
Glucose Metabolism and Aging of Hematopoietic Stem and Progenitor Cells

 , , and
, , and
Abstract
:1. Introduction
2. Aging and Senescence
3. Lessons Learned from Murine Models of Aging
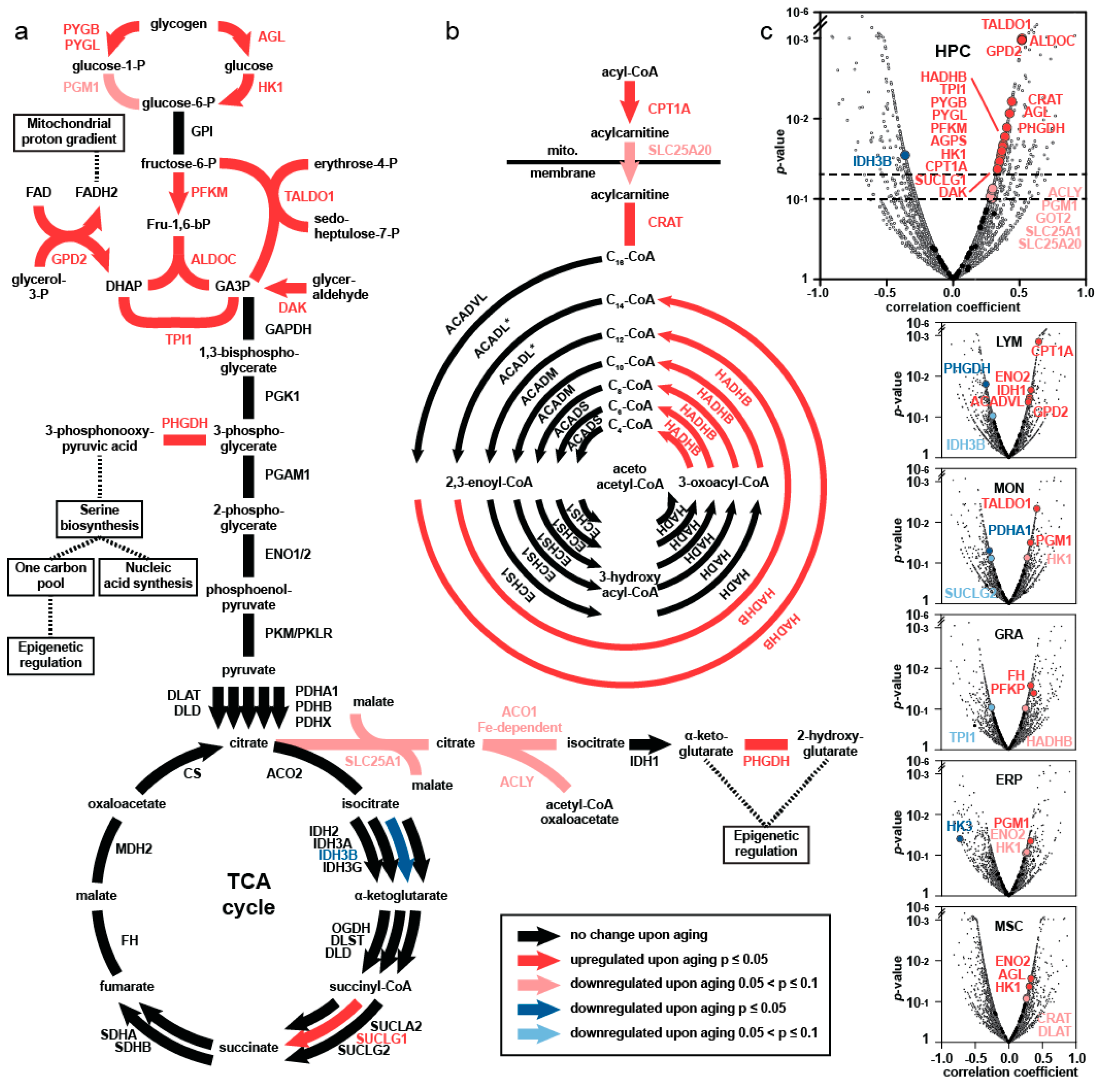
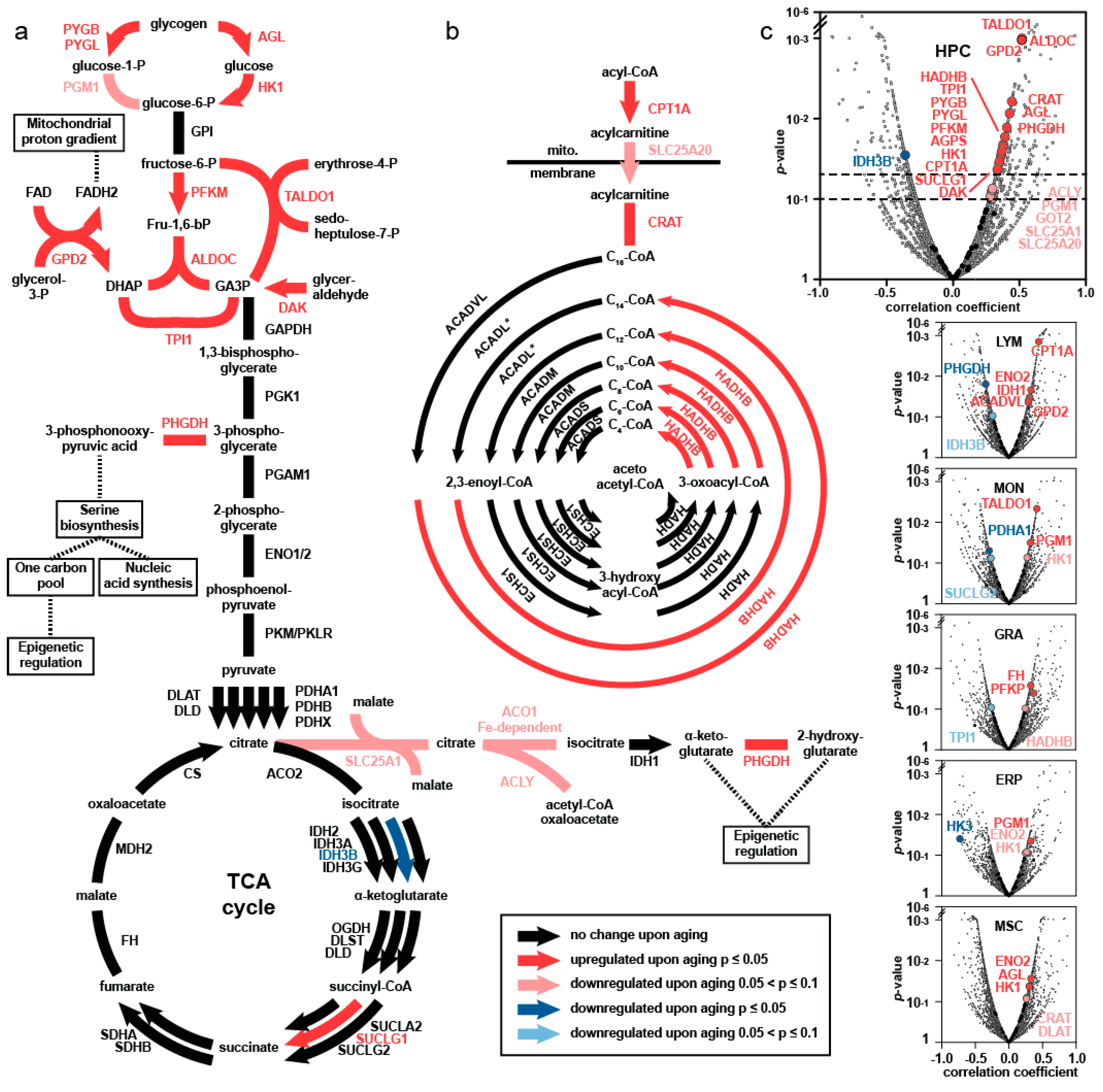
4. Comprehensive Proteomics Approach—Major Findings
5. Elevated Central Carbon Metabolism in HSPC upon Aging
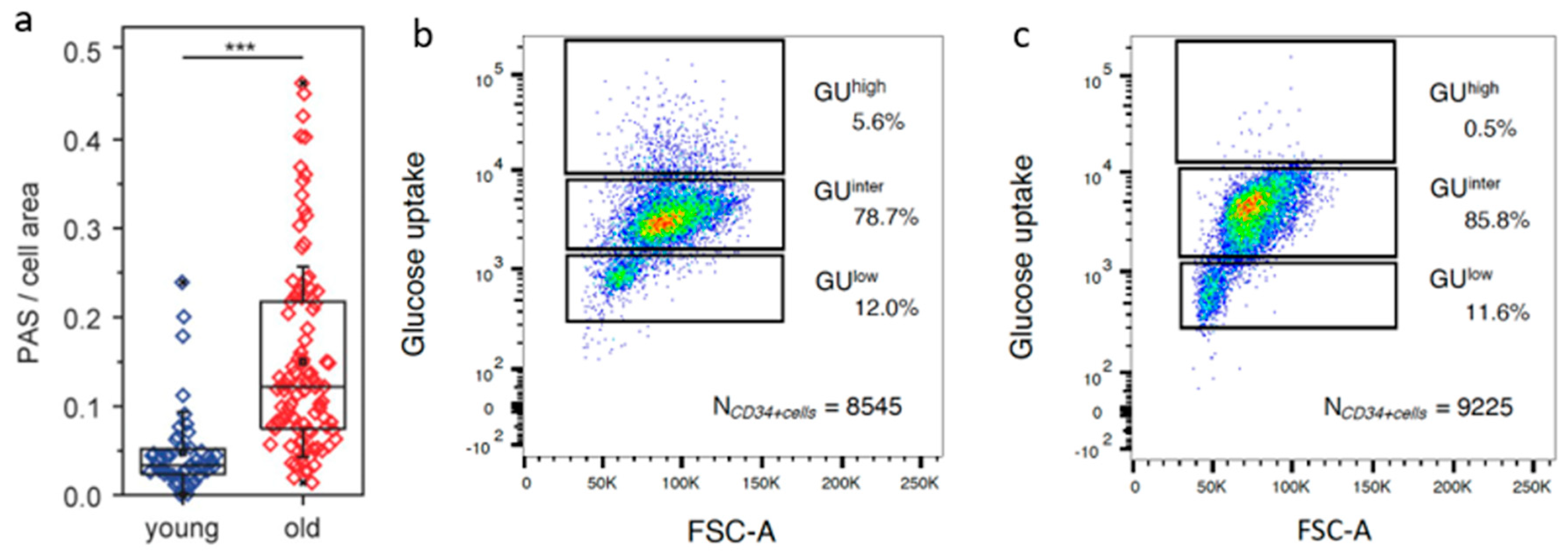
6. Separation of HSPC According to Carbon Metabolic Levels
7. Single-Cell Transcriptome Studies of HSPC with Different Levels of Carbon Metabolism
7.1. DNA Damage, Telomere Attrition, and Cell Cycle Arrest
7.2. Senescence-Associated Secretory Phenotype (SASP)
7.3. TP53, Apoptosis, and Pro-Survival Pathways
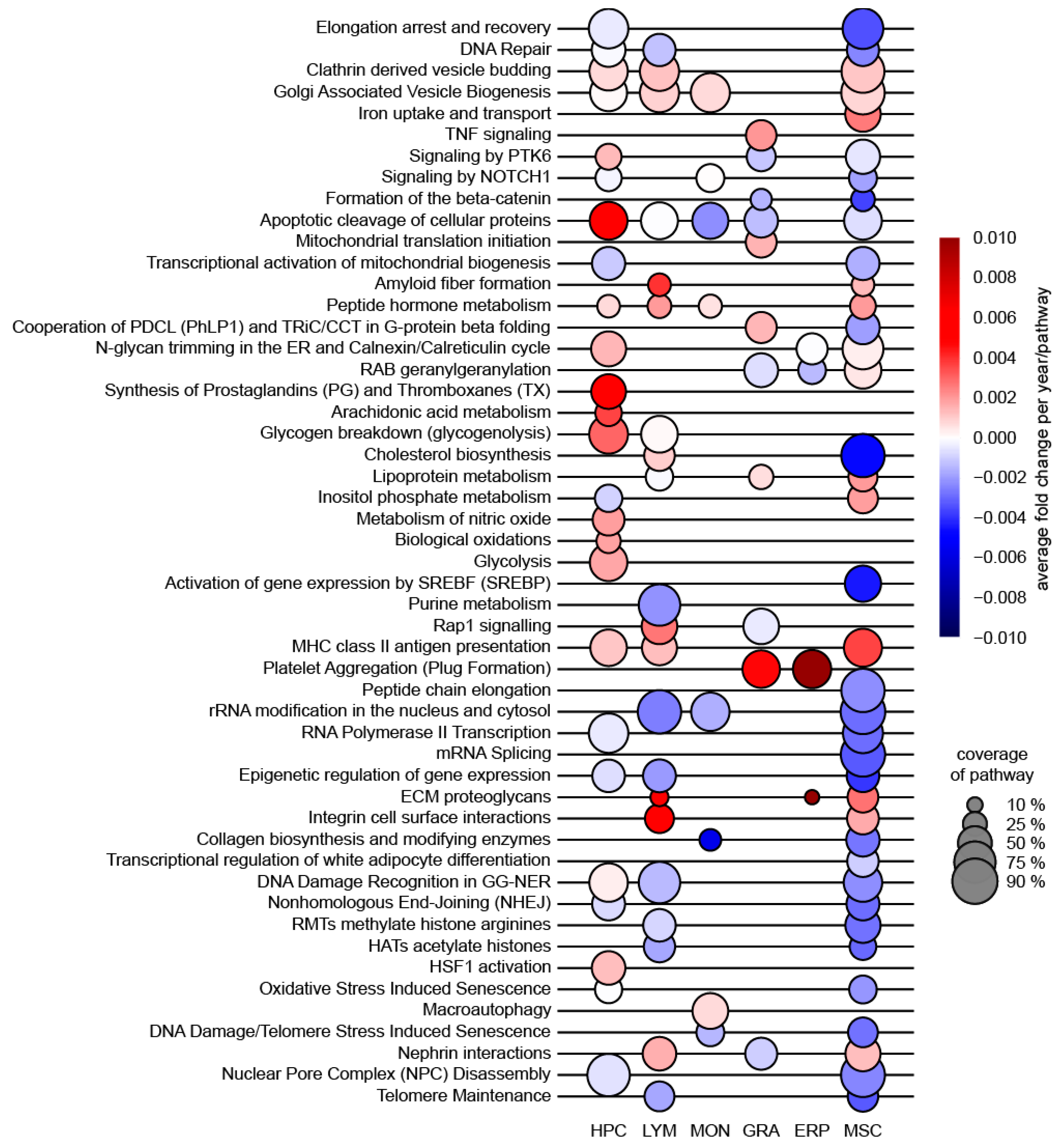
8. Gene Set Enrichment Analysis (GSEA)
9. GSEA Using “Aging Signature Gene Sets” as Reference
10. The Causative Role of Senescent Cells in Aging and Their Elimination
11. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iscove, N.N.; Nawa, K. Hematopoietic stem cells expand during serial transplantation in vivo without apparent exhaustion. Curr. Biol. 1997, 7, 805–808. [Google Scholar] [CrossRef] [Green Version]
- Schlessinger, D.; Van Zant, G. Does functional depletion of stem cells drive aging? Mech. Ageing Dev. 2001, 122, 1537–1553. [Google Scholar] [CrossRef]
- Ho, A.D.; Wagner, W.; Mahlknecht, U. Stem cells and ageing. EMBO Rep. 2005, 6, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Wandycz, A.M.; Akashi, K.; Globerson, A.; Weissman, I.L. The aging of hematopoietic stem cells. Nat. Med. 1996, 2, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- De Haan, G.; Nijhof, W.; Van Zant, G. Mouse strain-dependent changes in frequency and proliferation of hematopoietic stem cells during aging: Correlation between lifespan and cycling activity. Blood 1997, 89, 1543–1550. [Google Scholar] [CrossRef] [Green Version]
- Offner, F.; Kerre, T.; de Smedt, M.; Plum, J. Bone marrow CD34+cells generate fewer T cells in vitro with increasing age and following chemotherapy. Br. J. Haematol. 1999, 104, 801–808. [Google Scholar] [CrossRef]
