
Multicellular Modelling of Difficult-to-Treat Gastrointestinal Cancers: Current Possibilities and Challenges

 , ,
, ,  and
and
Abstract
1. Introduction
2. Difficult-to-Treat Gastrointestinal Cancers
2.1. Gastric Cancer
2.2. Pancreatic Ductal Adenocarcinoma
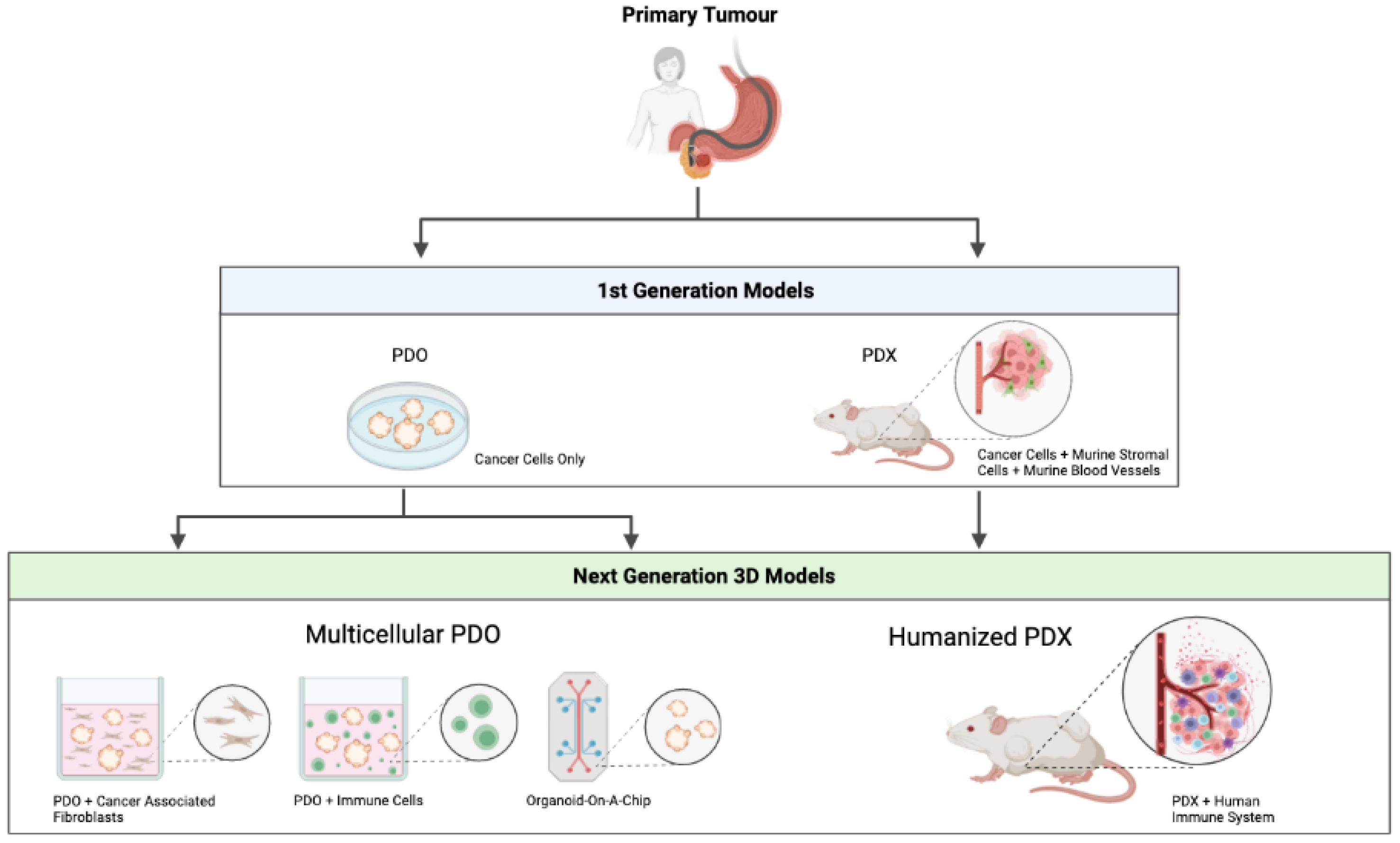
3. First Generation PDO/PDX Modelling
3.1. Patient-Derived Organoids (PDOs)
3.1.1. Gastric Cancer PDOs
3.1.2. PDAC PDOs
3.2. Patient-Derived Xenografts (PDXs)
3.2.1. Gastric Cancer PDXs
3.2.2. PDAC PDXs
4. Next Generation PDO/PDX Modelling
4.1. Multicellular PDO Models
4.1.1. PDO/Immune Cell Co-Cultures
4.1.2. PDO/CAF Co-Cultures
4.1.3. Organoid-on-a-Chip
4.2. Humanised PDX Models
4.3. MiniPDX Models
5. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Barsouk, A. Epidemiology of gastric cancer: Global trends, risk factors and prevention. Prz. Gastroenterol. 2019, 14, 26–38. [Google Scholar] [CrossRef]
- Liu, X.; Meltzer, S.J. Gastric Cancer in the Era of Precision Medicine. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Mashino, K.; Sadanaga, N.; Yamaguchi, H.; Tanaka, F.; Ohta, M.; Shibuta, K.; Inoue, H.; Mori, M. Expression of chemokine receptor CCR7 is associated with lymph node metastasis of gastric carcinoma. Cancer Res. 2002, 62, 2937–2941. [Google Scholar]
- Riihimaki, M.; Hemminki, A.; Sundquist, K.; Sundquist, J.; Hemminki, K. Metastatic spread in patients with gastric cancer. Oncotarget 2016, 7, 52307–52316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Deng, T.; Liu, R.; Bai, M.; Zhou, L.; Wang, X.; Li, S.; Wang, X.; Yang, H.; Li, J.; et al. Exosome-delivered EGFR regulates liver microenvironment to promote gastric cancer liver metastasis. Nat. Commun. 2017, 8, 15016. [Google Scholar] [CrossRef]
- Gullo, I.; Carneiro, F.; Oliveira, C.; Almeida, G.M. Heterogeneity in Gastric Cancer: From Pure Morphology to Molecular Classifications. Pathobiology 2018, 85, 50–63. [Google Scholar] [CrossRef]
- Ho, S.W.T.; Tan, P. Dissection of gastric cancer heterogeneity for precision oncology. Cancer Sci. 2019, 110, 3405–3414. [Google Scholar] [CrossRef]
- Laurén, P. The Two Histological Main Types of Gastric Carcinoma: Diffuse and so-called Intestinal-Type Carcinoma. An Attempt at a Histo-Clinical Classification. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef] [PubMed]
- WHO Classification of Tumours Editorial Board. Digestive System Tumours, 5th ed.; World Health Organization: Geneva, Switzerland, 2019; Volume 1.
- Bass, A.J.; Thorsson, V.; Shmulevich, I.; Reynolds, S.M.; Miller, M.; Bernard, B.; Hinoue, T.; Laird, P.W.; Curtis, C.; The Cancer Genome Atlas Research Network; et al. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. Available online: https://www.nature.com/articles/nature13480#supplementary-information (accessed on 3 February 2022).
- Ito, T.; Matoba, R.; Maekawa, H.; Sakurada, M.; Kushida, T.; Orita, H.; Wada, R.; Sato, K. Detection of gene mutations in gastric cancer tissues using a commercial sequencing panel. Mol. Clin. Oncol. 2019, 11, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Jang, B.-G.; Kim, W.H. Molecular Pathology of Gastric Carcinoma. Pathobiology 2011, 78, 302–310. [Google Scholar] [CrossRef]
- Nemtsova, M.V.; Kalinkin, A.I.; Kuznetsova, E.B.; Bure, I.V.; Alekseeva, E.A.; Bykov, I.I.; Khorobrykh, T.V.; Mikhaylenko, D.S.; Tanas, A.S.; Kutsev, S.I.; et al. Clinical relevance of somatic mutations in main driver genes detected in gastric cancer patients by next-generation DNA sequencing. Sci. Rep. 2020, 10, 504. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Jing, C.; Chang, X.; Ding, D.; Han, T.; Yang, J.; Lu, Z.; Hu, X.; Liu, Z.; Wang, J.; et al. Mutational landscape of gastric cancer and clinical application of genomic profiling based on target next-generation sequencing. J. Transl. Med. 2019, 17, 189. [Google Scholar] [CrossRef] [PubMed]
