
Therapeutic Effect of Melittin–dKLA Targeting Tumor-Associated Macrophages in Melanoma

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
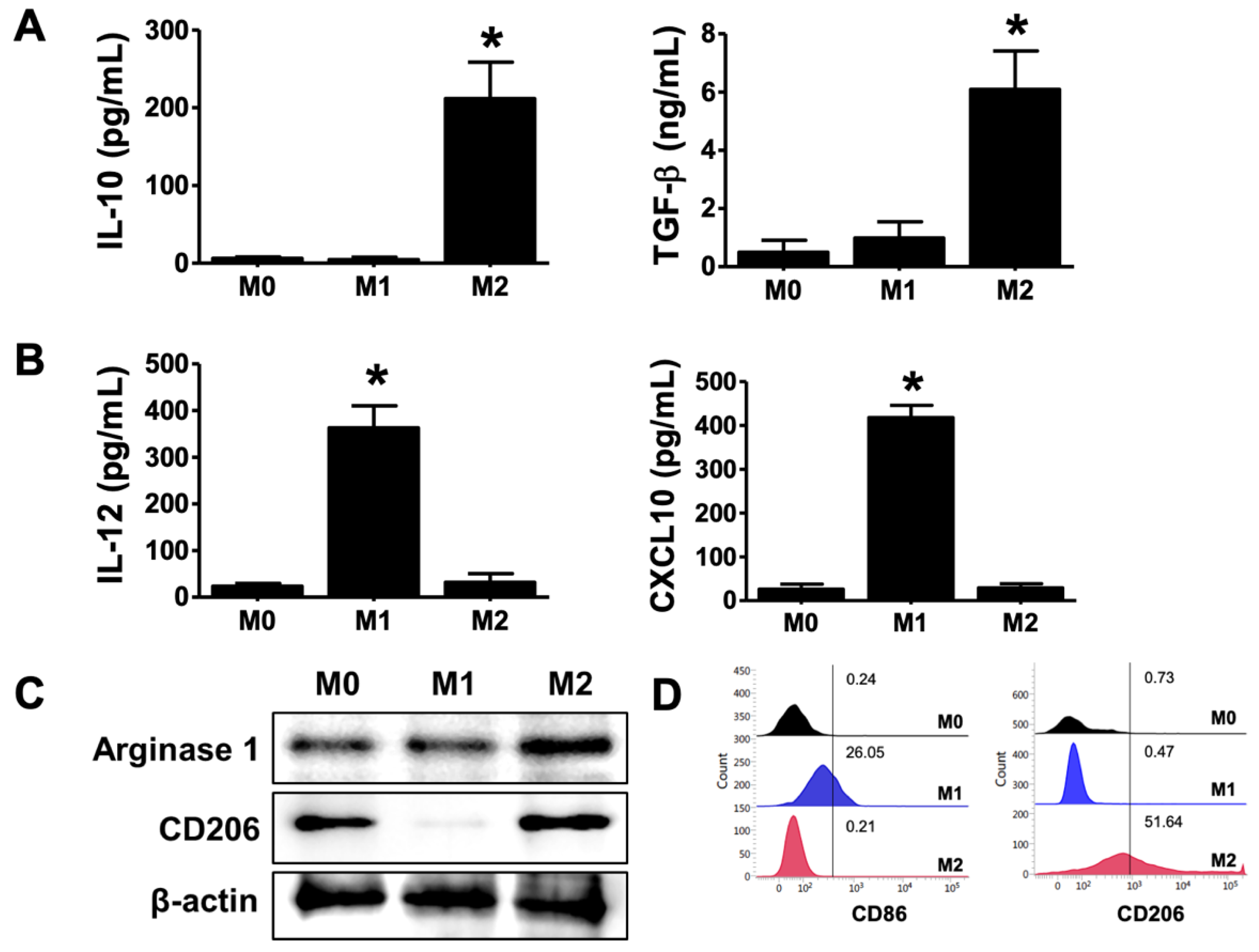
2.1. Differentiation of M2 Phenotype in THP-1-Derived Macrophages
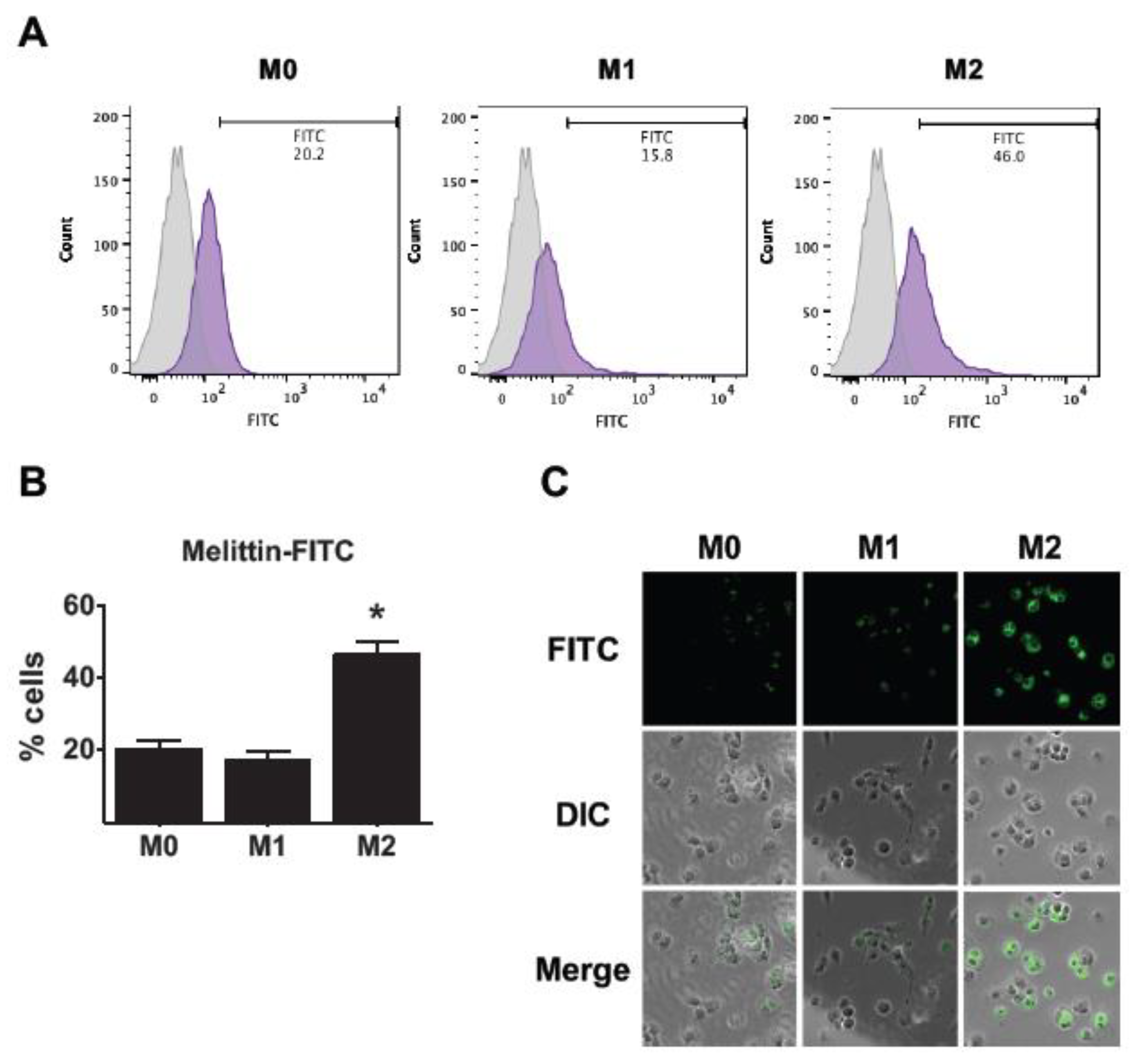
2.2. Affinity of Melittin in THP-1-Derived M2 Macrophages
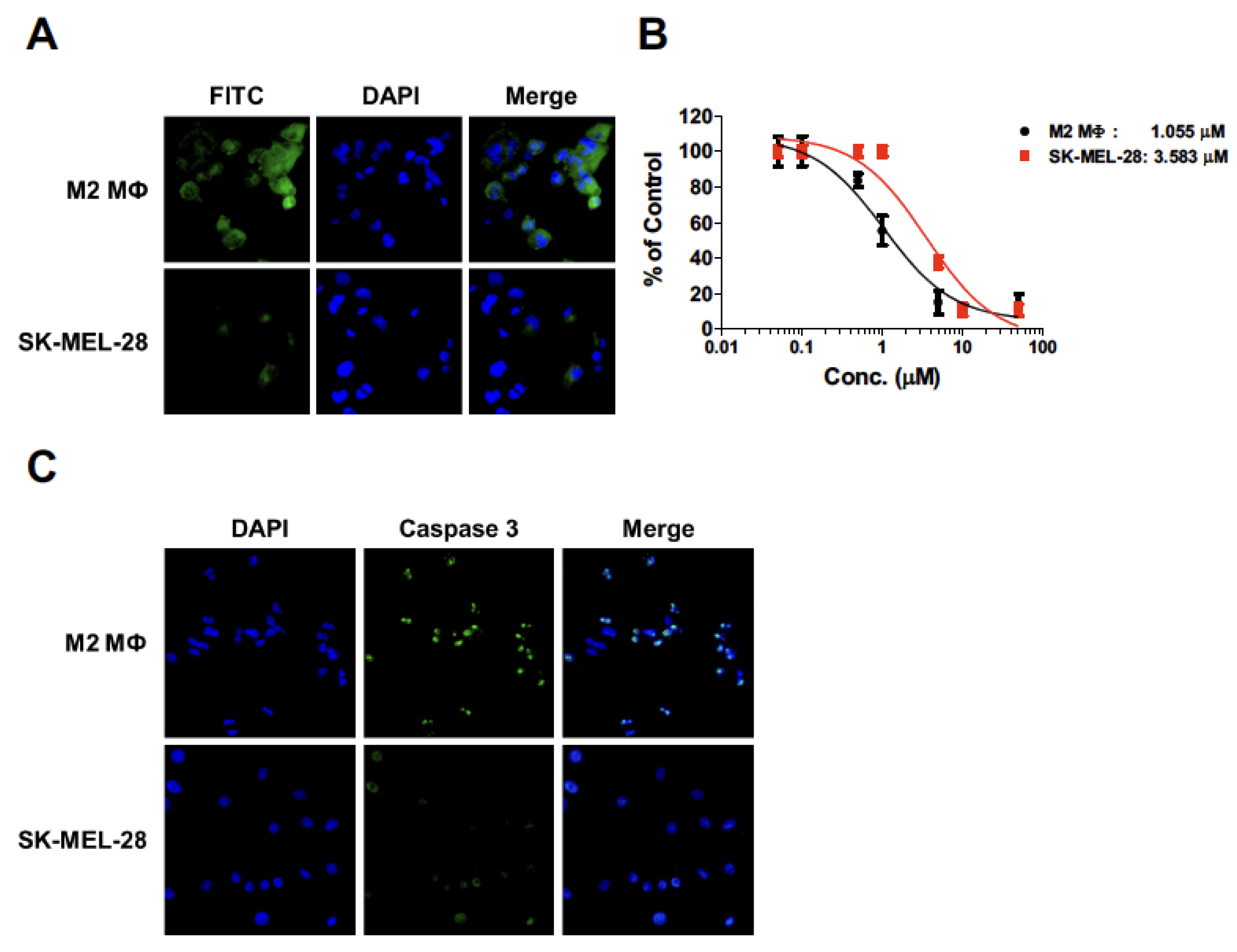
2.3. Apoptosis of THP-1-Derived M2 Macrophages by Melittin–dKLA
2.4. Cytotoxicity of Melittin–dKLA in THP-1-Derived M2 Macrophages and SK-MEL-28 Cells
2.5. Proliferation and Migration in SK-MEL-28 Cells by Conditioned Medium of THP-1-Derived M2 Macrophages Pretreated with Melittin–dKLA
2.6. Inhibition of Tumor Growth by Melittin–dKLA in Mouse Model of Melanoma
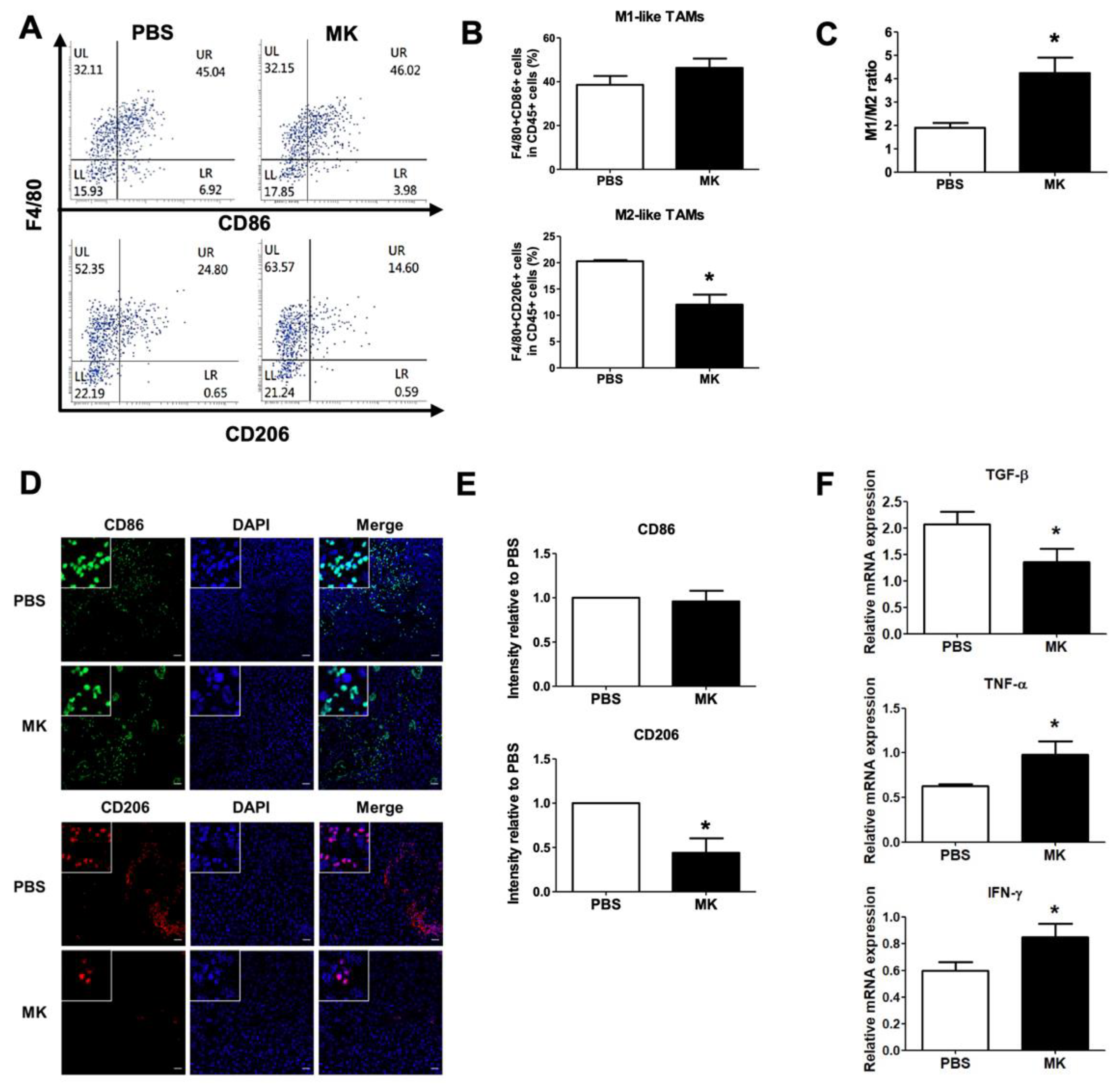
2.7. Reduction of M2-like TAMs by Melittin–dKLA in Mouse Model of Melanoma
3. Discussion
4. Materials and Methods
4.1. Cells and Mice
4.2. Differentiation of Macrophages and Preparation of Conditioned Medium
4.3. Tumor Inoculation and Animal Study
4.4. Tissue Cell Preparation and Flow Cytometry Analysis
4.5. Immunofluorescence Staining
4.6. ELISA
4.7. CCK-8 Assay
4.8. Wound Healing Assay
4.9. Western Blot Analysis
4.10. Quantitative Real-Time PCR
4.11. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, C.-E.; Esfandiari, A.; Ho, Y.-H.; Wang, N.; Mahdi, A.; Aptullahoglu, E.; Lovat, P.; Lunec, J. Targeting negative regulation of p53 by MDM2 and WIP1 as a therapeutic strategy in cutaneous melanoma. Br. J. Cancer 2018, 118, 495–508. [Google Scholar] [CrossRef] [Green Version]