- Sudo, K.; Ema, H.; Morita, Y.; Nakauchi, H. Age-associated characteristics of murine hematopoietic stem cells. J. Exp. Med. 2000, 192, 1273–1280. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Van Zant, G.; Szilvassy, S.J. Effects of aging on the homing and engraftment of murine hematopoietic stem and progenitor cells. Blood 2005, 106, 1479–1487. [Google Scholar] [CrossRef] [Green Version]
- Dykstra, B.; Olthof, S.; Schreuder, J.; Ritsema, M.; de Haan, G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J. Exp. Med. 2011, 208, 2691–2703. [Google Scholar] [CrossRef] [Green Version]
- Geiger, H.; de Haan, G.; Florian, M.C. The ageing haematopoietic stem cell compartment. Nat. Rev. Immunol. 2013, 13, 376–389. [Google Scholar] [CrossRef]
- Matteini, F.; Mulaw, M.A.; Florian, M.C. Aging of the hematopoietic stem cell niche: New tools to answer an old question. Front. Immunol. 2021, 12, 738204. [Google Scholar] [CrossRef] [PubMed]
- Gnani, D.; Crippa, S.; Della Volpe, L.; Rossella, V.; Conti, A.; Lettera, E.; Rivis, S.; Ometti, M.; Fraschini, G.; Bernardo, M.E.; et al. An early-senescence state in aged mesenchymal stromal cells contributes to hematopoietic stem and progenitor cell clonogenic impairment through the activation of a pro-inflammatory program. Aging Cell 2019, 18, e12933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oda, A.; Tezuka, T.; Ueno, Y.; Hosoda, S.; Amemiya, Y.; Notsu, C.; Kasahara, T.; Nishiyama, C.; Goitsuka, R. Niche-induced extramedullary hematopoiesis in the spleen is regulated by the transcription factor Tlx1. Sci. Rep. 2018, 8, 8308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottschling, S.; Saffrich, R.; Seckinger, A.; Krause, U.; Horsch, K.; Miesala, K.; Ho, A.D. Human mesenchymal stromal cells regulate initial self-renewing divisions of hematopoietic progenitor cells by a beta1-integrin-dependent mechanism. Stem Cells 2007, 25, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W.; Horn, P.; Bork, S.; Ho, A.D. Aging of hematopoietic stem cells is regulated by the stem cell niche. Exp. Gerontol. 2008, 43, 974–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bork, S.; Pfister, S.; Witt, H.; Horn, P.; Korn, B.; Ho, A.D.; Wagner, W. DNA methylation pattern changes upon long-term culture and aging of human mesenchymal stromal cells. Aging Cell. 2010, 9, 54–63. [Google Scholar] [CrossRef] [Green Version]
