
Fragments of rDNA Genes Scattered over the Human Genome Are Targets of Small RNAs

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
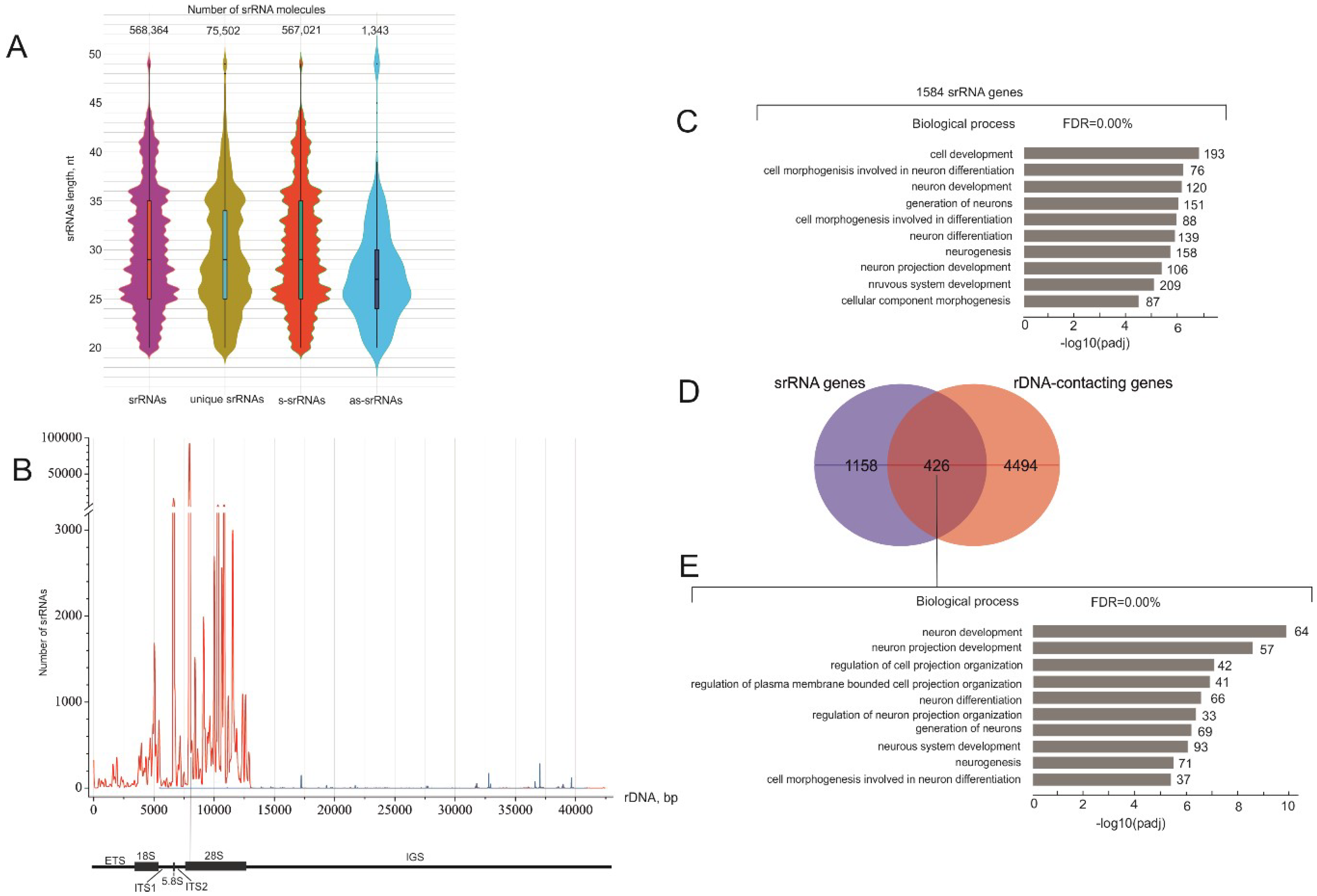
2.1. There Are Thousands of Unique srRNAs
2.2. Detection of srRNA Target Genes
2.3. Abundant srRNAs in the UNC45B Gene
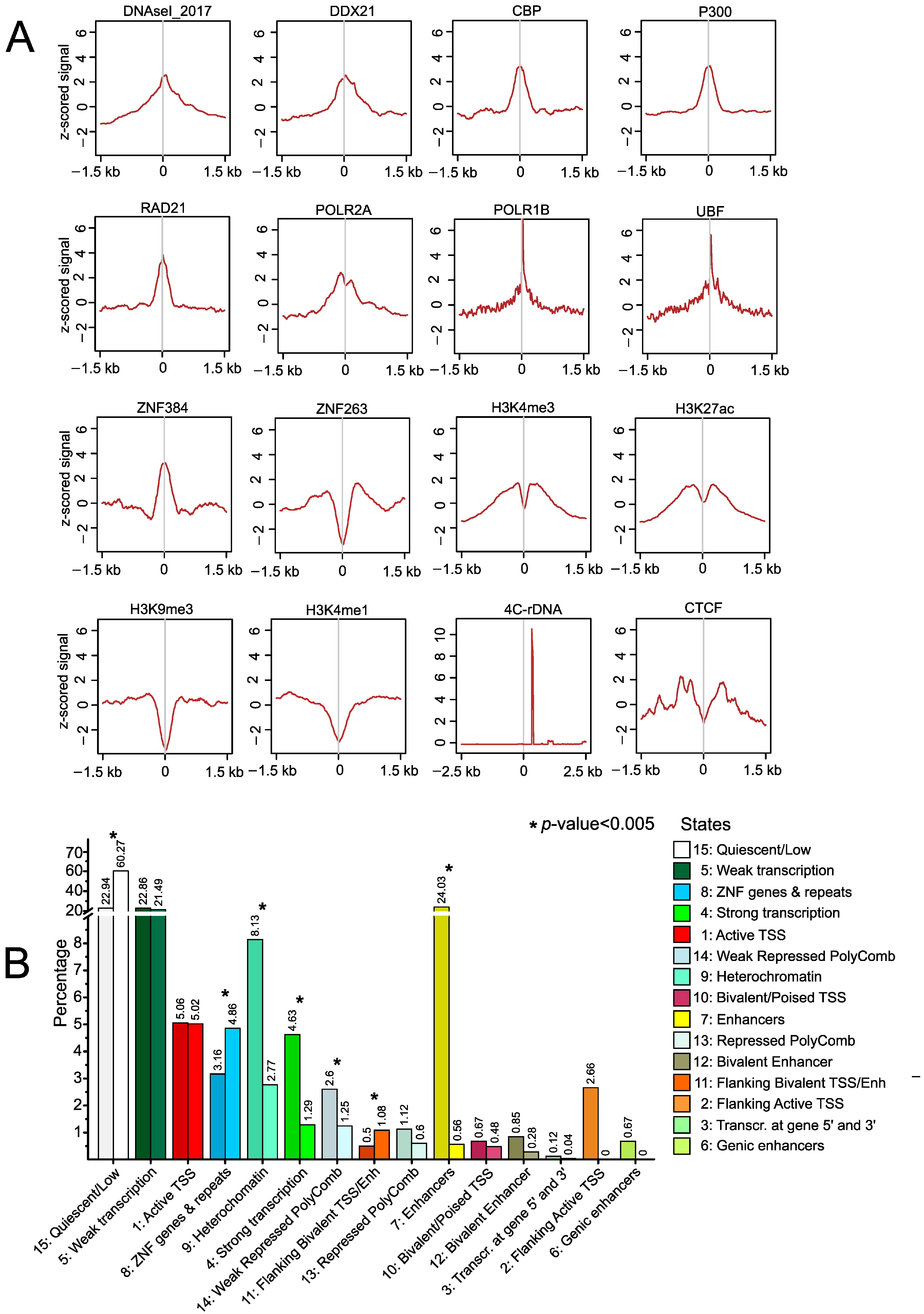
2.4. Epigenetic Features at srRNA Targets
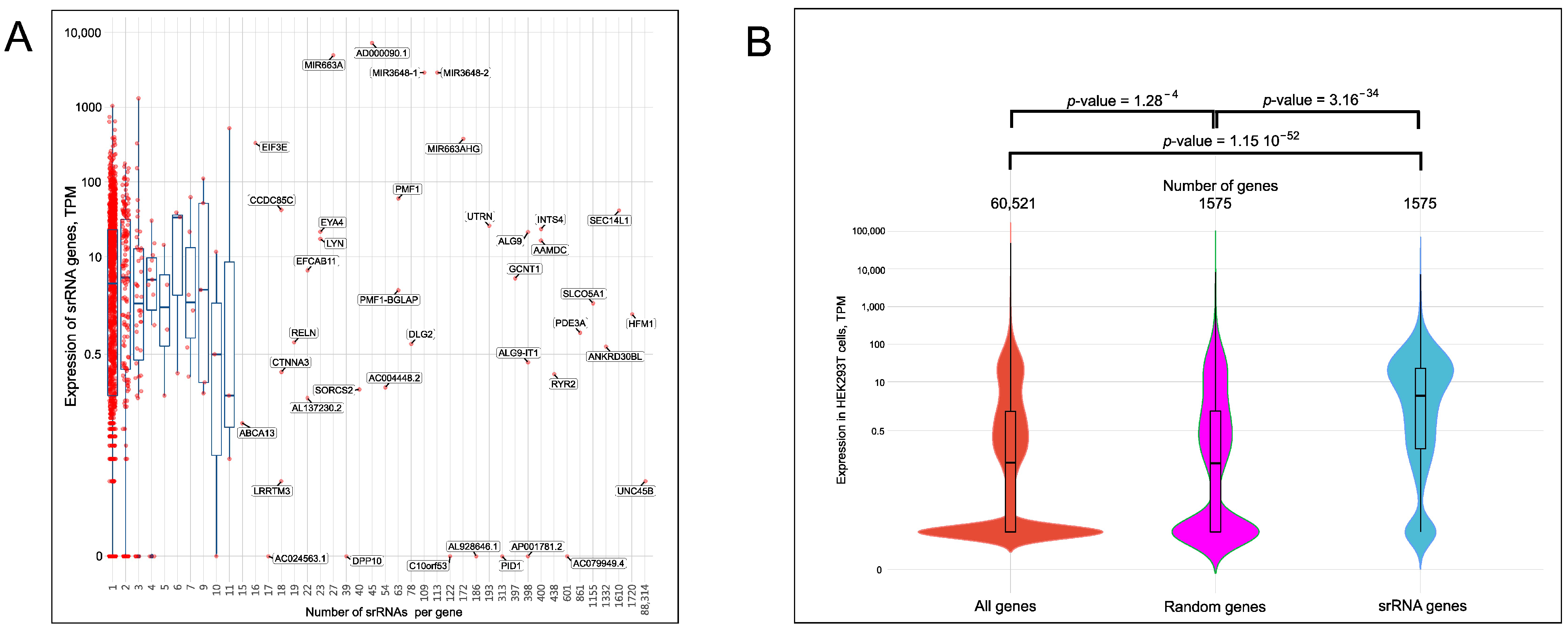
2.5. Expression of srRNA Target Genes
3. Discussion
4. Materials and Methods
4.1. Isolation of srRNAs
4.2. Genome-Wide Profiles
4.3. Epigenome Statistics
4.4. RNA-Seq Analysis
4.5. Transcription Start-Site Analysis
4.6. Permutation Analysis
4.7. Analysis of the Distribution of Genes versus Their Expression Levels by Violin Plots
- Test with subsets of the same size—Two gene subsets of equal size were created, and the gene expression values were shuffled randomly using Durstenfeld’s version of the Fisher–Yates shuffling algorithm [45]. Appropriate amounts of values were selected from the shuffled lists. The Mann–Whitney U-test was applied to test if these two randomly selected subsets originated from the same distribution. The procedure was performed 10,000 times. FDR = 0.0483 for subsets containing 1575 genes.
- Test with the full gene set and a smaller subset—A subset was created by randomly shuffling all gene expression values by Durstenfeld’s version of the Fisher–Yates shuffling algorithm and then selecting 1575 appropriate values from the shuffled list. The Mann–Whitney U-test was applied to test if the full expression set and created subset originate from the same distribution. The procedure was performed 100,000 times. FDR = 0.0453 for the subset with 1575 genes.