- Rigel, D.S.; Carucci, J.A. Malignant melanoma: Prevention, early detection, and treatment in the 21st century. CA Cancer J. Clin. 2000, 50, 215–236; quiz 237–240. [Google Scholar] [CrossRef] [PubMed]
- Caini, S.; Gandini, S.; Sera, F.; Raimondi, S.; Fargnoli, M.C.; Boniol, M.; Armstrong, B.K. Meta-analysis of risk factors for cutaneous melanoma according to anatomical site and clinico-pathological variant. Eur. J. Cancer 2009, 45, 3054–3063. [Google Scholar] [CrossRef] [PubMed]
- Rastrelli, M.; Tropea, S.; Rossi, C.R.; Alaibac, M. Melanoma: Epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo 2014, 28, 1005–1011. [Google Scholar] [PubMed]
- Paulson, K.G.; Gupta, D.; Kim, T.S.; Veatch, J.R.; Byrd, D.R.; Bhatia, S.; Wojcik, K.; Chapuis, A.G.; Thompson, J.A.; Madeleine, M.M.; et al. Age-Specific Incidence of Melanoma in the United States. JAMA Dermatol. 2020, 156, 57–64. [Google Scholar] [CrossRef] [PubMed]
- CChambers, A.F.; Groom, A.C.; Macdonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef]
- Al Samaraee, A.; Khout, H.; Barakat, T.; Fasih, T. Breast metastasis from a melanoma. Ochsner J. 2012, 12, 149–151. [Google Scholar]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Kerkar, S.P.; Restifo, N.P. Cellular Constituents of Immune Escape within the Tumor Microenvironment: Figure 1. Cancer Res. 2012, 72, 3125–3130. [Google Scholar] [CrossRef] [Green Version]
