
Self-Antigens Targeted by Regulatory T Cells in Type 1 Diabetes


Abstract
1. Introduction
2. T Cell Receptor–Peptide–MHC Interactions
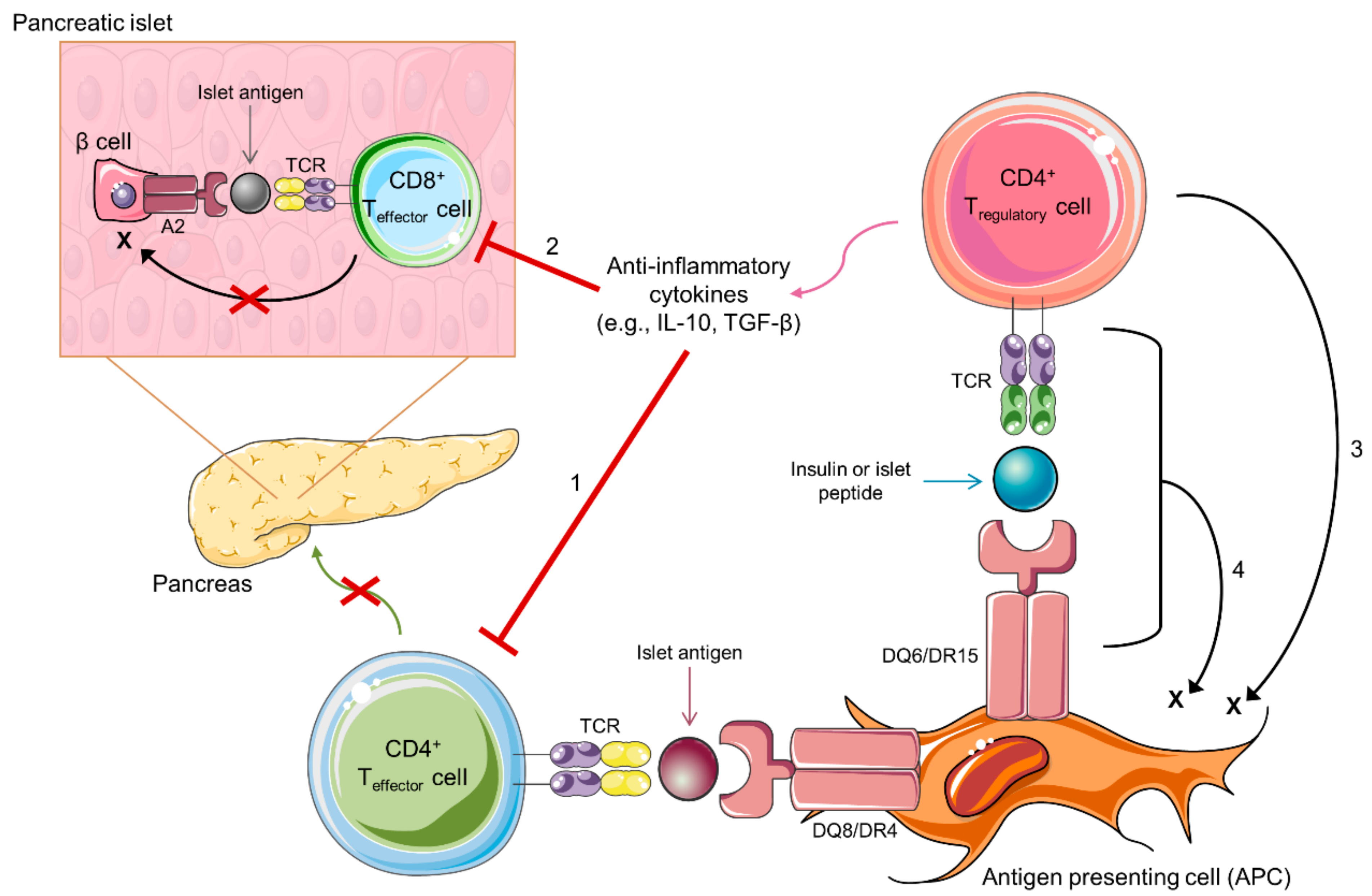
3. Immunologic Tolerance by Regulatory T Cells
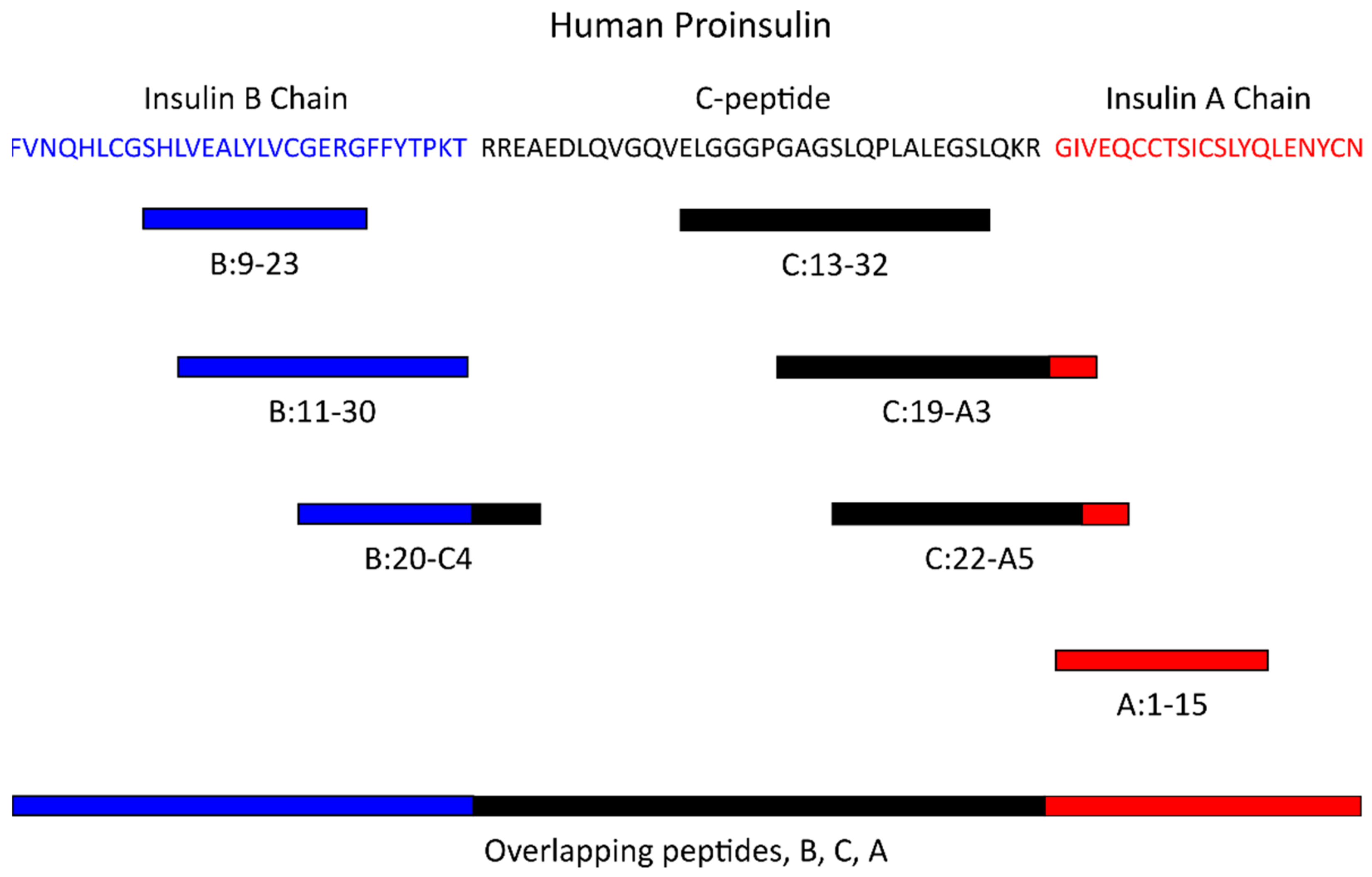
4. Antigen-Specific Tregs in Type 1 Diabetes
5. Structural Basis of a Treg T Cell Receptor Recognizing Peptide/MHC
6. Diabetes-Protective MHC Molecules Present Self-Antigens to Activate Tregs
7. Therapeutic Strategies to Enhance Tregs in Type 1 Diabetes
8. Challenges and Gaps in the Field
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Herold, K.; Eisenbarth, G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010, 464, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, A. Autoreactive T cells in type 1 diabetes. J. Clin. Investig. 2017, 127, 2881–2891. [Google Scholar] [CrossRef] [PubMed]
- Michels, A.W.; Redondo, M.J.; Atkinson, M.A. The pathogenesis, natural history, and treatment of type 1 diabetes: Time (thankfully) does not stand still. Lancet Diabetes Endocrinol. 2022, 10, 90–92. [Google Scholar] [CrossRef]
- Coppieters, K.T.; Dotta, F.; Amirian, N.; Campbell, P.D.; Kay, T.W.H.; Atkinson, M.A.; Roep, B.O.; von Herrath, M.G. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J. Exp. Med. 2012, 209, 51–60. [Google Scholar] [CrossRef]
- Gepts, W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes 1965, 14, 619–633. [Google Scholar] [CrossRef]
- Campbell-Thompson, M.; Fu, A.; Kaddis, J.S.; Wasserfall, C.; Schatz, D.A.; Pugliese, A.; Atkinson, M.A. Insulitis and β-Cell Mass in the Natural History of Type 1 Diabetes. Diabetes 2016, 65, 719–731. [Google Scholar] [CrossRef]
- Arif, S.; Leete, P.; Nguyen, V.; Marks, K.; Nor, N.M.; Estorninho, M.; Kronenberg-Versteeg, D.; Bingley, P.J.; Todd, J.A.; Guy, C.; et al. Blood and islet phenotypes indicate immunological heterogeneity in type 1 diabetes. Diabetes 2014, 63, 3835–3845. [Google Scholar] [CrossRef]
- Leete, P.; Willcox, A.; Krogvold, L.; Dahl-Jørgensen, K.; Foulis, A.K.; Richardson, S.J.; Morgan, N.G. Differential Insulitic Profiles Determine the Extent of β-Cell Destruction and the Age at Onset of Type 1 Diabetes. Diabetes 2016, 65, 1362–1369. [Google Scholar] [CrossRef]
- Hinman, R.M.; Cambier, J.C. Role of B lymphocytes in the pathogenesis of type 1 diabetes. Curr. Diab. Rep. 2014, 14, 543. [Google Scholar] [CrossRef]
- Mayer-Davis, E.J.; Lawrence, J.M.; Dabelea, D.; Divers, J.; Isom, S.; Dolan, L.; Imperatore, G.; Linder, B.; Marcovina, S.; Pettitt, D.J.; et al. Incidence Trends of Type 1 and Type 2 Diabetes among Youths, 2002–2012. N. Engl. J. Med. 2017, 376, 1419–1429. [Google Scholar] [CrossRef] [PubMed]
- Insel, R.A.; Dunne, J.L.; Atkinson, M.A.; Chiang, J.L.; Dabelea, D.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Krischer, J.P.; Lernmark, Å.; et al. Staging presymptomatic type 1 diabetes: A scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015, 38, 1964–1974. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.G.; Rewers, M.; Simell, O.; Simell, T.; Lempainen, J.; Steck, A.; Winkler, C.; Ilonen, J.; Veijola, R.; Knip, M.; et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 2013, 309, 2473–2479. [Google Scholar] [CrossRef]
- Simmons, K.M.; Michels, A.W. Type 1 diabetes: A predictable disease. World J. Diabetes 2015, 6, 380–390. [Google Scholar] [CrossRef]
- Herold, K.C.; Bundy, B.N.; Long, S.A.; Bluestone, J.A.; DiMeglio, L.A.; Dufort, M.J.; Gitelman, S.E.; Gottlieb, P.A.; Krischer, J.P.; Linsley, P.S.; et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 603–613. [Google Scholar] [CrossRef]
