
Further Delineation of Duplications of ARX Locus Detected in Male Patients with Varying Degrees of Intellectual Disability

 ,
,  ,
,  ,
,  , and
, and 
Abstract
1. Introduction
2. Duplications of the p23.1 Region of the X Chromosome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Extent (kb) | ChrX Genomic Coordinates (UCSC Release) | Duplicated Genes | Duplicated Enhancers | Clinical Signs/Reference |
|---|---|---|---|---|---|
| 505 | 41.1 | 24,992,915–25,033,979 (GRCh37/hg19) 24,974,798–25,015,862 (GRCh38/hg38) | ARX (full gene) | hs121, hs122, hs145 | Moderate Syndromic ID [32] |
| Case 1 | 540 | 24,861,402–25,398,496 (GRCh37/hg19) 24,843,285–25,380,379 (GRCh38/hg38) | ARX (full gene) | hs118, hs119, hs121, hs122, hs145 | No clinical signs [21] |
| Case 2 | 440.5 | 24,593,306–25,033,770 (GRCh37/hg19) 24,575,189–25,015,653 (GRCh38/hg38) | POLA1 (full gene) ARX (exons 2-5) | hs118, hs119, hs121, hs122, hs145 | Severe ID, Microphthalmia, growth retardation [21] |
| Case 3 | 630 | 24,537,027–25,163,704 (GRCh37/hg19) 24,518,910–25,145,587 (GRCh38/hg38) | PCYT1B (full gene); POLA1 (full gene); ARX (full gene), | hs118, hs119, hs121, hs122, hs145 | No clinical signs [21] |
| Case 4 | 580 | 24,650,157–25,230,368 (GRCh37/hg19) 24,632,040–25,212,251 (GRCh38/hg38) | POLA1 (full gene), ARX (full gene) | hs118, hs119, hs121, hs122, hs145 | autism, hyperactivity, delayed speech [21] |
| Case 5 | 720 | 24,542,008–25,542,728 (GRCh37/hg19) 24,523,891–25,524,611 (GRCh38/hg38) | PCYT1B (full gene); POLA1 (full gene); ARX (full gene), | hs118, hs119, hs121, hs122, hs145, hs123 | ID, psychiatric abnormalities, delayed speech [21] |
| Case 6 | 290 | 24,733,304–25,022,540 (GRCh37/hg19) 24,715,187–25,004,423 (GRCh38/hg38) | POLA1 (partial), ARX (partial) | hs118, hs119, hs121, hs122, hs145 | Developmental delay [21] |
| A150 | 3350 | 24,513,979–27,864,451 (GRCh37/hg19) 24,495,862–27,846,334 (GRCh38/hg38) | PCYT1B (full gene), POLA1 (full gene); ARX (full gene) & others | hs118, hs119, hs121, hs122, hs145, hs123 | Autism [30] |
| DECIPHER 277835 | 302 | 24,843,484–25,145,646 (GRCh38/hg38) | ARX (full gene) | hs118, hs119, hs121, hs122, hs145, hs123 | Moderate ID [30] |
| DECIPHER 268043 | 2300 | 24,810,754–27,125,219 (GRCh37/hg19) 24,792,637–27,107,102 (GRCh38/hg38) | ARX (full gene) &others | hs118, hs119, hs121, hs122, hs145, hs123 | ID; short stature [30] |
