancer Cells by Targeting HMGB1-RAGE-ERK1/2 Signaling. Int. J. Mol. Sci. 2022, 23, 3426. https://doi.org/10.3390/ijms23073426
Tanuma S-i, Oyama T, Okazawa M, Yamazaki H, Takao K, Sugita Y, Amano S, Abe T, Sakagami H. A Dual Anti-Inflammatory and Anti-Proliferative 3-Styrylchromone Derivative Synergistically Enhances the Anti-Cancer Effects of DNA-Damaging Agents on Colon Cancer Cells by Targeting HMGB1-RAGE-ERK1/2 Signaling. International Journal of Molecular Sciences. 2022; 23(7):3426. https://doi.org/10.3390/ijms23073426
Chicago/Turabian StyleTanuma, Sei-ichi, Takahiro Oyama, Miwa Okazawa, Hiroaki Yamazaki, Koichi Takao, Yoshiaki Sugita, Shigeru Amano, Takehiko Abe, and Hiroshi Sakagami. 2022. "A Dual Anti-Inflammatory and Anti-Proliferative 3-Styrylchromone Derivative Synergistically Enhances the Anti-Cancer Effects of DNA-Damaging Agents on Colon Cancer Cells by Targeting HMGB1-RAGE-ERK1/2 Signaling" International Journal of Molecular Sciences 23, no. 7: 3426. https://doi.org/10.3390/ijms23073426
APA StyleTanuma, S.-i., Oyama, T., Okazawa, M., Yamazaki, H., Takao, K., Sugita, Y., Amano, S., Abe, T., & Sakagami, H. (2022). A Dual Anti-Inflammatory and Anti-Proliferative 3-Styrylchromone Derivative Synergistically Enhances the Anti-Cancer Effects of DNA-Damaging Agents on Colon Cancer Cells by Targeting HMGB1-RAGE-ERK1/2 Signaling. International Journal of Molecular Sciences, 23(7), 3426. https://doi.org/10.3390/ijms23073426