- Ambrosi, T.H.; Marecic, O.; McArdle, A.; Sinha, R.; Gulati, G.S.; Tong, X.; Wang, Y.; Steininger, H.M.; Hoover, M.Y.; Koepke, L.S.; et al. Aged skeletal stem cells generate an inflammatory degenerative niche. Nature 2021, 597, 256–262. [Google Scholar] [CrossRef]
- Ambrosi, T.H.; Chan, C. Skeletal Stem Cells as the Developmental Origin of Cellular Niches for Hematopoietic Stem and Progenitor Cells. Curr. Top. Microbiol. Immunol. 2021, 434, 1–31. [Google Scholar]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Svendsen, A.F.; Yang, D.; Kim, K.; Lazare, S.; Skinder, N.; Zwart, E.; Mura-Meszaros, A.; Ausema, A.; von Eyss, B.; de Haan, G.; et al. A comprehensive transcriptome signature of murine hematopoietic stem cell aging. Blood 2021, 138, 439–451. [Google Scholar] [CrossRef]
- Rossi, D.J.; Bryder, D.; Zahn, J.M.; Ahlenius, H.; Sonu, R.; Wagers, A.J.; Weissman, I.L. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc. Natl. Acad. Sci. USA 2005, 102, 9194–9199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, W.W.; Price, E.A.; Schoo, D.; Beerman, I.; Maloney, W.J.; Rossi, D.J.; Schrier, S.L.; Weissmann, I.L. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc. Natl. Acad. Sci. USA 2011, 108, 20012–20017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennrich, M.L.; Romanov, N.; Horn, P.; Jaeger, S.; Eckstein, V.; Steeples, V.; Ye, F.; Ding, X.; Poisa-Beiro, L.; Lai, M.C.; et al. Cell-specific proteome analyses of human bone marrow reveal molecular features of age-dependent functional decline. Nat. Comm. 2018, 9, 4004. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a–positive senescent cells delay ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkland, J.L. Translating advances from the basic biology of aging into clinical application. Exp. Gerontol. 2013, 48, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [Green Version]
- Deursen, J.M. The role of senescent cells in aging. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular senescence: Defining a path forward. Cell 2019, 179, 814–827. [Google Scholar] [CrossRef]
- Roger, L.; Tomas, F.; Gire, V. Mechanisms and regulation of cellular senescence. Inter. J. Mol. Sci. 2021, 22, 13173. [Google Scholar] [CrossRef]