- Falleni, M.; Savi, F.; Tosi, D.; Agape, E.; Cerri, A.; Moneghini, L.; Bulfamante, G.P. M1 and M2 macrophages’ clinicopathological significance in cutaneous melanoma. Melanoma Res. 2017, 27, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Tucci, M.; Passarelli, A.; Mannavola, F.; Felici, C.; Stucci, L.S.; Cives, M.; Silvestris, F. Immune System Evasion as Hallmark of Melanoma Progression: The Role of Dendritic Cells. Front. Oncol. 2019, 9, 1148. [Google Scholar] [CrossRef] [Green Version]
- Schoppmann, S.F.; Birner, P.; Stöckl, J.; Kalt, R.; Ullrich, R.; Caucig, C.; Kriehuber, E.; Nagy, K.; Alitalo, K.; Kerjaschki, D. Tumor-Associated Macrophages Express Lymphatic Endothelial Growth Factors and Are Related to Peritumoral Lymphangiogenesis. Am. J. Pathol. 2002, 161, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Lissbrant, I.F.; Stattin, P.; Wikstrom, P.; Damber, J.E.; Egevad, L.; Bergh, A. Tumor associated macrophages in human prostate cancer: Relation to clinicopathological variables and survival. Int. J. Oncol. 2000, 17, 445–496. [Google Scholar] [CrossRef] [PubMed]
- Bardi, G.T.; Smith, M.A.; Hood, J.L. Melanoma exosomes promote mixed M1 and M2 macrophage polarization. Cytokine 2018, 105, 63–72. [Google Scholar] [CrossRef]
- Ceci, C.; Atzori, M.G.; Lacal, P.M.; Graziani, G. Targeting Tumor-Associated Macrophages to Increase the Efficacy of Immune Checkpoint Inhibitors: A Glimpse into Novel Therapeutic Approaches for Metastatic Melanoma. Cancers 2020, 12, 3401. [Google Scholar] [CrossRef]
- Carlino, M.S.; Larkin, J.; Long, G.V. Immune checkpoint inhibitors in melanoma. Lancet 2021, 398, 1002–1014. [Google Scholar] [CrossRef]