- Kemi, N.; Eskuri, M.; Kauppila, J.H. Tumour-stroma ratio and 5-year mortality in gastric adenocarcinoma: A systematic review and meta-analysis. Sci. Rep. 2019, 9, 16018. [Google Scholar] [CrossRef]
- Smyth, E.C.; Verheij, M.; Allum, W.; Cunningham, D.; Cervantes, A.; Arnold, D.; Committee, E.G. Gastric cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, v38–v49. [Google Scholar] [CrossRef] [PubMed]
- Coburn, N.; Cosby, R.; Klein, L.; Knight, G.; Malthaner, R.; Mamazza, J.; Mercer, C.D.; Ringash, J. Staging and surgical approaches in gastric cancer: A systematic review. Cancer Treat. Rev. 2018, 63, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Gravalos, C.; Jimeno, A. HER2 in gastric cancer: A new prognostic factor and a novel therapeutic target. Ann. Oncol. 2008, 19, 1523–1529. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Bang, Y.-J.; Feng-Yi, F.; Xu, J.M.; Lee, K.-W.; Jiao, S.-C.; Chong, J.L.; Lopez-Sanchez, R.I.; Price, T.; Gladkov, O.; et al. HER2 screening data from ToGA: Targeting HER2 in gastric and gastroesophageal junction cancer. Gastric Cancer 2015, 18, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Di Bartolomeo, M.; Niger, M.; Tirino, G.; Petrillo, A.; Berenato, R.; Laterza, M.M.; Pietrantonio, F.; Morano, F.; Antista, M.; Lonardi, S.; et al. Ramucirumab as Second-Line Therapy in Metastatic Gastric Cancer: Real-World Data from the RAMoss Study. Target. Oncol. 2018, 13, 227–234. [Google Scholar] [CrossRef]
- Siebenhuner, A.R.; De Dosso, S.; Helbling, D.; Astaras, C.; Szturz, P.; Moosmann, P.; Pederiva, S.; Winder, T.; Von Burg, P.; Borner, M. Advanced Gastric Cancer: Current Treatment Landscape and a Future Outlook for Sequential and Personalized Guide: Swiss Expert Statement Article. Oncol. Res. Treat. 2021, 44, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Puleo, F.; Nicolle, R.; Blum, Y.; Cros, J.; Marisa, L.; Demetter, P.; Quertinmont, E.; Svrcek, M.; Elarouci, N.; Iovanna, J.L.; et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018, 155, 1999–2013.e1993. [Google Scholar] [CrossRef]
- Chan-Seng-Yue, M.; Kim, J.C.; Wilson, G.W.; Ng, K.; Flores-Figueroa, E.; O’Kane, G.M.; Connor, A.A.; Denroche, R.E.; Grant, R.C.; McLeod, J.; et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 2020, 52, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, R.D.; Speers, C.; McGahan, C.E.; Renouf, D.J.; Schaeffer, D.F.; Kennecke, H.F. Prognostic factors and sites of metastasis in unresectable locally advanced pancreatic cancer. Cancer Med. 2015, 4, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Masiak-Segit, W.; Rawicz-Pruszyński, K.; Skórzewska, M.; Polkowski, W.P. Surgical treatment of pancreatic cancer. Pol. Prz. Chir. 2018, 90, 45–53. [Google Scholar] [CrossRef]
- Aung, K.L.; Fischer, S.E.; Denroche, R.E.; Jang, G.-H.; Dodd, A.; Creighton, S.; Southwood, B.; Liang, S.-B.; Chadwick, D.; Zhang, A.; et al. Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clin. Cancer Res. 2018, 24, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Ibarrola-Villava, M.; Cervantes, A.; Bardelli, A. Preclinical models for precision oncology. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 239–246. [Google Scholar] [CrossRef] [PubMed]