- Vaziri, H.; Dragowska, W.; Allsopp, R.C.; Thomas, T.E.; Harley, C.B.; Lansdorp, P.M. Evidence for a mitotic clock in human hematopoietic stem cells: Loss of telomeric DNA with age. Proc. Natl. Acad. Sci. USA 1994, 91, 9857–9860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, D.J.; Bryder, D.; Seita, J.; Nussenzweig, A.; Hoeijmakers, J.; Weissman, I.L. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 2007, 447, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casella, G.; Munk, R.; Kim, K.M.; Piao, Y.; De, S.; Abdelmohsen, K.; Gorospe, M. Transcriptome signature of cellular senescence. Nucleic Acid Res. 2019, 47, 7294–7305. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J.; Kapathi, P.; Lithgow, G.J.; Melow, S.; Newman, J.C.; Verdin, E. From discoveries in ageing research to therapeutics for healthy ageing. Nature 2019, 571, 183–192. [Google Scholar] [CrossRef] [Green Version]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Kollman, C.; Howe, C.W.S.; Anasetti, C.; Antin, J.H.; Davies, S.M.; Filipovich, A.H.; Hegland, J.; Kamani, N.; Kernan, N.A.; King, R.; et al. Donor characteristics as risk factors in recipients after transplantation of bone marrow from unrelated donors: The effect of donor age. Blood 2001, 98, 2043–2051. [Google Scholar] [CrossRef]
- Grover, A.; Sanjuan-Pla, A.; Thongjuea, S.; Carrelha, J.; Giustacchini, A.; Gambardella, A.; Macaulay, I.; Mancini, E.; Luis, T.C.; Mead, A.; et al. Single-cell RNA sequencing reveals molecular and functional platelet bias of aged haematopoietic stem cells. Nat. Commun. 2016, 7, 11075. [Google Scholar] [CrossRef]
- Noda, S.; Ichikawa, H.; Miyoshi, H. Hematopoietic stem cell aging is associated with functional decline and delayed cell cycle progression. Biochem. Biophys. Res. Commun. 2009, 383, 210–215. [Google Scholar] [CrossRef]
- Kowalczyk, M.S.; Tirosh, I.; Heckl, D.; Rao, T.N.; Dixit, A.; Haas, B.J.; Schneider, R.K.; Wagers, A.J.; Ebert, B.L.; Regev, A. Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res. 2015, 25, 1860–1872. [Google Scholar] [CrossRef] [Green Version]