- IImbert, C.; Montfort, A.; Fraisse, M.; Marcheteau, E.; Gilhodes, J.; Martin, E.; Bertrand, F.; Marcellin, M.; Burlet-Schiltz, O.; Peredo, A.G.; et al. Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1. Nat. Commun. 2020, 11, 437. [Google Scholar] [CrossRef]
- Eisenberg, D. Three-dimensional structure of membrane and surface proteins. Annu. Rev. Biochem. 1984, 53, 595–623. [Google Scholar] [CrossRef]
- Dempsey, C. The actions of melittin on membranes. Biochim. Biophys. Acta (BBA)—Rev. Biomembr. 1990, 1031, 143–161. [Google Scholar] [CrossRef]
- Ladokhin, A.; Selsted, M.; White, S. Sizing membrane pores in lipid vesicles by leakage of co-encapsulated markers: Pore formation by melittin. Biophys. J. 1997, 72, 1762–1766. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.-T.; Hung, W.-C.; Chen, F.-Y.; Huang, H.W. Mechanism and kinetics of pore formation in membranes by water-soluble amphipathic peptides. Proc. Natl. Acad. Sci. USA 2008, 105, 5087–5092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Jeong, H.; Bae, Y.; Shin, K.; Kang, S.; Kim, H.; Oh, J.; Bae, H. Targeting of M2-like tumor-associated macrophages with a melittin-based pro-apoptotic peptide. J. Immunother. Cancer 2019, 7, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef] [Green Version]
- Allavena, P.; Sica, A.; Solinas, G.; Porta, C.; Mantovani, A. The inflammatory micro-environment in tumor progression: The role of tumor-associated macrophages. Crit. Rev. Oncol. Hematol. 2008, 66, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- You, Z.; Shi, X.