| DECIPHER 250183 | 717 | 24,807,990–25,524,611 (GRCh37/hg19) 24,789,873–25,506,494 (GRCh38/hg38) | ARX (full coding region) | hs118, hs119, hs121, hs122, hs123, hs145 | Behavioral abnormality; ID; delayed speech [30] |
| DECIPHER 265145 | 580 | 24,632,040–25,212,251 (GRCh37/hg19) 24,613,923–25,194,134 (GRCh38/hg38) | POLA1 (full coding region) ARX (full coding region) | hs118, hs119, hs121, hs122, hs145 | attention deficit; hyperactivity; autism; delayed speech [30] |
| P1 | 438 | 24,887,676–25,325,777 (GRCh37/hg19) 24,869,559–25,307,660 (GRCh38/hg38) | ARX (full coding region) | hs118, hs119, hs121, hs122, hs145 | mild ID, speech delay and hypotonia [22] |
| P3 | 377 | 24,677,441–25,054,698 (GRCh37/hg19) 24,659,324–25,036,581 (GRCh38/hg38) | POLA1 (full coding region) ARX (full coding region) | hs118, hs119, hs121, hs122, hs145 | Developmental delay, growth retardation, delayed speech [22] |
| DECIPHER 266096 DP3 | 813 | 24,741,372–25,554,818 (GRCh37/hg19) 24,723,255–25,536,701 (GRCh38/hg38) | ARX (full coding region) | hs118, hs119, hs121, hs122, hs145, hs123 | hypotonia, microcephaly [22] |
| GE#1 | 803 | 24,828,871–25,631,863 (GRCh38/hg38) | ARX (full coding region) | hs118, hs119, hs121, hs122, hs145, hs123 | Moderate ID; psychomotor retardation, behavioural and developmental disorder [this report] |
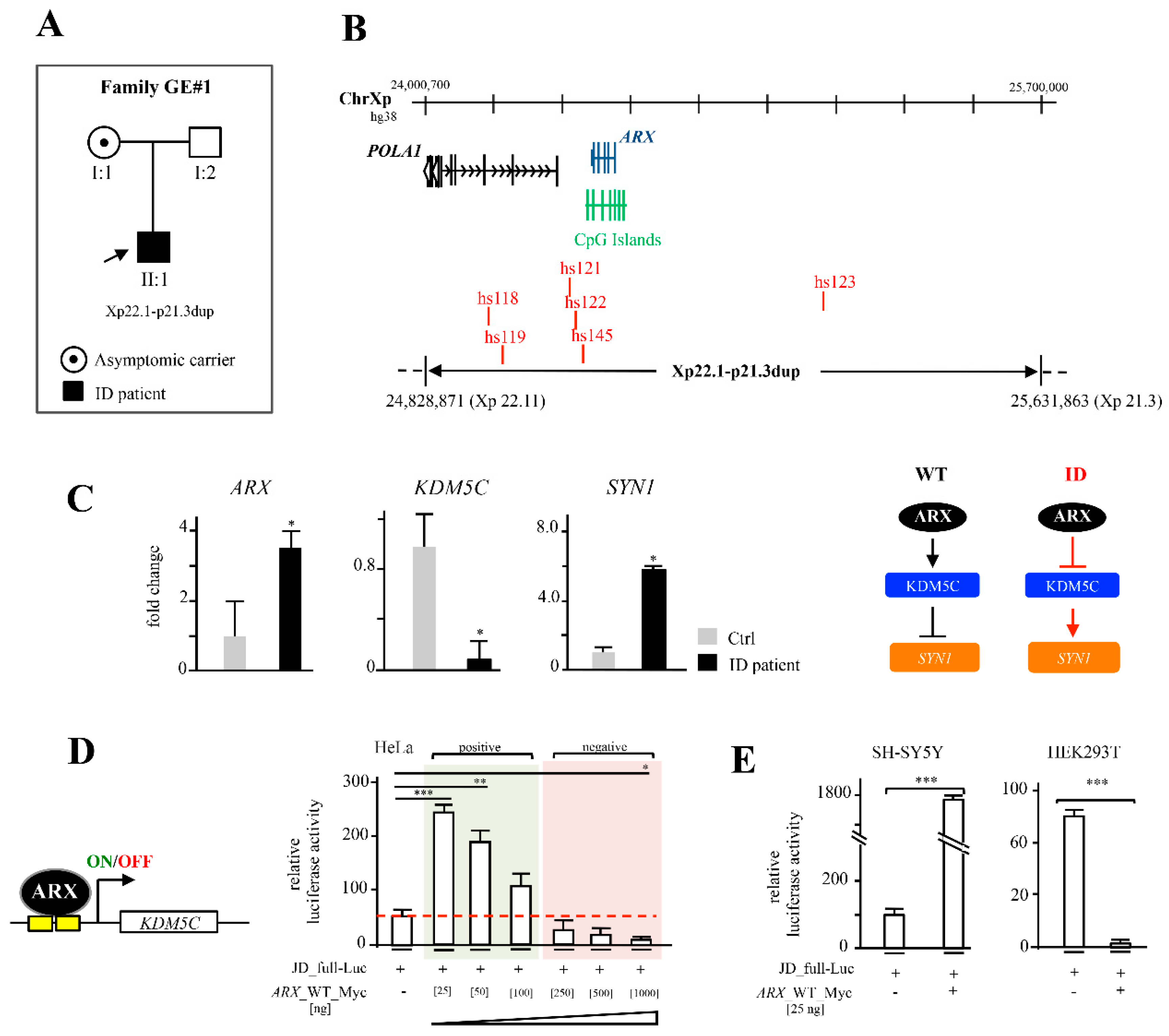
3. Identification of a Novel Xp22.11-p21.3—Duplication in a Male Child with Moderate ID and Analysis of Functional Implications
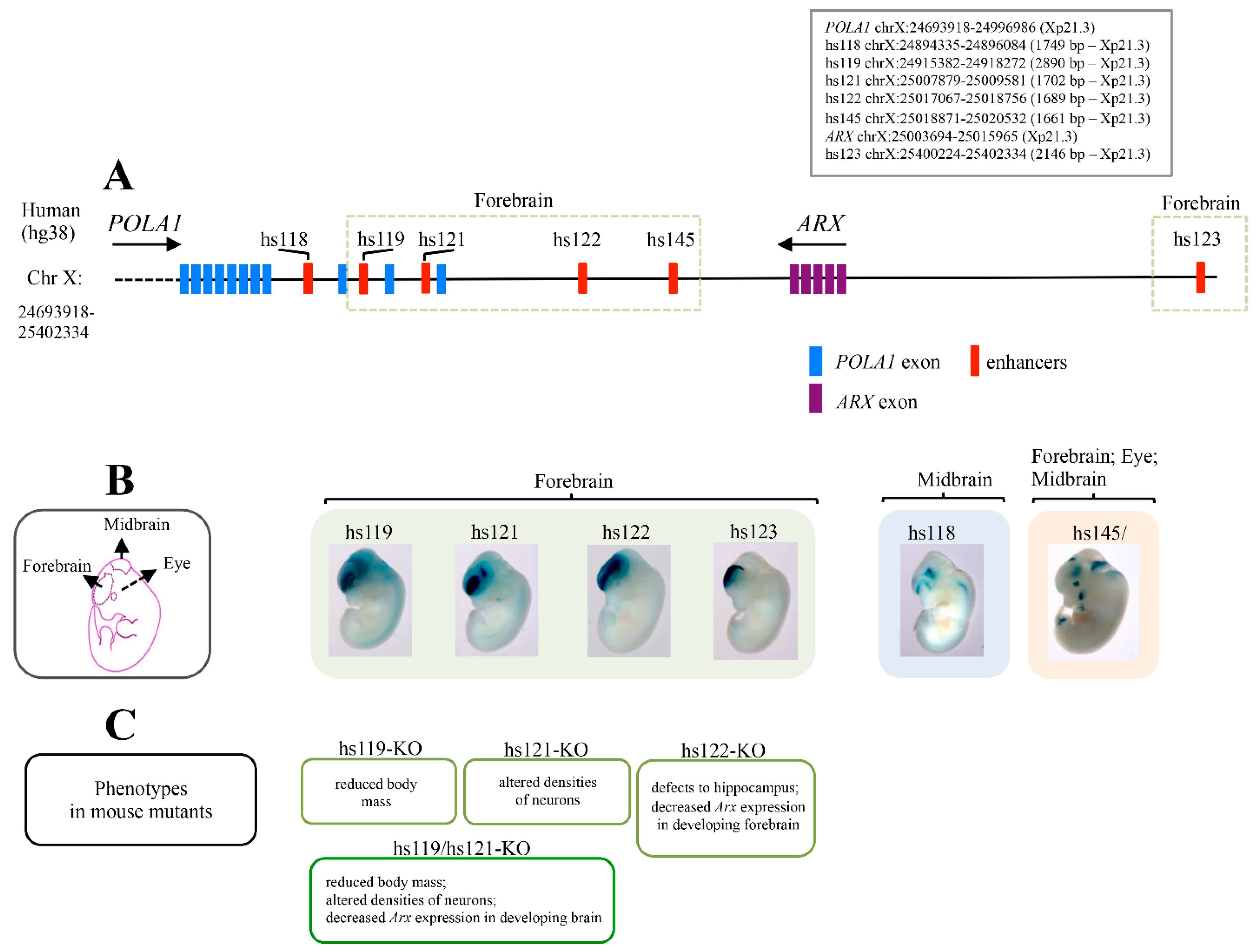
4. Ultraconserved ARX/Arx Brain Enhancers
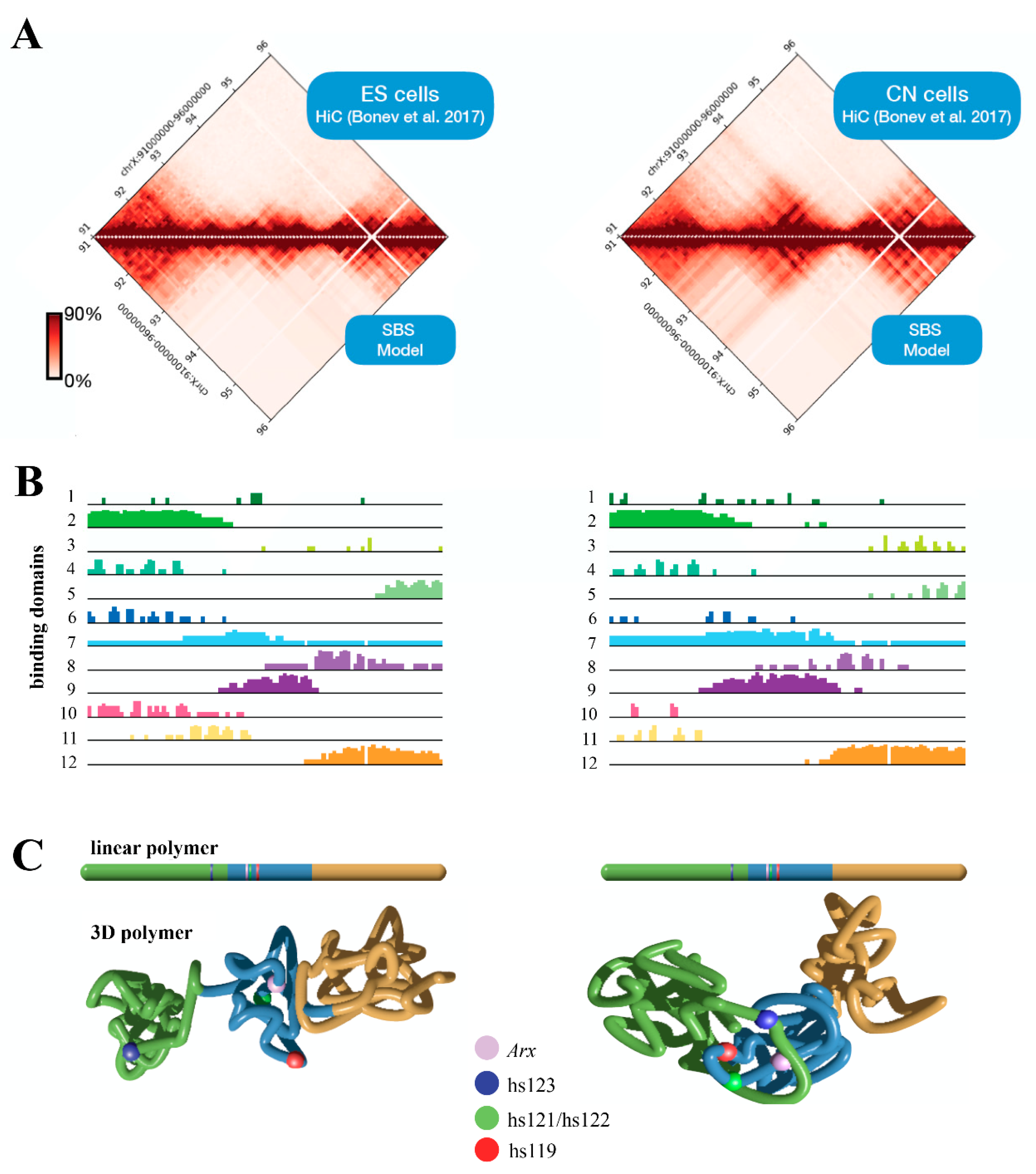
5. Three-Dimensional (3D) Structure of the Arx Locus in Embryonic Stem Cells (ESs) and Cortical Neurons (CNs)
6. Genotype-Phenotype Correlation
7. Discussion & Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Strømme, P.; Mangelsdorf, M.E.; Shaw, M.A.; Lower, K.M.; Lewis, S.M.; Bruyere, H.; Lütcherath, V.; Gedeon, A.K.; Wallace, R.H.; Scheffer, I.E.; et al. Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat. Genet. 2002, 30, 441–445. [Google Scholar] [CrossRef]
- Colombo, E.; Galli, R.; Cossu, G.; Gécz, J.; Broccoli, V. Mouse orthologue of ARX; a gene mutated in several X-linked forms of mental retardation and epilepsy; is a marker of adult neural stem cells and forebrain GABAergic neurons. Dev. Dyn. 2004, 231, 631–639. [Google Scholar] [CrossRef]