- McMillin, D.W.; Negri, J.M.; Mitsiades, C.S. The role of tumour–stromal interactions in modifying drug response: Challenges and opportunities. Nat. Rev. Drug Discov. 2013, 12, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.-Z.; Han, R.-R.; Qiu, G.-Z.; Ju, X.-C.; Lou, G.; Jin, W.-L. Organoids: An intermediate modeling platform in precision oncology. Cancer Lett. 2018, 414, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Barker, N.; Huch, M.; Kujala, P.; Van De Wetering, M.; Snippert, H.J.; Van Es, J.H.; Sato, T.; Stange, D.E.; Begthel, H.; van den Born, M.; et al. Lgr5+ve Stem Cells Drive Self-Renewal in the Stomach and Build Long-Lived Gastric Units In Vitro. Cell Stem Cell 2010, 6, 25–36. [Google Scholar] [CrossRef]
- Broutier, L.; Andersson-Rolf, A.; Hindley, C.J.; Boj, S.F.; Clevers, H.; Koo, B.-K.; Huch, M. Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nat. Protoc. 2016, 11, 1724–1743. [Google Scholar] [CrossRef] [PubMed]
- Nuciforo, S.; Fofana, I.; Matter, M.S.; Blumer, T.; Calabrese, D.; Boldanova, T.; Piscuoglio, S.; Wieland, S.; Ringnalda, F.; Schwank, G.; et al. Organoid Models of Human Liver Cancers Derived from Tumor Needle Biopsies. Cell Rep. 2018, 24, 1363–1376. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Lin, M.; Rao, M.; Thompson, H.; Hirai, K.; Choi, M.; Georgakis, G.V.; Sasson, A.R.; Bucobo, J.C.; Tzimas, D.; et al. Development of Patient-Derived Gastric Cancer Organoids from Endoscopic Biopsies and Surgical Tissues. Ann. Surg. Oncol. 2018, 25, 2767–2775. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Koo, B.-K. Modeling mouse and human development using organoid cultures. Development 2015, 142, 3113–3125. [Google Scholar] [CrossRef]
- Miyabayashi, K.; Baker, L.A.; Deschenes, A.; Traub, B.; Caligiuri, G.; Plenker, D.; Alagesan, B.; Belleau, P.; Li, S.; Kendall, J.; et al. Intraductal Transplantation Models of Human Pancreatic Ductal Adenocarcinoma Reveal Progressive Transition of Molecular Subtypes. Cancer Discov. 2020, 10, 1566–1589. [Google Scholar] [CrossRef]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Bartfeld, S.; Bayram, T.; van de Wetering, M.; Huch, M.; Begthel, H.; Kujala, P.; Vries, R.; Peters, P.J.; Clevers, H. In Vitro Expansion of Human Gastric Epithelial Stem Cells and Their Responses to Bacterial Infection. Gastroenterology 2015, 148, 126–136.e126. [Google Scholar] [CrossRef] [PubMed]
- Seidlitz, T.; Merker, S.R.; Rothe, A.; Zakrzewski, F.; von Neubeck, C.; Grutzmann, K.; Sommer, U.; Schweitzer, C.; Scholch, S.; Uhlemann, H.; et al. Human gastric cancer modelling using organoids. Gut 2019, 68, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.H.N.; Siu, H.C.; Law, S.; Ho, S.L.; Yue, S.S.K.; Tsui, W.Y.; Chan, D.; Chan, A.S.; Ma, S.; Lam, K.O.; et al. A Comprehensive Human Gastric Cancer Organoid Biobank Captures Tumor Subtype Heterogeneity and Enables Therapeutic Screening. Cell Stem Cell 2018, 23, 882–897.e811. [Google Scholar] [CrossRef] [PubMed]
- Steele, N.G.; Chakrabarti, J.; Wang, J.; Biesiada, J.; Holokai, L.; Chang, J.; Nowacki, L.M.; Hawkins, J.; Mahe, M.; Sundaram, N.; et al. An Organoid-Based Preclinical Model of Human Gastric Cancer. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 161–184. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Calin, G.A.; Croce, C.M. MicroRNAs in Cancer. Annu. Rev. Med. 2009, 60, 167–179. [Google Scholar] [CrossRef]