-B.; DuRaine, G.; Haudenschild, D.; Tepper, C.G.; Lo, S.H.; Gandour-Edwards, R.; White, R.W.D.V.; Reddi, A.H. Interleukin-17 Receptor-Like Gene Is a Novel Antiapoptotic Gene Highly Expressed in Androgen-Independent Prostate Cancer. Cancer Res. 2006, 66, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Lan, C.; Huang, X.; Lin, S.; Huang, H.; Cai, Q.; Wan, T.; Lu, J.; Liu, J. Expression of M2-Polarized Macrophages is Associated with Poor Prognosis for Advanced Epithelial Ovarian Cancer. Technol. Cancer Res. Treat. 2013, 12, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Yu, Y.; He, X.; Niu, N.; Li, X.; Zhang, R.; Hu, J.; Ma, J.; Yu, X.; Sun, Y.; et al. Tumor-associated macrophages induce invasion and poor prognosis in human gastric cancer in a cyclooxygenase-2/MMP9-dependent manner. Am. J. Transl. Res. 2019, 11, 6040–6054. [Google Scholar]
- Sumitomo, R.; Hirai, T.; Fujita, M.; Murakami, H.; Otake, Y.; Huang, C.L. M2 tumor-associated macrophages promote tumor progression in non-small-cell lung cancer. Exp. Ther. Med. 2019, 18, 4490–4498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Mo, C.; Wang, Y.; Wei, D.; Xiao, H. Anti-tumour strategies aiming to target tumour-associated macrophages. Immunology 2013, 138, 93–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngambenjawong, C.; Gustafson, H.H.; Pun, S.H. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv. Drug Deliv. Rev. 2017, 114, 206–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paull, B.R.; Yunginger, J.W.; Gleich, G.J. Melittin: An allergen of honeybee venom. J. Allergy Clin. Immunol. 1977, 59, 334–338. [Google Scholar] [CrossRef]