- Kirschner, K.; Chandra, T.; Kiselev, V.; Flores-Santa Cruz, D.; Macaulay, I.C.; Park, H.J.; Li, J.; Kent, D.G.; Kumar, R.; Pask, D.C.; et al. Proliferation drives aging-related functional decline in a subpopulation of the hematopoietic stem cell compartment. Cell Rep. 2017, 19, 1503–1511. [Google Scholar] [CrossRef] [Green Version]
- Chambers, S.M.; Shaw, C.A.; Gatza, C.; Fisk, C.J.; Donehower, L.A.; Goodell, M.A. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007, 5, e201. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Mehta, A.; de Boer, C.G.; Kowalczyk, M.S.; Lee, K.; Farouq, D.; Regev, A.; Baltimore, D. Heterogeneous responses of hematopoietic stem cells to inflammatory stimuli are altered with age. Cell Rep. 2018, 25, 2992–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flach, J.; Bakker, S.T.; Mohrin, M.; Conroy, P.C.; Pietras, E.M.; Reynaud, D.; Alvarez, S.; Diolaiti, M.E.; Ugarte, F.; Forsberg, E.C.; et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 2014, 512, 198–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerman, I.; Bock, C.; Garrison, B.S.; Smith, Z.D.; Gu, H.; Meissner, A.; Rossi, D.J. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell 2013, 12, 413–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerman, I.; Rossi, D.J. Epigenetic control of stem cell potential during homeostasis, aging, and disease. Cell Stem Cell 2015, 16, 613–625. [Google Scholar] [CrossRef] [Green Version]
- Wahlestedt, M.; Norddahl, G.L.; Sten, G.; Ugale, A.; Frisk, M.M.; Mattsson, R.; Deierborg, T.; Sigvardsson, M.; Bryder, D. An epigenetic component of hematopoietic stem cell aging amenable to reprogramming into a young state. Blood 2013, 121, 4257–4264. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Luo, M.; Jeong, M.; Rodriguez, B.; Xia, Z.; Hannah, R.; Wang, H.; Le, T.; Faull, K.F.; Chen, R.; et al. Epigenomic Profiling of Young and Aged HSCs Reveals Concerted Changes during Aging that Reinforce Self-Renewal. Cell Stem Cell 2014, 14, 673–688. [Google Scholar] [CrossRef] [Green Version]
- Poisa-Beiro, L.; Landry, T.J.; Sauer, S.; Yamamoto, A.; Eckstein, V.; Romanov, N.; Raffel, S.; Hoffmann, G.F.; Bork, P.; Benes, V.; et al. Glycogen accumulation, central carbon metabolism, and aging of hematopoietic stem and progenitor cells. Sci. Rep. 2020, 10, 11597. [Google Scholar] [CrossRef]