- Tisch, R.; McDevitt, H. Insulin-dependent diabetes mellitus. Cell 1996, 85, 291–297. [Google Scholar] [CrossRef]
- Wicker, L.S.; Miller, B.J.; Fischer, P.A.; Pressey, A.; Peterson, L.B. Genetic control of diabetes and insulitis in the nonobese diabetic mouse. Pedigree analysis of a diabetic H-2nod/b heterozygote. J. Immunol. 1989, 142, 781–784. [Google Scholar]
- Wicker, L.S.; Appel, M.C.; Dotta, F.; Pressey, A.; Miller, B.J.; DeLarato, N.H.; Fischer, P.A.; Boltz, R.C.; Peterson, L.B. Autoimmune syndromes in major histocompatibility complex (MHC) congenic strains of nonobese diabetic (NOD) mice. The NOD MHC is dominant for insulitis and cyclophosphamide-induced diabetes. J. Exp. Med. 1992, 176, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Verdaguer, J.; Averill, N.; Santamaria, P. A mechanism for the major histocompatibility complex-linked resistance to autoimmunity. J. Exp. Med. 1997, 186, 1059–1075. [Google Scholar] [CrossRef] [PubMed]
- Lühder, F.; Katz, J.; Benoist, C.; Mathis, D. Major histocompatibility complex class II molecules can protect from diabetes by positively selecting T cells with additional specificities. J. Exp. Med. 1998, 187, 379–387. [Google Scholar] [CrossRef]
- Tsai, S.; Serra, P.; Clemente-Casares, X.; Yamanouchi, J.; Thiessen, S.; Slattery, R.M.; Elliott, J.F.; Santamaria, P. Antidiabetogenic MHC class II promotes the differentiation of MHC-promiscuous autoreactive T cells into FOXP3+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2013, 110, 3471–3476. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Clayton, D.G.; Concannon, P.; Akolkar, B.; Cooper, J.D.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Nierras, C.; et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009, 41, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Deutsch, A.J.; Lenz, T.L.; Onengut-Gumuscu, S.; Han, B.; Chen, W.M.; Howson, J.M.M.; Todd, J.A.; De Bakker, P.I.W.; Rich, S.S.; et al. Additive and interaction effects at three amino acid positions in HLA-DQ and HLA-DR molecules drive type 1 diabetes risk. Nat. Genet. 2015, 47, 898–905. [Google Scholar] [CrossRef]
- Robertson, C.C.; Inshaw, J.R.J.; Onengut-Gumuscu, S.; Chen, W.-M.; Santa Cruz, D.F.; Yang, H.; Cutler, A.J.; Crouch, D.J.M.; Farber, E.; Bridges, S.L.; et al. Fine-mapping, trans-ancestral and genomic analyses identify causal variants, cells, genes and drug targets for type 1 diabetes. Nat. Genet. 2021, 53, 962–971. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Petersen, J.; Rossjohn, J.; Fugger, L. HLA variation and disease. Nat. Rev. Immunol. 2018, 18, 325–339. [Google Scholar] [CrossRef]
- Erlich, H.; Valdes, A.M.; Noble, J.; Carlson, J.A.; Varney, M.; Concannon, P.; Mychaleckyj, J.C.; Todd, J.A.; Bonella, P.; Fear, A.L.; et al. HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: Analysis of the type 1 diabetes genetics consortium families. Diabetes 2008, 57, 1084–1092. [Google Scholar] [CrossRef]
- Pugliese, A.; Boulware, D.; Yu, L.; Babu, S.; Steck, A.K.; Becker, D.; Rodriguez, H.; Di Meglio, L.; Evans-Molina, C.; Harrison, L.C.; et al. HLA-DRB1∗15:01-DQA1∗01:02-DQB1∗06:02 haplotype protects autoantibody-positive relatives from type 1 diabetes throughout the stages of disease progression. Diabetes 2016, 65, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Simmons, K.M.; Mitchell, A.M.; Alkanani, A.A.; McDaniel, K.A.; Baschal, E.E.; Armstrong, T.; Pyle, L.; Yu, L.; Michels, A.W. Failed Genetic Protection: Type 1 Diabetes in the Presence of HLA-DQB1*06:02. Diabetes 2020, 69, 1763–1769. [Google Scholar] [CrossRef]
- Nakayama, M.; Simmons, K.M.; Michels, A.W. Molecular Interactions Governing Autoantigen Presentation in Type 1 Diabetes. Curr. Diab. Rep. 2015, 15, 113. [Google Scholar] [CrossRef][Green Version]