- Sun, G.-L.; Li, Z.; Wang, W.-Z.; Chen, Z.; Zhang, L.; Li, Q.; Wei, S.; Li, B.-W.; Xu, J.-H.; Chen, L.; et al. miR-324-3p promotes gastric cancer development by activating Smad4-mediated Wnt/beta-catenin signaling pathway. J. Gastroenterol. 2018, 53, 725–739. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Li, Z.; Wang, W.; Xia, Y.; He, Z.; Li, B.; Wang, S.; Huang, X.; Sun, G.; Xu, J.; et al. MIR-1265 regulates cellular proliferation and apoptosis by targeting calcium binding protein 39 in gastric cancer and, thereby, impairing oncogenic autophagy. Cancer Lett. 2019, 449, 226–236. [Google Scholar] [CrossRef]
- Wu, W.; Yang, Z.; Xiao, X.; An, T.; Li, B.; Ouyang, J.; Li, H.; Wang, C.; Zhang, Y.; Zhang, H.; et al. A thioredoxin reductase inhibitor ethaselen induces growth inhibition and apoptosis in gastric cancer. J. Cancer 2020, 11, 3013–3019. [Google Scholar] [CrossRef]
- Haag, G.M. OPPOSITE: Outcome Prediction of Systemic Treatment in Esophagogastric Carcinoma. Available online: https://clinicaltrials.gov/ct2/show/NCT03429816 (accessed on 17 January 2022).
- Boj, S.F.; Hwang, C.-I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid Models of Human and Mouse Ductal Pancreatic Cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Driehuis, E.; van Hoeck, A.; Moore, K.; Kolders, S.; Francies, H.E.; Gulersonmez, M.C.; Stigter, E.C.A.; Burgering, B.; Geurts, V.; Gracanin, A.; et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. USA 2019, 116, 26580–26590. [Google Scholar] [CrossRef] [PubMed]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschenes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Bockorny, B.; Paul, I.; Akshinthala, D.; Frappart, P.-O.; Gandarilla, O.; Bose, A.; Sanchez-Gonzalez, V.; Rouse, E.E.; Lehoux, S.D.; et al. PDX-derived organoids model in vivo drug response and secrete biomarkers. JCI Insight 2020, 5, e135544. [Google Scholar] [CrossRef] [PubMed]
- Juiz, N.; Elkaoutari, A.; Bigonnet, M.; Gayet, O.; Roques, J.; Nicolle, R.; Iovanna, J.; Dusetti, N. Basal-like and classical cells coexist in pancreatic cancer revealed by single-cell analysis on biopsy-derived pancreatic cancer organoids from the classical subtype. FASEB J. 2020, 34, 12214–12228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, L.; Fan, S.; Zhang, M.; Fu, H.; Liu, Y.; Yin, X.; Chen, H.; Xie, L.; Zhang, J.; et al. Patient-Derived Gastric Carcinoma Xenograft Mouse Models Faithfully Represent Human Tumor Molecular Diversity. PLoS ONE 2015, 10, e0134493. [Google Scholar] [CrossRef]
- Hernandez, M.C.; Bergquist, J.R.; Leiting, J.L.; Ivanics, T.; Yang, L.; Smoot, R.L.; Nagorney, D.M.; Truty, M.J. Patient-Derived Xenografts Can Be Reliably Generated from Patient Clinical Biopsy Specimens. J. Gastrointest. Surg. 2019, 23, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.M. Patient-derived orthotopic xenografts: Better mimic of metastasis than subcutaneous xenografts. Nat. Rev. Cancer 2015, 15, 451–452. [Google Scholar] [CrossRef] [PubMed]
- Foygel, K.; Wang, H.; Machtaler, S.; Lutz, A.M.; Chen, R.; Pysz, M.; Lowe, A.W.; Tian, L.; Carrigan, T.; Brentnall, T.A.; et al. Detection of Pancreatic Ductal Adenocarcinoma in Mice by Ultrasound Imaging of Thymocyte Differentiation Antigen 1. Gastroenterology 2013, 145, 885–894.e883. [Google Scholar] [CrossRef]
- Choi, Y.Y.; Lee, J.E.; Kim, H.; Sim, M.H.; Kim, K.-K.; Lee, G.; Kim, H.I.; An, J.Y.; Hyung, W.J.; Kim, C.-B.; et al. Establishment and characterisation of patient-derived xenografts as paraclinical models for gastric cancer. Sci. Rep. 2016, 6, 22172. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lu, J.; Tang, J.; Chen, S.; He, K.; Jiang, X.; Jiang, W.; Teng, L. Establishment of patient-derived gastric cancer xenografts: A useful tool for preclinical evaluation of targeted therapies involving alterations in HER-2, MET and FGFR2 signaling pathways. BMC Cancer 2017, 17, 191. [Google Scholar] [CrossRef] [PubMed]