- Shyh-Chang, N.; Daley, G.Q.; Cantley, L.C. Stem cell metabolism in tissue development and aging. Development 2013, 140, 2535–2547. [Google Scholar] [CrossRef] [Green Version]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O.H. Über den Stoffwechsel der Carcinomzelle. Wien. Klein. Wochenschr. 1925, 4, 534–536. [Google Scholar] [CrossRef]
- Smith, B.; Schafer, X.L.; Ambeskovic, A.; Spencer, C.M.; Land, H.; Munger, J. Addiction to coupling of the Warburg effect with glutamine catabolism in cancer cells. Cell Rep. 2016, 17, 821–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poisa-Beiro, L.; Landry, J.; Rafffel, S.; Eckstein, V.; Gavin, A.C.; Tanaka, M.; Benes, V.; Ho, A.D. Elevated central carbon metabolism–a hallmark for senescent cells in aging human hematopoietic stem cell compartment. Blood 2021, 138, 1088. [Google Scholar] [CrossRef]
- Denchi, E.L.; de Lange, T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 2007, 448, 1068–1071. [Google Scholar] [CrossRef]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [Green Version]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F. Living on a break: Cellular senescence as a DNA damage response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA damage response activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Baar, M.P.; Brandt, R.M.; Putavet, D.A.; Klein, J.D.; Derks, K.W.; Bourgeois, B.R.; Stryeck, S.; Rijksen, Y.; Willigenburg, H.V.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147. [Google Scholar] [CrossRef] [Green Version]
- Ben-Porath, I.; Weinberg, R.A. The signals and pathways activating cellular senescence. Int. J. Biochem. Cell Biol. 2005, 37, 961–976. [Google Scholar] [CrossRef] [PubMed]
- Deursen, J.M. Senolytic therapies for healthy longevity. Science 2019, 364, 636–637. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 958–961. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y.; Hoare, M.; Narita, M. Spatial and temporal control of senescence. Trends Cell Biol. 2017, 27, 820–832. [Google Scholar] [CrossRef] [Green Version]