- Colombo, E.; Collombat, P.; Colasante, G.; Bianchi, M.; Long, J.; Mansouri, A.; Rubenstein, J.L.; Broccoli, V. Inactivation of Arx; the murine ortholog of the X-linked lissencephaly with ambiguous genitalia gene; leads to severe disorganization of the ventral telencephalon with impaired neuronal migration and differentiation. J. Neurosci. 2007, 27, 4786–4798. [Google Scholar] [CrossRef]
- Friocourt, G.; Poirier, K.; Rakić, S.; Parnavelas, J.G.; Chelly, J. The role of ARX in cortical development. Eur J. Neurosci. 2006, 23, 869–876. [Google Scholar] [CrossRef]
- Friocourt, G.; Kanatani, S.; Tabata, H.; Yozu, M.; Takahashi, T.; Antypa, M.; Raguénès, O.; Chelly, J.; Férec, C.; Nakajima, K.; et al. Cell-autonomous roles of ARX in cell proliferation and neuronal migration during corticogenesis. J. Neurosci. 2008, 28, 5794–5805. [Google Scholar] [CrossRef]
- Seufert, D.W.; Prescott, N.L.; El-Hodiri, H.M. Xenopus aristaless-related homeobox (xARX) gene product functions as both a transcriptional activator and repressor in forebrain development. Dev. Dyn. 2005, 232, 313–324. [Google Scholar] [CrossRef]
- Fulp, C.T.; Cho, G.; Marsh, E.D.; Nasrallah, I.M.; Labosky, P.A.; Golden, J.A. Identification of Arx transcriptional targets in the developing basal forebrain. Hum. Mol. Genet. 2008, 17, 3740–3760. [Google Scholar] [CrossRef]
- Laperuta, C.; Spizzichino, L.; D′Adamo, P.; Monfregola, J.; Maiorino, A.; D′Eustacchio, A.; Ventruto, V.; Neri, G.; D’Urso, M.; Chiurazzi, P.; et al. MRX87 family with Aristaless X dup24bp mutation and implication for polyalanine expansions. BMC Med. Genet. 2007, 8, 25. [Google Scholar] [CrossRef]
- Poeta, L.; Fusco, F.; Drongitis, D.; Shoubridge, C.; Manganelli, G.; Filosa, S.; Paciolla, M.; Courtney, M.; Collombat, P.; Lioi, M.B.; et al. A regulatory path associated with X-linked intellectual disability and epilepsy links KDM5C to the polyalanine expansions in ARX. Am. J. Hum. Genet. 2013, 92, 114–125. [Google Scholar] [CrossRef]
- Shoubridge, C.; Tan, M.H.; Seiboth, G.; Gécz, J. ARX homeodomain mutations abolish DNA binding and lead to a loss of transcriptional repression. Hum. Mol. Genet. 2012, 21, 1639–1647. [Google Scholar] [CrossRef]
- Fullston, T.; Brueton, L.; Willis, T.; Philip, S.; MacPherson, L.; Finnis, M.; Gecz, J.; Morton, J. Ohtahara syndrome in a family with an ARX protein truncation mutation (c.81C>G/p.Y27X). Eur. J. Hum. Genet. 2010, 18, 157–162. [Google Scholar] [CrossRef]