- Grazia Roncarolo, M.; Gregori, S.; Battaglia, M.; Bacchetta, R.; Fleischhauer, K.; Levings, M.K. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol. Rev. 2006, 212, 28–50. [Google Scholar] [CrossRef]
- Groux, H.; O’Garra, A.; Bigler, M.; Rouleau, M.; Antonenko, S.; de Vries, J.E.; Roncarolo, M.G. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 1997, 389, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Grossman, W.J.; Verbsky, J.W.; Tollefsen, B.L.; Kemper, C.; Atkinson, J.P.; Ley, T.J. Differential expression of granzymes A and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood 2004, 104, 2840–2848. [Google Scholar] [CrossRef] [PubMed]
- Solé, P.; Santamaria, P. Re-Programming Autoreactive T Cells Into T-Regulatory Type 1 Cells for the Treatment of Autoimmunity. Front. Immunol. 2021, 12, 684240. [Google Scholar] [CrossRef] [PubMed]
- Lindley, S.; Dayan, C.M.; Bishop, A.; Roep, B.O.; Peakman, M.; Tree, T.I.M. Defective suppressor function in CD4(+)CD25(+) T-cells from patients with type 1 diabetes. Diabetes 2005, 54, 92–99. [Google Scholar] [CrossRef]
- Brusko, T.M.; Wasserfall, C.H.; Clare-Salzler, M.J.; Schatz, D.A.; Atkinson, M.A. Functional defects and the influence of age on the frequency of CD4+ CD25+ T-cells in type 1 diabetes. Diabetes 2005, 54, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Haseda, F.; Imagawa, A.; Murase-Mishiba, Y.; Terasaki, J.; Hanafusa, T. CD4+ CD45RA− FoxP3high activated regulatory T cells are functionally impaired and related to residual insulin-secreting capacity in patients with type 1 diabetes. Clin. Exp. Immunol. 2013, 173, 207–216. [Google Scholar] [CrossRef]
- Hull, C.M.; Peakman, M.; Tree, T.I.M. Regulatory T cell dysfunction in type 1 diabetes: what’s broken and how can we fix it? Diabetologia 2017, 60, 1839–1850. [Google Scholar] [CrossRef]
- Danke, N.A.; Koelle, D.M.; Yee, C.; Beheray, S.; Kwok, W.W. Autoreactive T Cells in Healthy Individuals. J. Immunol. 2004, 172, 5967–5972. [Google Scholar] [CrossRef]
- Yang, J.; Danke, N.A.; Berger, D.; Reichstetter, S.; Reijonen, H.; Greenbaum, C.; Pihoker, C.; James, E.A.; Kwok, W.W. Islet-Specific Glucose-6-Phosphatase Catalytic Subunit-Related Protein-Reactive CD4 + T Cells in Human Subjects. J. Immunol. 2006, 176, 2781–2789. [Google Scholar] [CrossRef]
- Nakayama, M.; McDaniel, K.; Fitzgerald-Miller, L.; Kiekhaefer, C.; Snell-Bergeon, J.K.; Davidson, H.W.; Rewers, M.; Yu, L.; Gottlieb, P.; Kappler, J.W.; et al. Regulatory vs. inflammatory cytokine T-cell responses to mutated insulin peptides in healthy and type 1 diabetic subjects. Proc. Natl. Acad. Sci. USA 2015, 112, 4429–4434. [Google Scholar] [CrossRef]
- Arif, S.; Tree, T.I.; Astill, T.P.; Tremble, J.M.; Bishop, A.J.; Dayan, C.M.; Roep, B.O.; Peakman, M. Autoreactive T cell responses show proinflammatory polarization in diabetes but a regulatory phenotype in health. J. Clin. Investig. 2004, 113, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Arif, S.; Pujol-Autonell, I.; Kamra, Y.; Williams, E.; Yusuf, N.; Domingo-Vila, C.; Shahrabi, Y.; Pollock, E.; Khatri, L.; Peakman, M.; et al. Mapping T Cell Responses to Native and Neo-Islet Antigen Epitopes in at Risk and Type 1 Diabetes Subjects. Front. Immunol. 2021, 12, 675746. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.M.; Alkanani, A.A.; McDaniel, K.A.; Pyle, L.; Waugh, K.; Steck, A.K.; Nakayama, M.; Yu, L.; Gottlieb, P.A.; Rewers, M.J.; et al. T-cell responses to hybrid insulin peptides prior to type 1 diabetes development. Proc. Natl. Acad. Sci. USA 2021, 118, e2019129118. [Google Scholar] [CrossRef]