- Son, D.J.; Lee, J.W.; Lee, Y.H.; Song, H.S.; Kil Lee, C.; Hong, J.T. Therapeutic application of anti-arthritis, pain-releasing, and anti-cancer effects of bee venom and its constituent compounds. Pharmacol. Ther. 2007, 115, 246–270. [Google Scholar] [CrossRef]
- Jo, M.; Park, M.H.; Kollipara, P.S.; An, B.J.; Song, H.S.; Han, S.B.; Kim, J.H.; Song, M.J.; Hong, J.T. Anti-cancer effect of bee venom toxin and melittin in ovarian cancer cells through induction of death receptors and inhibition of JAK2/STAT3 pathway. Toxicol. Appl. Pharmacol. 2012, 258, 72–81. [Google Scholar] [CrossRef]
- Soman, N.R.; Baldwin, S.L.; Hu, G.; Marsh, J.N.; Lanza, G.M.; Heuser, J.E.; Arbeit, J.M.; Wickline, S.A.; Schlesinger, P.H. Molecularly targeted nanocarriers deliver the cytolytic peptide melittin specifically to tumor cells in mice, reducing tumor growth. J. Clin. Investig. 2009, 119, 2830–2842. [Google Scholar] [CrossRef]
- Lee, C.; Bae, S.-J.S.; Joo, H.; Bae, H. Melittin suppresses tumor progression by regulating tumor-associated macrophages in a Lewis lung carcinoma mouse model. Oncotarget 2017, 8, 54951–54965. [Google Scholar] [CrossRef] [Green Version]
- Foillard, S.; Jin, Z.-H.; Garanger, E.; Boturyn, D.; Favrot, M.-C.; Coll, J.-L.; Dumy, P. Synthesis and Biological Characterisation of Targeted Pro-Apoptotic Peptide. ChemBioChem 2008, 9, 2326–2332. [Google Scholar] [CrossRef]