- Ghosal, G.; Chen, J. DNA damage tolerance: A double-edged sword guarding the genome. Transl. Cancer Res. 2013, 2, 107–129. [Google Scholar]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Kirkland, J.L. The Achilles‘heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Kirkland, J.L. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Liu, Y.; Liu, Y.; Zheng, P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci. Signal 2009, 2, ra75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogenious mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbig, U.; Ferreira, M.; Condel, L.; Carey, D.; Sedivy, J.M. Cellular senescence in aging primates. Science 2006, 311, 1257. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.P.; Kauser, K.; Campisi, J.; Beauséjour, C.M. Secretion ofvascular endothelial growth factor by primary human fibroblasts atsenescence. J. Biol. Chem. 2006, 281, 29568–29574. [Google Scholar] [CrossRef] [Green Version]
- Kirkland, J.L.; Tchkonia, T. Clinical strategies and animal models for developing senolytic agents. Exp. Gerontol. 2014, 14, 291–295. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Wang, J.; Shao, L.; Laberge, R.M.; Zhuo, D. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budamagunta, V.; Foster, T.C.; Zhou, D. Cellular senescence in lymphoid organs and immunosenescence. Aging 2021, 13, 19920–19941. [Google Scholar] [CrossRef] [PubMed]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Odavya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Comm. 2016, 10, 11190. [Google Scholar] [CrossRef]
- Nakamura-Ishizu, A.; Ito, K.; Suda, T. Hematopoietic stem cell metabolism during development and aging. Dev. Cell 2020, 54, 239–255. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAP KAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laberge, R.-M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.J.; Madrigal-Matute, J.; Scheibye-Knudsen, M.; Fang, E.; Aon, M.; González-Reyes, J.A.; Cortassa, S.; Kaushik, S.; Gonzalez-Freire, M.; Patel, B. Effects of Sex, Strain, and Energy Intake on Hallmarks of Aging in Mice. Cell Metab. 2016, 23, 1093–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| CD34+ Subsets | Young (n = 3) in % | Old (n = 7) in % | p |
|---|---|---|---|
| GUhigh | 1.7 ± 1.5 | 5.4 ± 3.5 | 0.02 |
| GUinter | 66.5 ± 36.9 | 66.4 ± 22.5 | ns |
| GUlow | 31.8 ± 36.7 | 28.2 ± 21.7 | ns |