- Delong, T.; Wiles, T.A.; Baker, R.L.; Bradley, B.; Barbour, G.; Reisdorph, R.; Armstrong, M.; Powell, R.L.; Reisdorph, N.; Kumar, N.; et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science 2016, 351, 711–714. [Google Scholar] [CrossRef]
- Baker, R.L.; Rihanek, M.; Hohenstein, A.C.; Nakayama, M.; Michels, A.; Gottlieb, P.A.; Haskins, K.; Delong, T. Hybrid Insulin Peptides Are Autoantigens in Type 1 Diabetes. Diabetes 2019, 68, 1830–1840. [Google Scholar] [CrossRef] [PubMed]
- Arribas-Layton, D.; Guyer, P.; Delong, T.; Dang, M.; Chow, I.-T.; Speake, C.; Greenbaum, C.J.; Kwok, W.W.; Baker, R.L.; Haskins, K.; et al. Hybrid Insulin Peptides are Recognized by Human T Cells in the Context of DRB1*04:01. Diabetes 2020. [Google Scholar] [CrossRef] [PubMed]
- Wiles, T.A.; Hohenstein, A.; Landry, L.G.; Dang, M.; Powell, R.; Guyer, P.; James, E.A.; Nakayama, M.; Haskins, K.; Delong, T.; et al. Characterization of Human CD4 T Cells Specific for a C-Peptide/C-Peptide Hybrid Insulin Peptide. Front. Immunol. 2021, 12, 668680. [Google Scholar] [CrossRef] [PubMed]
- Tree, T.I.M.; Lawson, J.; Edwards, H.; Skowera, A.; Arif, S.; Roep, B.O.; Peakman, M. Naturally Arising Human CD4 T-Cells That Recognize Islet Autoantigens and Secrete Interleukin-10 Regulate Proinflammatory T-Cell Responses via Linked Suppression. Diabetes 2010, 59, 1451–1460. [Google Scholar] [CrossRef]
- Chujo, D.; Nguyen, T.-S.; Foucat, E.; Blankenship, D.; Banchereau, J.; Nepom, G.T.; Chaussabel, D.; Ueno, H. Adult-onset type 1 diabetes patients display decreased IGRP-specific Tr1 cells in blood. Clin. Immunol. 2015, 161, 270–277. [Google Scholar] [CrossRef]
- Spence, A.; Purtha, W.; Tam, J.; Dong, S.; Kim, Y.; Ju, C.; Sterling, T.; Nakayama, M.; Robinson, W.H.; Bluestone, J.A.; et al. Revealing the specificity of regulatory T cells in murine autoimmune diabetes. Proc. Natl. Acad. Sci. USA 2018, 115, 5265–5270. [Google Scholar] [CrossRef]
- Beringer, D.X.; Kleijwegt, F.S.; Wiede, F.; van der Slik, A.R.; Loh, K.L.; Petersen, J.; Dudek, N.L.; Duinkerken, G.; Laban, S.; Joosten, A.; et al. T cell receptor reversed polarity recognition of a self-antigen major histocompatibility complex. Nat. Immunol. 2015, 16, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Gras, S.; Chadderton, J.; Del Campo, C.M.; Farenc, C.; Wiede, F.; Josephs, T.M.; Sng, X.Y.X.; Mirams, M.; Watson, K.A.; Tiganis, T.; et al. Reversed T Cell Receptor Docking on a Major Histocompatibility Class I Complex Limits Involvement in the Immune Response. Immunity 2016, 45, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Yang, J.; James, E.; Chow, I.T.; Reijonen, H.; Kwok, W.W. Increased islet antigen-specific regulatory and effector CD4+ T cells in healthy individuals with the type 1 diabetes-protective haplotype. Sci. Immunol. 2020, 5, eaax8767. [Google Scholar] [CrossRef]
- Ooi, J.D.; Petersen, J.; Tan, Y.H.; Huynh, M.; Willett, Z.J.; Ramarathinam, S.H.; Eggenhuizen, P.J.; Loh, K.L.; Watson, K.A.; Gan, P.Y.; et al. Dominant protection from HLA-linked autoimmunity by antigen-specific regulatory T cells. Nature 2017, 545, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Brand, D.; Zheng, S.G. Targeting IL-2: An unexpected effect in treating immunological diseases. Signal Transduct. Target. Ther. 2018, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros-Tato, A. Beyond regulatory T cells: The potential role for IL-2 to deplete T-follicular helper cells and treat autoimmune diseases. Immunotherapy 2014, 6, 1207–1220. [Google Scholar] [CrossRef] [PubMed]