- Javadpour, M.M.; Juban, M.M.; Lo, W.-C.J.; Bishop, S.M.; Alberty, J.B.; Cowell, S.M.; Becker, C.L.; McLaughlin, M.L. De Novo Antimicrobial Peptides with Low Mammalian Cell Toxicity. J. Med. Chem. 1996, 39, 3107–3113. [Google Scholar] [CrossRef]
- Kim, H.Y.; Kim, S.; Youn, H.; Chung, J.-K.; Shin, D.H.; Lee, K. The cell penetrating ability of the proapoptotic peptide, KLAKLAKKLAKLAK fused to the N-terminal protein transduction domain of translationally controlled tumor protein, MIIYRDLISH. Biomaterials 2011, 32, 5262–5268. [Google Scholar] [CrossRef] [PubMed]
- Mai, J.C.; Mi, Z.; Kim, S.H.; Ng, B.; Robbins, P.D. A proapoptotic peptide for the treatment of solid tumors. Cancer Res. 2001, 61, 7709–7712. [Google Scholar] [PubMed]
- Tian, Y.; Guo, Y.; Zhu, P.; Zhang, D.; Liu, S.; Tang, M.; Wang, Y.; Jin, Z.; Li, D.; Yan, D.; et al. TRIM59 loss in M2 macrophages promotes melanoma migration and invasion by upregulating MMP-9 and Madcam1. Aging 2019, 11, 8623–8641. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, B.; Bai, X.; Cohen, C.; Zhong, H.; Kilroy, S.; Louis, G.; Moses, M.; Arbiser, J.L. Malignant Transformation of Melanocytes to Melanoma by Constitutive Activation of Mitogen-activated Protein Kinase Kinase (MAPKK) Signaling. J. Biol. Chem. 2003, 278, 9790–9795. [Google Scholar] [CrossRef] [Green Version]
- VValastyan, S.; Weinberg, R.A. Tumor Metastasis: Molecular Insights and Evolving Paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; You, M.J.; Yang, Y.; Hu, D.; Tian, C. The Role of Tumor-Associated Macrophages in Leukemia. Acta Haematol. 2020, 143, 112–117. [Google Scholar] [CrossRef]