| Gene Symbols | GUhigh vs. GUlow p Values | Old vs. Young p Values |
|---|---|---|
| (a) Cell cycle arrest | ||
| TERF2 | *** | 0.073 |
| POT1 | 0.082 | * |
| CHEK1 | *** | 0.633 |
| CHEK2 | *** | 0.717 |
| TP53 | * | * |
| CDKN1A (P21 CIP1) | *** | 0.498 |
| CDKN2A (P15 INK4A) | 0.244 | 0.103 |
| RB1 | *** | 0.193 |
| (b) Senescence associated secretory phenotype | ||
| NOTCH1 | *** | 0.530 |
| CEBPA | *** | ** |
| CEBPB | *** | 0.281 |
| NFKB1 | * | 0.451 |
| NFKB2 | * | 0.330 |
| IL1B | *** | * |
| IL1R1 | 0.081 | 0.101 |
| IL1R2 | ** | na |
| (c) Apoptosis and pro-survival | ||
| BCL2 | *** | 0.0899 |
| BCL2L1 | *** | 0.681 |
| BCL3 | *** | 0.18 |
| MCL1 | *** | * |
| GS TITLE (HALLMARK ANALYSIS) | SIZE | ES | NES | NOM p-val | FDR q-val | RANK AT MAX | LEADING EDGE | |
|---|---|---|---|---|---|---|---|---|
| a | G2M CHECKPOINT | 198 | 0.59 | 2.39 | 0.000 | 0.000 | 2893 | tags = 49%, list = 13%, signal = 56% |
| b | MTORC1 SIGNALING | 198 | 0.43 | 1.92 | 0.000 | 0.003 | 4682 | tags = 45%, list = 22%, signal = 57% |
| c | INFLAMMA-TORY RESPONSE | 172 | 0.40 | 1.75 | 0.008 | 0.010 | 5872 | tags = 47%, list = 27%, signal = 64% |
| d | APOPTOSIS | 152 | 0.34 | 1.55 | 0.014 | 0.030 | 4055 | tags = 30%, ist = 19%, signal = 37% |
| e | SVENDSEN et al. (2021) | 176 | 0.46 | 2.03 | 0.000 | 0.000 | 4453 | tags = 49%, list = 13%, signal = 56% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poisa-Beiro, L.; Landry, J.J.M.; Raffel, S.; Tanaka, M.; Zaugg, J.; Gavin, A.-C.; Ho, A.D. Glucose Metabolism and Aging of Hematopoietic Stem and Progenitor Cells. Int. J. Mol. Sci. 2022, 23, 3028. https://doi.org/10.3390/ijms23063028
Poisa-Beiro L, Landry JJM, Raffel S, Tanaka M, Zaugg J, Gavin A-C, Ho AD. Glucose Metabolism and Aging of Hematopoietic Stem and Progenitor Cells. International Journal of Molecular Sciences. 2022; 23(6):3028. https://doi.org/10.3390/ijms23063028
Chicago/Turabian StylePoisa-Beiro, Laura, Jonathan J. M. Landry, Simon Raffel, Motomu Tanaka, Judith Zaugg, Anne-Claude Gavin, and Anthony D. Ho. 2022. "Glucose Metabolism and Aging of Hematopoietic Stem and Progenitor Cells" International Journal of Molecular Sciences 23, no. 6: 3028. https://doi.org/10.3390/ijms23063028