- Long, S.A.; Rieck, M.; Sanda, S.; Bollyky, J.B.; Samuels, P.L.; Goland, R.; Ahmann, A.; Rabinovitch, A.; Aggarwal, S.; Phippard, D.; et al. Rapamycin/IL-2 combination therapy in patients with type 1 diabetes augments Tregs yet transiently impairs β-cell function. Diabetes 2012, 61, 2340–2348. [Google Scholar] [CrossRef]
- Rosenzwajg, M.; Churlaud, G.; Mallone, R.; Six, A.; Dérian, N.; Chaara, W.; Lorenzon, R.; Long, S.A.; Buckner, J.H.; Afonso, G.; et al. Low-dose interleukin-2 fosters a dose-dependent regulatory T cell tuned milieu in T1D patients. J. Autoimmun. 2015, 58, 48–58. [Google Scholar] [CrossRef]
- Yu, A.; Snowhite, I.; Vendrame, F.; Rosenzwajg, M.; Klatzmann, D.; Pugliese, A.; Malek, T.R. Selective IL-2 responsiveness of regulatory T cells through multiple intrinsic mechanisms supports the use of low-dose IL-2 therapy in type 1 diabetes. Diabetes 2015, 64, 2172–2183. [Google Scholar] [CrossRef]
- Waldron-Lynch, F.; Kareclas, P.; Irons, K.; Walker, N.M.; Mander, A.; Wicker, L.S.; Todd, J.A.; Bond, S. Rationale and study design of the Adaptive study of IL-2 dose on regulatory T cells in type 1 diabetes (DILT1D): A non-randomised, open label, adaptive dose finding trial. BMJ Open 2014, 4, e005559. [Google Scholar] [CrossRef]
- Todd, J.A.; Evangelou, M.; Cutler, A.J.; Pekalski, M.L.; Walker, N.M.; Stevens, H.E.; Porter, L.; Smyth, D.J.; Rainbow, D.B.; Ferreira, R.C.; et al. Regulatory T Cell Responses in Participants with Type 1 Diabetes after a Single Dose of Interleukin-2: A Non-Randomised, Open Label, Adaptive Dose-Finding Trial. PLoS Med. 2016, 13, e1002139. [Google Scholar] [CrossRef] [PubMed]
- Seelig, E.; Howlett, J.; Porter, L.; Truman, L.; Heywood, J.; Kennet, J.; Arbon, E.L.; Anselmiova, K.; Walker, N.M.; Atkar, R.; et al. The DILfrequency study is an adaptive trial to identify optimal IL-2 dosing in patients with type 1 diabetes. JCI Insight 2018, 3, e99306. [Google Scholar] [CrossRef] [PubMed]
- Hartemann, A.; Bensimon, G.; Payan, C.A.; Jacqueminet, S.; Bourron, O.; Nicolas, N.; Fonfrede, M.; Rosenzwajg, M.; Bernard, C.; Klatzmann, D. Low-dose interleukin 2 in patients with type 1 diabetes: A phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2013, 1, 295–305. [Google Scholar] [CrossRef]
- Rosenzwajg, M.; Salet, R.; Lorenzon, R.; Tchitchek, N.; Roux, A.; Bernard, C.; Carel, J.-C.; Storey, C.; Polak, M.; Beltrand, J.; et al. Low-dose IL-2 in children with recently diagnosed type 1 diabetes: A Phase I/II randomised, double-blind, placebo-controlled, dose-finding study. Diabetologia 2020, 63, 1808–1821. [Google Scholar] [CrossRef] [PubMed]
- Bluestone, J.A.; Buckner, J.H.; Fitch, M.; Gitelman, S.E.; Gupta, S.; Hellerstein, M.K.; Herold, K.C.; Lares, A.; Lee, M.R.; Li, K.; et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci. Transl. Med. 2015, 7, 315ra189. [Google Scholar] [CrossRef]
- Putnam, A.L.; Brusko, T.M.; Lee, M.R.; Liu, W.; Szot, G.L.; Ghosh, T.; Atkinson, M.A.; Bluestone, J.A. Expansion of human regulatory T-cells from patients with type 1 diabetes. Diabetes 2009, 58, 652–662. [Google Scholar] [CrossRef]
- Dong, S.; Hiam-Galvez, K.J.; Mowery, C.T.; Herold, K.C.; Gitelman, S.E.; Esensten, J.H.; Liu, W.; Lares, A.P.; Leinbach, A.S.; Lee, M.; et al. The effect of low-dose IL-2 and Treg adoptive cell therapy in patients with type 1 diabetes. JCI Insight 2021, 6, e147474. [Google Scholar] [CrossRef]
- Sockolosky, J.T.; Trotta, E.; Parisi, G.; Picton, L.; Su, L.L.; Le, A.C.; Chhabra, A.; Silveria, S.L.; George, B.M.; King, I.C.; et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science 2018, 359, 1037–1042. [Google Scholar] [CrossRef]