- Blumer, T.; Fofana, I.; Matter, M.S.; Wang, X.; Montazeri, H.; Calabrese, D.; Coto-Llerena, M.; Boldanova, T.; Nuciforo, S.; Kancherla, V.; et al. Hepatocellular Carcinoma Xenografts Established from Needle Biopsies Preserve the Characteristics of the Originating Tumors. Hepatol. Commun. 2019, 3, 971–986. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Cao, S.; Mashl, R.J.; Mo, C.-K.; Zaccaria, S.; Wendl, M.C.; Davies, S.R.; Bailey, M.H.; Primeau, T.M.; Hoog, J.; et al. Comprehensive characterization of 536 patient-derived xenograft models prioritizes candidates for targeted treatment. Nat. Commun. 2021, 12, 5086. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.-Y. Patient-derived xenografts as compatible models for precision oncology. Lab. Anim. Res. 2020, 36, 14. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Lin, W.; Huang, Y.; Chen, X.; Wang, H.; Teng, L. The Essential Factors of Establishing Patient-derived Tumor Model. J. Cancer 2021, 12, 28–37. [Google Scholar] [CrossRef]
- Morton, J.J.; Bird, G.H.; Keysar, S.B.; Astling, D.P.; Lyons, T.R.; Anderson, R.T.; Glogowska, M.J.; Estes, P.S.; Eagles, J.R.; Le, P.N.; et al. XactMice: Humanizing mouse bone marrow enables microenvironment reconstitution in a patient-derived xenograft model of head and neck cancer. Oncogene 2016, 35, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, T.; Yanagihara, K.; Iino, Y.; Komatsu, T.; Ochiai, A.; Sekine, S.; Taniguchi, H.; Katai, H.; Kinoshita, T.; Ohtsu, A. Establishment of Novel Gastric Cancer Patient-Derived Xenografts and Cell Lines: Pathological Comparison between Primary Tumor, Patient-Derived, and Cell-Line Derived Xenografts. Cells 2019, 8, 585. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tian, T.; Li, Z.; Tang, Z.; Wang, L.; Wu, J.; Li, Y.; Dong, B.; Li, Y.; Li, N.; et al. Establishment and characterization of patient-derived tumor xenograft using gastroscopic biopsies in gastric cancer. Sci. Rep. 2015, 5, 8542. [Google Scholar] [CrossRef]
- Chen, Z.; Huang, W.; Tian, T.; Zang, W.; Wang, J.; Liu, Z.; Li, Z.; Lai, Y.; Jiang, Z.; Gao, J.; et al. Characterization and validation of potential therapeutic targets based on the molecular signature of patient-derived xenografts in gastric cancer. J. Hematol. Oncol. 2018, 11, 20. [Google Scholar] [CrossRef]
- Wang, X.; Fu, R.; Hu, Y.; Du, H.; Li, S.; Li, Z.; Liu, Y.; Li, Q.; Zhang, L.; Ji, J. EGFR gene status predicts response and survival benefit in a preclinical gastric cancer trial treating patient-derived xenografts with cetuximab. Oncol. Rep. 2017, 38, 2387–2393. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Okines, A.F.; Cunningham, D. Trastuzumab in gastric cancer. Eur. J. Cancer 2010, 46, 1949–1959. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.J.; Tong, P.J.; Zhu, M.Y. The combinational therapy of trastuzumab and cetuximab inhibits tumor growth in a patient-derived tumor xenograft model of gastric cancer. Clin. Transl. Oncol. 2016, 18, 507–514. [Google Scholar] [CrossRef]
- Rubio-Viqueira, B.; Jimeno, A.; Cusatis, G.; Zhang, X.; Iacobuzio-Donahue, C.; Karikari, C.; Shi, C.; Danenberg, K.; Danenberg, P.V.; Kuramochi, H.; et al. An In vivo Platform for Translational Drug Development in Pancreatic Cancer. Clin. Cancer Res. 2006, 12, 4652–4661. [Google Scholar] [CrossRef] [PubMed]