- Kato, M.; Saitoh, S.; Kamei, A.; Shiraishi, H.; Ueda, Y.; Akasaka, M.; Tohyama, J.; Akasaka, N.; Hayasaka, K.A. A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome). Am. J. Hum. Genet. 2007, 81, 361–366. [Google Scholar] [CrossRef]
- Quillé, M.L.; Carat, S.; Quéméner-Redon, S.; Hirchaud, E.; Baron, D.; Benech, C.; Guihot, J.; Placet, M.; Mignen, O.; Férec, C.; et al. High-throughput analysis of promoter occupancy reveals new targets for Arx; a gene mutated in mental retardation and interneuronopathies. PLoS ONE 2011, 6, e25181. [Google Scholar] [CrossRef]
- Poeta, L.; Padula, A.; Attianese, B.; Valentino, M.; Verrillo, L.; Filosa, S.; Shoubridge, C.; Barra, A.; Schwartz, C.E.; Christensen, J.; et al. Histone demethylase KDM5C is a SAHA-sensitive central hub at the crossroads of transcriptional axes involved in multiple neurodevelopmental disorders. Hum. Mol. Genet. 2019, 28, 4089–4102. [Google Scholar] [CrossRef]
- Poeta, L.; Padula, A.; Lioi, M.B.; van Bokhoven, H.; Miano, M.G. Analysis of a Set of KDM5C Regulatory Genes Mutated in Neurodevelopmental Disorders Identifies Temporal Coexpression Brain Signatures. Genes 2021, 12, 1088. [Google Scholar] [CrossRef] [PubMed]
- Biressi, S.; Messina, G.; Collombat, P.; Tagliafico, E.; Monteverde, S.; Benedetti, L.; Cusella De Angelis, M.G.; Mansouri, A.; Ferrari, S.; Tajbakhsh, S.; et al. The homeobox gene Arx is a novel positive regulator of embryonic myogenesis. Cell Death Differ. 2008, 15, 94–104. [Google Scholar] [CrossRef][Green Version]
- McKenzie, O.; Ponte, I.; Mangelsdorf, M.; Finnis, M.; Colasante, G.; Shoubridge, C.; Stifani, S.; Gécz, J.; Broccoli, V. Aristaless-related homeobox gene; the gene responsible for West syndrome and related disorders; is a Groucho/transducin-like enhancer of split dependent transcriptional repressor. Neuroscience 2007, 146, 236–247. [Google Scholar] [CrossRef]
- Cho, I.T.; Lim, Y.; Golden, J.A.; Cho, G. Aristaless Related Homeobox (ARX) Interacts with β-Catenin; BCL9; and P300 to Regulate Canonical Wnt Signaling. PLoS ONE 2017, 12, e0170282. [Google Scholar] [CrossRef]
- Kitamura, K.; Yanazawa, M.; Sugiyama, N.; Miura, H.; Iizuka-Kogo, A.; Kusaka, M.; Omichi, K.; Suzuki, R.; Kato-Fukui, Y.; Kamiirisa, K.; et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat. Genet. 2002, 32, 359–369. [Google Scholar] [CrossRef]