- Serr, I.; Scherm, M.G.; Zahm, A.M.; Schug, J.; Flynn, V.K.; Hippich, M.; Kälin, S.; Becker, M.; Achenbach, P.; Nikolaev, A.; et al. A miRNA181a/NFAT5 axis links impaired T cell tolerance induction with autoimmune type 1 diabetes. Sci. Transl. Med. 2018, 10, eaag1782. [Google Scholar] [CrossRef]
- Scherm, M.G.; Serr, I.; Zahm, A.M.; Schug, J.; Bellusci, S.; Manfredini, R.; Salb, V.K.; Gerlach, K.; Weigmann, B.; Ziegler, A.-G.; et al. miRNA142-3p targets Tet2 and impairs Treg differentiation and stability in models of type 1 diabetes. Nat. Commun. 2019, 10, 5697. [Google Scholar] [CrossRef]
- Serr, I.; Fürst, R.W.; Ott, V.B.; Scherm, M.G.; Nikolaev, A.; Gökmen, F.; Kälin, S.; Zillmer, S.; Bunk, M.; Weigmann, B.; et al. miRNA92a targets KLF2 and the phosphatase PTEN signaling to promote human T follicular helper precursors in T1D islet autoimmunity. Proc. Natl. Acad. Sci. USA 2016, 113, E6659–E6668. [Google Scholar] [CrossRef] [PubMed]
- Garavelli, S.; Bruzzaniti, S.; Tagliabue, E.; Prattichizzo, F.; Di Silvestre, D.; Perna, F.; La Sala, L.; Ceriello, A.; Mozzillo, E.; Fattorusso, V.; et al. Blood Co-Circulating Extracellular microRNAs and Immune Cell Subsets Associate with Type 1 Diabetes Severity. Int. J. Mol. Sci. 2020, 21, 477. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, G.; Ventriglia, G.; Stabilini, A.; Socci, C.; Morsiani, C.; Laurenzi, A.; Nigi, L.; Formichi, C.; Mfarrej, B.; Petrelli, A.; et al. Regulatory T-cells from pancreatic lymphnodes of patients with type-1 diabetes express increased levels of microRNA miR-125a-5p that limits CCR2 expression. Sci. Rep. 2017, 7, 6897. [Google Scholar] [CrossRef] [PubMed]
- Herold, K.C.; Hagopian, W.; Auger, J.A.; Poumian-Ruiz, E.; Taylor, L.; Donaldson, D.; Gitelman, S.E.; Harlan, D.M.; Xu, D.; Zivin, R.A.; et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N. Engl. J. Med. 2002, 346, 1692–1698. [Google Scholar] [CrossRef]
- Keymeulen, B.; Vandemeulebroucke, E.; Ziegler, A.G.; Mathieu, C.; Kaufman, L.; Hale, G.; Gorus, F.; Goldman, M.; Walter, M.; Candon, S.; et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N. Engl. J. Med. 2005, 352, 2598–2608. [Google Scholar] [CrossRef]
- Sherry, N.; Hagopian, W.; Ludvigsson, J.; Jain, S.M.; Wahlen, J.; Ferry, R.J.; Bode, B.; Aronoff, S.; Holland, C.; Carlin, D.; et al. Teplizumab for treatment of type 1 diabetes (Protégé study): 1-year results from a randomised, placebo-controlled trial. Lancet 2011, 378, 487–497. [Google Scholar] [CrossRef]
- Sims, E.K.; Bundy, B.N.; Stier, K.; Serti, E.; Lim, N.; Long, S.A.; Geyer, S.M.; Moran, A.; Greenbaum, C.J.; Evans-Molina, C.; et al. Teplizumab improves and stabilizes beta cell function in antibody-positive high-risk individuals. Sci. Transl. Med. 2021, 13, eabc8980. [Google Scholar] [CrossRef]
- Rigby, M.R.; DiMeglio, L.A.; Rendell, M.S.; Felner, E.I.; Dostou, J.M.; Gitelman, S.E.; Patel, C.M.; Griffin, K.J.; Tsalikian, E.; Gottlieb, P.A.; et al. Targeting of memory T cells with alefacept in new-onset type 1 diabetes (T1DAL study): 12 month results of a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Diabetes Endocrinol. 2013, 1, 284–294. [Google Scholar] [CrossRef]
- Rigby, M.R.; Harris, K.M.; Pinckney, A.; DiMeglio, L.A.; Rendell, M.S.; Felner, E.I.; Dostou, J.M.; Gitelman, S.E.; Griffin, K.J.; Tsalikian, E.; et al. Alefacept provides sustained clinical and immunological effects in new-onset type 1 diabetes patients. J. Clin. Investig. 2015, 125, 3285–3296. [Google Scholar] [CrossRef]
- Haller, M.J.; Gitelman, S.E.; Gottlieb, P.A.; Michels, A.W.; Rosenthal, S.M.; Shuster, J.J.; Zou, B.; Brusko, T.M.; Hulme, M.A.; Wasserfall, C.H.; et al. Anti-thymocyte globulin/G-CSF treatment preserves β cell function in patients with established type 1 diabetes. J. Clin. Investig. 2015, 125, 448–455. [Google Scholar] [CrossRef]