- Delitto, D.; Pham, K.; Vlada, A.C.; Sarosi, G.A.; Thomas, R.M.; Behrns, K.E.; Liu, C.; Hughes, S.J.; Wallet, S.M.; Trevino, J.G. Patient-Derived Xenograft Models for Pancreatic Adenocarcinoma Demonstrate Retention of Tumor Morphology through Incorporation of Murine Stromal Elements. Am. J. Pathol. 2015, 185, 1297–1303. [Google Scholar] [CrossRef]
- Jung, J.; Lee, C.H.; Seol, H.S.; Choi, Y.S.; Kim, E.; Lee, E.J.; Rhee, J.-K.; Singh, S.R.; Jun, E.S.; Han, B.; et al. Generation and molecular characterization of pancreatic cancer patient-derived xenografts reveals their heterologous nature. Oncotarget 2016, 7, 62533–62546. [Google Scholar] [CrossRef]
- Garcia, P.L.; Council, L.N.; Christein, J.D.; Arnoletti, J.P.; Heslin, M.J.; Gamblin, T.L.; Richardson, J.H.; Bjornsti, M.-A.; Yoon, K.J. Development and Histopathological Characterization of Tumorgraft Models of Pancreatic Ductal Adenocarcinoma. PLoS ONE 2013, 8, e78183. [Google Scholar] [CrossRef] [PubMed]
- Torphy, R.J.; Tignanelli, C.J.; Kamande, J.W.; Moffitt, R.A.; Loeza, S.G.H.; Soper, S.A.; Yeh, J.J. Circulating Tumor Cells as a Biomarker of Response to Treatment in Patient-Derived Xenograft Mouse Models of Pancreatic Adenocarcinoma. PLoS ONE 2014, 9, e89474. [Google Scholar] [CrossRef]
- Garrido-Laguna, I.; Uson, M.; RajeshKumar, N.V.; Tan, A.C.; De Oliveira, E.; Karikari, C.; Villaroel, M.C.; Salomon, A.; Taylor, G.; Sharma, R.; et al. Tumor Engraftment in Nude Mice and Enrichment in Stroma- Related Gene Pathways Predict Poor Survival and Resistance to Gemcitabine in Patients with Pancreatic Cancer. Clin. Cancer Res. 2011, 17, 5793–5800. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Rubio-Viqueira, B.; RajeshKumar, N.; Chan, A.; Solomon, A.; Hidalgo, M. A Fine-Needle Aspirate–Based Vulnerability Assay Identifies Polo-Like Kinase 1 as a Mediator of Gemcitabine Resistance in Pancreatic Cancer. Mol. Cancer Ther. 2010, 9, 311–318. [Google Scholar] [CrossRef]
- Miller, A.L.; Fehling, S.C.; Garcia, P.L.; Gamblin, T.L.; Council, L.N.; van Waardenburg, R.C.; Yang, E.S.; Bradner, J.E.; Yoon, K.J. The BET inhibitor JQ1 attenuates double-strand break repair and sensitizes models of pancreatic ductal adenocarcinoma to PARP inhibitors. eBioMedicine 2019, 44, 419–430. [Google Scholar] [CrossRef]
- Nicolle, R.; Blum, Y.; Marisa, L.; Loncle, C.; Gayet, O.; Moutardier, V.; Turrini, O.; Giovannini, M.; Bian, B.; Bigonnet, M.; et al. Pancreatic Adenocarcinoma Therapeutic Targets Revealed by Tumor-Stroma Cross-Talk Analyses in Patient-Derived Xenografts. Cell Rep. 2017, 21, 2458–2470. [Google Scholar] [CrossRef] [PubMed]
- Nicolle, R.; Gayet, O.; Duconseil, P.; Vanbrugghe, C.; Roques, J.; Bigonnet, M.; Blum, Y.; Elarouci, N.; Armenoult, L.; Ayadi, M.; et al. A transcriptomic signature to predict adjuvant gemcitabine sensitivity in pancreatic adenocarcinoma. Ann. Oncol. 2021, 32, 250–260. [Google Scholar] [CrossRef]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; Van De Haar, J.; Fanchi, L.F.; Slagter, M.; Van Der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598.e12. [Google Scholar] [CrossRef]
- Nozaki, K.; Mochizuki, W.; Matsumoto, Y.; Matsumoto, T.; Fukuda, M.; Mizutani, T.; Watanabe, M.; Nakamura, T. Co-culture with intestinal epithelial organoids allows efficient expansion and motility analysis of intraepithelial lymphocytes. J. Gastroenterol. 2016, 51, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, J.; Holokai, L.; Syu, L.; Steele, N.; Chang, J.; Dlugosz, A.; Zavros, Y. Mouse-Derived Gastric Organoid and Immune Cell Co-culture for the Study of the Tumor Microenvironment. Methods Mol. Biol. 2018, 1817, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Holokai, L.; Chakrabarti, J.; Lundy, J.; Croagh, D.; Adhikary, P.; Richards, S.S.; Woodson, C.; Steele, N.; Kuester, R.; Scott, A.; et al. Murine- and Human-Derived Autologous Organoid/Immune Cell Co-Cultures as Pre-Clinical Models of Pancreatic Ductal Adenocarcinoma. Cancers 2020, 12, 3816. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Zhou, L.; Lu, J.; Wang, Y.; Liu, C.; You, L.; Guo, J. Stroma-Targeting Therapy in Pancreatic Cancer: One Coin with Two Sides? Front. Oncol. 2020, 10, 576399. [Google Scholar] [CrossRef]