- Poirier, K.; Van Esch, H.; Friocourt, G.; Saillour, Y.; Bahi, N.; Backer, S.; Souil, E.; Castelnau-Ptakhine, L.; Beldjord, C.; Francis, F.; et al. Neuroanatomical distribution of ARX in brain and its localisation in GABAergic neurons. Brain Res. Mol. Brain Res. 2004, 122, 35–46. [Google Scholar] [CrossRef]
- Popovici, C.; Busa, T.; Boute, O.; Thuresson, A.C.; Perret, O.; Sigaudy, S.; Södergren, T.; Andrieux, J.; Moncla, A.; Philip, N. Whole ARX gene duplication is compatible with normal intellectual development. Am. J. Med. Genet. A 2014, 164A, 2324–2327. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, M.; Manning, E.; Shoubridge, C.; Krecsmarik, M.; Hawkins, T.A.; Giacomotto, J.; Zhao, T.; Mueller, T.; Bader, P.I.; Cheung, S.W.; et al. Copy number variants in patients with intellectual disability affect the regulation of ARX transcription factor gene. Hum. Genet. 2015, 134, 1163–1182. [Google Scholar] [CrossRef] [PubMed]
- Bejerano, G.; Pheasant, M.; Makunin, I.; Stephen, S.; Kent, W.J.; Mattick, J.S.; Haussler, D. Ultraconserved elements in the human genome. Science 2004, 304, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Dickel, D.E.; Ypsilanti, A.R.; Pla, R.; Zhu, Y.; Barozzi, I.; Mannion, B.J.; Khin, Y.S.; Fukuda-Yuzawa, Y.; Plajzer-Frick, I.; Pickle, C.S.; et al. Ultraconserved Enhancers Are Required for Normal Development. Cell 2018, 172, 491–499. [Google Scholar] [CrossRef]
- Li, R.; Liu, Y.; Li, T.; Li, C. 3Disease Browser: A Web server for integrating 3D genome and disease-associated chromosome rearrangement data. Sci. Rep. 2016, 6, 34651. [Google Scholar] [CrossRef] [PubMed]
- Franke, M.; Ibrahim, D.M.; Andrey, G.; Schwarzer, W.; Heinrich, V.; Schöpflin, R.; Kraft, K.; Kempfer, R.; Jerković, I.; Chan, W.L.; et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 2016, 538, 265–269. [Google Scholar] [CrossRef]
- Bianco, S.; Lupiáñez, D.G.; Chiariello, A.M.; Annunziatella, C.; Kraft, K.; Schöpflin, R.; Wittler, L.; Andrey, G.; Vingron, M.; Pombo, A.; et al. Polymer physics predicts the effects of structural variants on chromatin architecture. Nat. Genet. 2018, 50, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Thorson, L.; Bryke, C.; Rice, G.; Artzer, A.; Schilz, C.; Israel, J.; Huber, S.; Laffin, J.; Raca, G. Clinical and molecular characterization of overlapping interstitial Xp21-p22 duplications in two unrelated individuals. Am. J. Med. Genet. Part A 2010, 152A, 904–915. [Google Scholar] [CrossRef]