- Haller, M.J.; Schatz, D.A.; Skyler, J.S.; Krischer, J.P.; Bundy, B.N.; Miller, J.L.; Atkinson, M.A.; Becker, D.J.; Baidal, D.; DiMeglio, L.A.; et al. Low-Dose Anti-Thymocyte Globulin (ATG) Preserves β-Cell Function and Improves HbA1c in New-Onset Type 1 Diabetes. Diabetes Care 2018, 41, 1917–1925. [Google Scholar] [CrossRef] [PubMed]
- Michels, A.W.; Ostrov, D.A.; Zhang, L.; Nakayama, M.; Fuse, M.; McDaniel, K.; Roep, B.O.; Gottlieb, P.A.; Atkinson, M.A.; Eisenbarth, G.S. Structure-based selection of small molecules to alter allele-specific MHC class II antigen presentation. J. Immunol. 2011, 187, 5921–5930. [Google Scholar] [CrossRef] [PubMed]
- Ostrov, D.A.; Alkanani, A.; McDaniel, K.A.; Case, S.; Baschal, E.E.; Pyle, L.; Ellis, S.; Pöllinger, B.; Seidl, K.J.; Shah, V.N.; et al. Methyldopa blocks MHC class II binding to disease-specific antigens in autoimmune diabetes. J. Clin. Investig. 2018, 128, 1888–1902. [Google Scholar] [CrossRef] [PubMed]
- Ostrov, D.A.; Gottlieb, P.A.; Michels, A.W. Rationally designed small molecules to prevent type 1 diabetes. Curr. Opin. Endocrinol. Diabetes. Obes. 2019, 26, 90–95. [Google Scholar] [CrossRef]
- Diabetes Prevention Trial—Type 1 Diabetes Study Group. Effects of insulin in relatives of patients with type 1 diabetes mellitus. N. Engl. J. Med. 2002, 346, 1685–1691. [Google Scholar] [CrossRef]
- Näntö-Salonen, K.; Kupila, A.; Simell, S.; Siljander, H.; Salonsaari, T.; Hekkala, A.; Korhonen, S.; Erkkola, R.; Sipilä, J.I.; Haavisto, L.; et al. Nasal insulin to prevent type 1 diabetes in children with HLA genotypes and autoantibodies conferring increased risk of disease: A double-blind, randomised controlled trial. Lancet 2008, 372, 1746–1755. [Google Scholar] [CrossRef]
- Writing Committee for the Type 1 Diabetes TrialNet Oral Insulin Study Group; Krischer, J.P.; Schatz, D.A.; Bundy, B.; Skyler, J.S.; Greenbaum, C.J. Effect of Oral Insulin on Prevention of Diabetes in Relatives of Patients With Type 1 Diabetes: A Randomized Clinical Trial. JAMA 2017, 318, 1891–1902. [Google Scholar] [CrossRef]
- Bonifacio, E.; Ziegler, A.-G.; Klingensmith, G.; Schober, E.; Bingley, P.J.; Rottenkolber, M.; Theil, A.; Eugster, A.; Puff, R.; Peplow, C.; et al. Effects of high-dose oral insulin on immune responses in children at high risk for type 1 diabetes: The Pre-POINT randomized clinical trial. JAMA 2015, 313, 1541–1549. [Google Scholar] [CrossRef]
- Assfalg, R.; Knoop, J.; Hoffman, K.L.; Pfirrmann, M.; Zapardiel-Gonzalo, J.M.; Hofelich, A.; Eugster, A.; Weigelt, M.; Matzke, C.; Reinhardt, J.; et al. Oral insulin immunotherapy in children at risk for type 1 diabetes in a randomised controlled trial. Diabetologia 2021, 64, 1079–1092. [Google Scholar] [CrossRef]
- Ludvigsson, J.; Faresjö, M.; Hjorth, M.; Axelsson, S.; Chéramy, M.; Pihl, M.; Vaarala, O.; Forsander, G.; Ivarsson, S.; Johansson, C.; et al. GAD treatment and insulin secretion in recent-onset type 1 diabetes. N. Engl. J. Med. 2008, 359, 1909–1920. [Google Scholar] [CrossRef]
- Wherrett, D.K.; Bundy, B.; Becker, D.J.; DiMeglio, L.A.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Marks, J.B.; et al. Antigen-based therapy with glutamic acid decarboxylase (GAD) vaccine in patients with recent-onset type 1 diabetes: A randomised double-blind trial. Lancet 2011, 378, 319–327. [Google Scholar] [CrossRef]
- Hannelius, U.; Beam, C.A.; Ludvigsson, J. Efficacy of GAD-alum immunotherapy associated with HLA-DR3-DQ2 in recently diagnosed type 1 diabetes. Diabetologia 2020, 63, 2177–2181. [Google Scholar] [CrossRef] [PubMed]