- Bertaux-Skeirik, N.; Centeno, J.; Feng, R.; Schumacher, M.A.; Shivdasani, R.A.; Zavros, Y. Co-culture of Gastric Organoids and Immortalized Stomach Mesenchymal Cells. Methods Mol. Biol. 2016, 1422, 23–31. [Google Scholar] [CrossRef]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Tsai, S.; McOlash, L.; Palen, K.; Johnson, B.; Duris, C.; Yang, Q.; Dwinell, M.B.; Hunt, B.; Evans, D.B.; Gershan, J.; et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 2018, 18, 335. [Google Scholar] [CrossRef] [PubMed]
- Park, S.E.; Georgescu, A.; Huh, D. Organoids-on-a-chip. Science 2019, 364, 960–965. [Google Scholar] [CrossRef]
- Sidar, B.; Jenkins, B.R.; Huang, S.; Spence, J.R.; Walk, S.T.; Wilking, J.N. Long-term flow through human intestinal organoids with the gut organoid flow chip (GOFlowChip). Lab Chip 2019, 19, 3552–3562. [Google Scholar] [CrossRef] [PubMed]
- Cherne, M.D.; Sidar, B.; Sebrell, T.A.; Sanchez, H.S.; Heaton, K.; Kassama, F.J.; Roe, M.M.; Gentry, A.B.; Chang, C.B.; Walk, S.T.; et al. A Synthetic Hydrogel, VitroGel((R)) ORGANOID-3, Improves Immune Cell-Epithelial Interactions in a Tissue Chip Co-Culture Model of Human Gastric Organoids and Dendritic Cells. Front. Pharmacol. 2021, 12, 707891. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J.; et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 2005, 174, 6477–6489. [Google Scholar] [CrossRef] [PubMed]
- Joo, S.-Y.; Choi, B.-K.; Kang, M.J.; Jung, D.Y.; Park, K.S.; Park, J.B.; Choi, G.S.; Joh, J.-W.; Kwon, C.H.D.; Jung, G.O.; et al. Development of functional human immune system with the transplantations of human fetal liver/thymus tissues and expanded hematopoietic stem cells in RAG2−/−γ(c)−/− MICE. Transplant. Proc. 2009, 41, 1885–1890. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Choi, B.; Kim, S.Y.; Yang, J.-H.; Roh, C.R.; Lee, K.-Y.; Kim, S.J. Co-transplantation of fetal bone tissue facilitates the development and reconstitution in human B cells in humanized NOD/SCID/IL-2Rgammanull (NSG) mice. J. Clin. Immunol. 2011, 31, 699–709. [Google Scholar] [CrossRef]
- Morton, J.J.; Bird, G.; Refaeli, Y.; Jimeno, A. Humanized Mouse Xenograft Models: Narrowing the Tumor–Microenvironment Gap. Cancer Res. 2016, 76, 6153–6158. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.-X.; Wang, J.-Y.; Li, H.-Z.; Guo, H.-R.; Dunmall, L.S.C.; Zhang, Z.-X.; Cheng, Z.-G.; Gao, D.-L.; Dong, J.-Z.; Wang, Z.-D.; et al. Promising xenograft animal model recapitulating the features of human pancreatic cancer. World J. Gastroenterol. 2020, 26, 4802–4816. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Facciponte, J.; Jin, M.; Shen, Q.; Lin, Q. Humanized NOD-SCID IL2rg−/− mice as a preclinical model for cancer research and its potential use for individualized cancer therapies. Cancer Lett. 2014, 344, 13–19. [Google Scholar] [CrossRef]