- Wu, L.; Liu, J.; Lv, W.; Wen, J.; Xia, Y.; Liang, D. An Xp21.3p11.4 duplication observed in a boy with intellectual deficiency and speech delay and his asymptomatic mother. Birth Defects Res. A Clin. Mol. Teratol. 2013, 97, 467–470. [Google Scholar] [CrossRef]
- Egger, G.; Roetzer, K.M.; Noor, A.; Lionel, A.C.; Mahmood, H.; Schwarzbraun, T.; Boright, O.; Mikhailov, A.; Marshall, C.R.; Windpassinger, C.; et al. Identification of risk genes for autism spectrum disorder through copy number variation analysis in Austrian families. Neurogenetics 2014, 15, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Piccione, M.; Sanfilippo, C.; Cavani, S.; Salatiello, P.; Malacarne, M.; Pierluigi, M.; Fichera, M.; Luciano, D.; Corsello, G. Molecular and clinical characterization of a small duplication Xp in a human female with psychiatric disorders. J. Genet. 2011, 90, 473–477. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Abidi, F.; Fullston, T.; Choma, M.K.; Boucher, C.A.; Shepherd, L.; Willatt, L.; Parkin, G.; Smith, R.; Futreal, P.A.; Shaw, M.; et al. Fine-scale survey of X chromosome copy number variants and indels underlying intellectual disability. Am. J. Hum. Genet. 2010, 87, 173–188. [Google Scholar]
- Starokadomskyy, P.; Gemelli, T.; Rios, J.J.; Xing, C.; Wang, R.C.; Li, H.; Pokatayev, V.; Dozmorov, I.; Khan, S.; Miyata, N.; et al. DNA polymerase-α regulates the activation of type I interferons through cytosolic RNA:DNA synthesis. Nat. Immunol. 2016, 17, 495–504. [Google Scholar] [CrossRef] [PubMed]
- D’haene, E.; Vergult, S. Interpreting the impact of noncoding structural variation in neurodevelopmental disorders. Genet. Med. 2021, 23, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Shen, E.; Shulha, H.; Weng, Z.; Akbarian, S. Regulation of histone H3K4 methylation in brain development and disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130514. [Google Scholar] [CrossRef] [PubMed]
- Visel, A.; Taher, L.; Girgis, H.; May, D.; Golonzhka, O.; Hoch, R.V.; McKinsey, G.L.; Pattabiraman, K.; Silberberg, S.N.; Blow, M.J.; et al. A high-resolution enhancer atlas of the developing telencephalon. Cell 2013, 152, 895–908. [Google Scholar] [CrossRef] [PubMed]