- Ludvigsson, J.; Sumnik, Z.; Pelikanova, T.; Nattero Chavez, L.; Lundberg, E.; Rica, I.; Martínez-Brocca, M.A.; Ruiz de Adana, M.; Wahlberg, J.; Katsarou, A.; et al. Intralymphatic Glutamic Acid Decarboxylase With Vitamin D Supplementation in Recent-Onset Type 1 Diabetes: A Double-Blind, Randomized, Placebo-Controlled Phase IIb Trial. Diabetes Care 2021, 44, 1604–1612. [Google Scholar] [CrossRef] [PubMed]
- Alhadj Ali, M.; Liu, Y.-F.; Arif, S.; Tatovic, D.; Shariff, H.; Gibson, V.B.; Yusuf, N.; Baptista, R.; Eichmann, M.; Petrov, N.; et al. Metabolic and immune effects of immunotherapy with proinsulin peptide in human new-onset type 1 diabetes. Sci. Transl. Med. 2017, 9, eaaf7779. [Google Scholar] [CrossRef]
- Liu, Y.-F.; Powrie, J.; Arif, S.; Yang, J.H.M.; Williams, E.; Khatri, L.; Joshi, M.; Lhuillier, L.; Fountoulakis, N.; Smith, E.; et al. Immune and Metabolic Effects of Antigen-Specific Immunotherapy Using Multiple β-Cell Peptides in Type 1 Diabetes. Diabetes 2022. [Google Scholar] [CrossRef] [PubMed]
- Campbell-Thompson, M.; Wasserfall, C.; Kaddis, J.; Albanese-O’Neill, A.; Staeva, T.; Nierras, C.; Moraski, J.; Rowe, P.; Gianani, R.; Eisenbarth, G.; et al. Network for Pancreatic Organ Donors with Diabetes (nPOD): Developing a tissue biobank for type 1 diabetes. Diabetes. Metab. Res. Rev. 2012, 28, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Kaestner, K.H.; Powers, A.C.; Naji, A.; HPAP Consortium; Atkinson, M.A. NIH Initiative to Improve Understanding of the Pancreas, Islet, and Autoimmunity in Type 1 Diabetes: The Human Pancreas Analysis Program (HPAP). Diabetes 2019, 68, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Hagopian, W.A.; Lernmark, A.; Rewers, M.J.; Simell, O.G.; She, J.-X.; Ziegler, A.G.; Krischer, J.P.; Akolkar, B. TEDDY—The Environmental Determinants of Diabetes in the Young: An observational clinical trial. Ann. N. Y. Acad. Sci. 2006, 1079, 320–326. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Method | Autoantigen Target(s) [HLA(s)] | Primary Outcome | References |
|---|---|---|---|
| Tetramer staining | IGRP [DR3, DR4] | IL-10 | [39] |
| Cytokine ELISPOT | Insulin B:9–23; mimotopes [DQ8] | IL-10 | [40] |
| HIPs [DQ8] | IL-10 | [42,43] | |
| Proinsulin, IA-2 [DR4] | IL-10 | [41] | |
| Cloning, cytokine ELISA; flow cytometry | Proinsulin, IA-2 | IL-10; | |
| [DR3, DR4] | granzyme A, B; | [48] | |
| Treg suppression | |||
| Cytokine beads; flow cytometry | GAD65, PPI, IGRP | IL-10; IP-10 | [49] |
| Cloning; cytokine ELISA; flow cytometry; TCR-peptide-MHC crystallization | Proinsulin [DR4] | IL-10, granzyme B; TCR-peptide-MHCdocking | [51] |
| Tetramer staining, cloning, flow cytometry | GAD, IGRP, PPI, ZnT8 [DR3, DR4, DR15, DQ6] | IL-10; Treg suppression | [53] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitchell, A.M.; Michels, A.W. Self-Antigens Targeted by Regulatory T Cells in Type 1 Diabetes. Int. J. Mol. Sci. 2022, 23, 3155. https://doi.org/10.3390/ijms23063155
Mitchell AM, Michels AW. Self-Antigens Targeted by Regulatory T Cells in Type 1 Diabetes. International Journal of Molecular Sciences. 2022; 23(6):3155. https://doi.org/10.3390/ijms23063155
Chicago/Turabian StyleMitchell, Angela M., and Aaron W. Michels. 2022. "Self-Antigens Targeted by Regulatory T Cells in Type 1 Diabetes" International Journal of Molecular Sciences 23, no. 6: 3155. https://doi.org/10.3390/ijms23063155
APA StyleMitchell, A. M., & Michels, A. W. (2022). Self-Antigens Targeted by Regulatory T Cells in Type 1 Diabetes. International Journal of Molecular Sciences, 23(6), 3155. https://doi.org/10.3390/ijms23063155