- Rios-Doria, J.; Stevens, C.; Maddage, C.; Lasky, K.; Koblish, H.K. Characterization of human cancer xenografts in humanized mice. J. Immunother. Cancer 2020, 8, e000416. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Rodriguez, I.; Schalper, K.A.; ONate, C.; Azpilikueta, A.; Rodriguez-Ruiz, M.E.; Morales-Kastresana, A.; Labiano, S.; Perez-Gracia, J.L.; Martín-Algarra, S.; et al. Nivolumab and Urelumab Enhance Antitumor Activity of Human T Lymphocytes Engrafted in Rag2-/-IL2Rgammanull Immunodeficient Mice. Cancer Res. 2015, 75, 3466–3478. [Google Scholar] [CrossRef] [PubMed]
- Ames, E.; Canter, R.J.; Grossenbacher, S.K.; Mac, S.; Chen, M.; Smith, R.C.; Hagino, T.; Perez-Cunningham, J.; Sckisel, G.D.; Urayama, S.; et al. NK Cells Preferentially Target Tumor Cells with a Cancer Stem Cell Phenotype. J. Immunol. 2015, 195, 4010–4019. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, W.; Long, Y.; Liu, H.; Cheng, J.; Guo, L.; Li, R.; Meng, C.; Yu, S.; Zhao, Q.; et al. Characterization of drug responses of mini patient-derived xenografts in mice for predicting cancer patient clinical therapeutic response. Cancer Commun. 2018, 38, 60. [Google Scholar] [CrossRef]
- Ge, Y.; Zhang, X.; Liang, W.; Tang, C.; Gu, D.; Shi, J.; Wei, X. OncoVee-MiniPDX-Guided Anticancer Treatment for Gastric Cancer Patients with Synchronous Liver Metastases: A Retrospective Cohort Analysis. Front. Oncol. 2021, 11, 757383. [Google Scholar] [CrossRef]
- Zhu, X.; Zhu, Y.; Chen, N.; Tang, C.; Shi, J. The drugs screened by OncoVeeTM-Mini-PDX have significantly benefited the patient with HER2-positive advanced gastric cancer. J. Oncol. Pharm. Pract. 2022, in press. [CrossRef] [PubMed]
- Nollmann, F.I.; Ruess, D.A. Targeting Mutant KRAS in Pancreatic Cancer: Futile or Promising? Biomedicines 2020, 8, 281. [Google Scholar] [CrossRef] [PubMed]
- Grossman, J.E.; Muthuswamy, L.; Huang, L.; Akshinthala, D.; Perea, S.; Gonzalez, R.S.; Tsai, L.L.; Cohen, J.; Bockorny, B.; Bullock, A.J.; et al. Organoid Sensitivity Correlates with Therapeutic Response in Patients with Pancreatic Cancer. Clin. Cancer Res. 2022, 28, 708–718. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Features | PDO | PDX | Multicellular PDO | Humanised PDX |
|---|---|---|---|---|
| Patient tumour recapitulation | + | + | ++(+) * | ++ |
| Presence of TME | − | + | −/+ | ++ |
| Model stability | ++ | ++ | ? | ? |
| Establishment time | ++ | − | ++ | − |
| Scalability | ++ | + | ? | − |
| Costs | low | high | low | very high |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hakuno, S.K.; Michiels, E.; Kuhlemaijer, E.B.; Rooman, I.; Hawinkels, L.J.A.C.; Slingerland, M. Multicellular Modelling of Difficult-to-Treat Gastrointestinal Cancers: Current Possibilities and Challenges. Int. J. Mol. Sci. 2022, 23, 3147. https://doi.org/10.3390/ijms23063147
Hakuno SK, Michiels E, Kuhlemaijer EB, Rooman I, Hawinkels LJAC, Slingerland M. Multicellular Modelling of Difficult-to-Treat Gastrointestinal Cancers: Current Possibilities and Challenges. International Journal of Molecular Sciences. 2022; 23(6):3147. https://doi.org/10.3390/ijms23063147
Chicago/Turabian StyleHakuno, Sarah K., Ellis Michiels, Eleonore B. Kuhlemaijer, Ilse Rooman, Lukas J. A. C. Hawinkels, and Marije Slingerland. 2022. "Multicellular Modelling of Difficult-to-Treat Gastrointestinal Cancers: Current Possibilities and Challenges" International Journal of Molecular Sciences 23, no. 6: 3147. https://doi.org/10.3390/ijms23063147
APA StyleHakuno, S. K., Michiels, E., Kuhlemaijer, E. B., Rooman, I., Hawinkels, L. J. A. C., & Slingerland, M. (2022). Multicellular Modelling of Difficult-to-Treat Gastrointestinal Cancers: Current Possibilities and Challenges. International Journal of Molecular Sciences, 23(6), 3147. https://doi.org/10.3390/ijms23063147