- Pennacchio, L.A.; Ahituv, N.; Moses, A.M.; Prabhakar, S.; Nobrega, M.A.; Shoukry, M.; Minovitsky, S.; Dubchak, I.; Holt, A.; Lewis, K.D.; et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature 2006, 444, 499–502. [Google Scholar] [CrossRef]
- Snetkova, V.; Ypsilanti, A.R.; Akiyama, J.A.; Mannion, B.J.; Plajzer-Frick, I.; Novak, C.S.; Harrington, A.N.; Pham, Q.T.; Kato, M.; Zhu, Y.; et al. Ultraconserved enhancer function does not require perfect sequence conservation. Nat. Genet. 2021, 53, 521–528. [Google Scholar] [CrossRef]
- Snetkova, V.; Pennacchio, L.A.; Visel, A.; Dickel, D.E. Perfect and imperfect views of ultraconserved sequences. Nat. Rev. Genet. 2021, 23, 182–194. [Google Scholar] [CrossRef]
- Bonev, B.; Mendelson Cohen, N.; Szabo, Q.; Fritsch, L.; Papadopoulos, G.L.; Lubling, Y.; Xu, X.; Lv, X.; Hugnot, J.P.; Tanay, A.; et al. Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 2017, 171, 557–572. [Google Scholar] [CrossRef]
- Barbieri, M.; Chotalia, M.; Fraser, J.; Lavitas, L.M.; Dostie, J.; Pombo, A.; Nicodemi, M. Complexity of chromatin folding is captured by the strings and binders switch model. Proc. Natl. Acad. Sci. USA 2012, 109, 16173–16178. [Google Scholar] [CrossRef] [PubMed]
- Chiariello, A.M.; Esposito, A.; Annunziatella, C.; Bianco, S.; Fiorillo, L.; Prisco, A.; Nicodemi, M. A Polymer Physics Investigation of the Architecture of the Murine Orthologue of the 7q11.23 Human Locus. Front. Neurosci. 2017, 11, 559. [Google Scholar] [CrossRef] [PubMed]
- Chiariello, A.M.; Bianco, S.; Oudelaar, A.M.; Esposito, A.; Annunziatella, C.; Fiorillo, L.; Conte, M.; Corrado, A.; Prisco, A.; Larke, M.; et al. A Dynamic Folded Hairpin Conformation Is Associated with α-Globin Activation in Erythroid Cells. Cell Rep. 2020, 30, 2125–2135. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, L.; Musella, F.; Conte, M.; Kempfer, R.; Chiariello, A.M.; Bianco, S.; Kukalev, A.; Irastorza-Azcarate, I.; Esposito, A.; Abraham, A.; et al. Comparison of the Hi-C, GAM and SPRITE methods using polymer models of chromatin. Nat. Methods 2021, 18, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Klingler, E.; Francis, F.; Jabaudon, D.; Cappello, S. Mapping the molecular and cellular complexity of cortical malformations. Science 2021, 371, eaba4517. [Google Scholar] [CrossRef] [PubMed]