- Georgoudaki, A.-M.; Prokopec, K.E.; Boura, V.F.; Hellqvist, E.; Sohn, S.; Östling, J.; Dahan, R.; Harris, R.A.; Rantalainen, M.; Klevebring, D.; et al. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep. 2016, 15, 2000–2011. [Google Scholar] [CrossRef] [Green Version]
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In vivo imaging reveals a tumor-associated macrophage–mediated resistance pathway in anti–PD-1 therapy. Sci. Transl. Med. 2017, 9, 9. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, I.-H.; Jeong, C.; Yang, J.; Park, S.-H.; Hwang, D.-S.; Bae, H. Therapeutic Effect of Melittin–dKLA Targeting Tumor-Associated Macrophages in Melanoma. Int. J. Mol. Sci. 2022, 23, 3094. https://doi.org/10.3390/ijms23063094
Han I-H, Jeong C, Yang J, Park S-H, Hwang D-S, Bae H. Therapeutic Effect of Melittin–dKLA Targeting Tumor-Associated Macrophages in Melanoma. International Journal of Molecular Sciences. 2022; 23(6):3094. https://doi.org/10.3390/ijms23063094
Chicago/Turabian StyleHan, Ik-Hwan, Chanmi Jeong, Juwon Yang, Seung-Hyeok Park, Deok-Sang Hwang, and Hyunsu Bae. 2022. "Therapeutic Effect of Melittin–dKLA Targeting Tumor-Associated Macrophages in Melanoma" International Journal of Molecular Sciences 23, no. 6: 3094. https://doi.org/10.3390/ijms23063094
APA StyleHan, I.-H., Jeong, C., Yang, J., Park, S.-H., Hwang, D.-S., & Bae, H. (2022). Therapeutic Effect of Melittin–dKLA Targeting Tumor-Associated Macrophages in Melanoma. International Journal of Molecular Sciences, 23(6), 3094. https://doi.org/10.3390/ijms23063094