- Klopocki, E.; Ott, C.E.; Benatar, N.; Ullmann, R.; Mundlos, S.; Lehmann, K. A microduplication of the long range SHH limb regulator (ZRS) is associated with triphalangeal thumb-polysyndactyly syndrome. J. Med. Genet. 2008, 45, 370–375. [Google Scholar] [CrossRef]
- Phillips-Cremins, J.E.; Sauria, M.E.; Sanyal, A.; Gerasimova, T.I.; Lajoie, B.R.; Bell, J.S.; Ong, C.T.; Hookway, T.A.; Guo, C.; Sun, Y.; et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 2013, 153, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Drongitis, D.; Caterino, M.; Verrillo, L.; Santonicola, P.; Costanzo, M.; Poeta, L.; Attianese, B.; Barra, A.; Terrone, G.; Lioi, M.B.; et al. Deregulation of microtubule organization and RNA metabolism in Arx models for Lissencephaly and developmental epileptic encephalopathy. Hum. Mol. Genet. 2022, ddac028. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poeta, L.; Malacarne, M.; Padula, A.; Drongitis, D.; Verrillo, L.; Lioi, M.B.; Chiariello, A.M.; Bianco, S.; Nicodemi, M.; Piccione, M.; et al. Further Delineation of Duplications of ARX Locus Detected in Male Patients with Varying Degrees of Intellectual Disability. Int. J. Mol. Sci. 2022, 23, 3084. https://doi.org/10.3390/ijms23063084
Poeta L, Malacarne M, Padula A, Drongitis D, Verrillo L, Lioi MB, Chiariello AM, Bianco S, Nicodemi M, Piccione M, et al. Further Delineation of Duplications of ARX Locus Detected in Male Patients with Varying Degrees of Intellectual Disability. International Journal of Molecular Sciences. 2022; 23(6):3084. https://doi.org/10.3390/ijms23063084
Chicago/Turabian StylePoeta, Loredana, Michela Malacarne, Agnese Padula, Denise Drongitis, Lucia Verrillo, Maria Brigida Lioi, Andrea M. Chiariello, Simona Bianco, Mario Nicodemi, Maria Piccione, and et al. 2022. "Further Delineation of Duplications of ARX Locus Detected in Male Patients with Varying Degrees of Intellectual Disability" International Journal of Molecular Sciences 23, no. 6: 3084. https://doi.org/10.3390/ijms23063084
APA StylePoeta, L., Malacarne, M., Padula, A., Drongitis, D., Verrillo, L., Lioi, M. B., Chiariello, A. M., Bianco, S., Nicodemi, M., Piccione, M., Salzano, E., Coviello, D., & Miano, M. G. (2022). Further Delineation of Duplications of ARX Locus Detected in Male Patients with Varying Degrees of Intellectual Disability. International Journal of Molecular Sciences, 23(6), 3084. https://doi.org/10.3390/ijms23063084