4.8. Availability of Data Sources and Applied Scripts
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, J.T. Lessons from X-chromosome inactivation: Long ncRNA as guides and tethers to the epigenome. Genes Dev. 2009, 23, 1831–1842. [Google Scholar] [CrossRef] [PubMed]
- Kapranov, P.; Cheng, P.J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermüller, J.; Hofacker, I.L.; et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.A.; Aravin, A.A. Non-Coding RNAs in Transcriptional Regulation. Curr. Mol. Biol. Rep. 2015, 1, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed]
- Ozata, D.M.; Gainetdinov, I.; Zoch, A.; Zamore, P.D. PIWI-interacting RNAs: Small RNAs with big functions. Nat. Rev. Genet. 2019, 20, 89–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lin, H. Roles of piRNAs in transposon and pseudogene regulation of germline mRNAs and lncRNAs. Genome Biol. 2021, 22, 27. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Zhou, B.; Zhang, F.; Tu, Y.; Hu, Y.; Zhang, B.; Zhai, Q. Profiling and identification of small rDNA-derived RNAs and their potential biological functions. PLoS ONE 2013, 8, e56842. [Google Scholar] [CrossRef]
- Lambert, M.; Benmoussa, A.; Provost, P. Small non-coding RNAs derived from eukaryotic ribosomal RNA. Noncoding RNA 2019, 5, 16. [Google Scholar] [CrossRef]
- Li, S. Human 28s rRNA 5’ terminal derived small RNA inhibits ribosomal protein mRNA levels. bioRxiv 2019, 618520. [Google Scholar] [CrossRef]
- Tchurikov, N.A.; Fedoseeva, D.M.; Klushevskaya, E.S.; Slovohotov, I.Y.; Chechetkin, V.R.; Kravatsky, Y.V.; Kretova, O.V. rDNA Clusters make contact with genes that are involved in differentiation and cancer and change contacts after heat shock treatment. Cells 2019, 8, 1393. [Google Scholar] [CrossRef]
- Tchurikov, N.A.; Klushevskaya, E.S.; Fedoseeva, D.M.; Alembekov, I.R.; Kravatskaya, G.I.; Chechetkin, V.R.; Kravatsky, Y.V.; Kretova, O.V. Dynamics of whole-genome contacts of nucleoli in Drosophila cells suggests a role for rDNA genes in global epigenetic regulation. Cells 2020, 9, 2587. [Google Scholar] [CrossRef] [PubMed]
- Macias, S.; Plass, M.; Stajuda, A.; Michlewski, G.; Eyras, E.; Cáceres, J.F. DGCR8 HITS-CLIP reveals novel functions for the Microprocessor. Nat. Struct. Mol. Biol. 2012, 19, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Sun, Y.; Yang, X.; Wu, Z.; Guo, K.; Niu, X.; Wang, Q.; Ruan, J.; Bu, W.; Gao, S. Two featured series of rRNA-derived RNA fragments (rRFs) constitute a novel class of small RNAs. PLoS ONE 2017, 12, e0176458. [Google Scholar] [CrossRef] [PubMed]
- Shaw, G.; Morse, S.; Ararat, M.; Graham, F.L. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 2002, 16, 869–971. [Google Scholar] [CrossRef] [PubMed]
- Katoh, N.; Kuroda, K.; Tomikawa, J.; Ogata-Kawata, H.; Ozaki, R.; Ochiai, A.; Kitade, M.; Takeda, S.; Nakabayashi, K.; Hata, K. Reciprocal changes of H3K27ac and H3K27me3 at the promoter regions of the critical genes for endometrial decidualization. Epigenomics 2018, 10, 1243–1257. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.S.; Cattoglio, C.; Darzacq, X.; Tjian, R. Recent evidence that TADs and chromatin loops are dynamic structures. Nucleus 2018, 9, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.M.; Ernst, J.; Wilder, S.P.; Kundaje, A.; Harris, R.S.; Libbrecht, M.; Giardine, B.; Ellenbogen, P.M.; Bilmes, J.A.; Birney, E.; et al. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res. 2013, 41, 827–841. [Google Scholar] [CrossRef]
- Savic, N.; Bär, D.; Leone, S.; Frommel, S.C.; Weber, F.A.; Vollenweider, E.; Ferrari, E.; Ziegler, U.; Kaech, A.; Shakhova, O.; et al. lncRNA maturation to initiate heterochromatin formation in the nucleolus is required for exit from pluripotency in ESCs. Cell Stem Cell 2014, 15, 720–734. [Google Scholar] [CrossRef] [PubMed]
- Tchurikov, N.A.; Kretova, O.V.; Fedoseeva, D.M.; Sosin, D.V.; Grachev, S.A.; Serebraykova, M.V.; Romanenko, S.A.; Vorobieva, N.V.; Kravatsky, Y.V. DNA double-strand breaks coupled with PARP1 and HNRNPA2B1 binding sites flank coordinately expressed domains in human chromosomes. PLoS Genet. 2013, 9, e1003429. [Google Scholar] [CrossRef]
- Tchurikov, N.A.; Fedoseeva, D.M.; Sosin, D.V.; Snezhkina, A.V.; Melnikova, N.V.; Kudryavtseva, A.V.; Kravatsky, Y.V.; Kretova, O.V. Hot spots of DNA double-strand breaks and genomic contacts of human rDNA units are involved in epigenetic regulation. J. Mol. Cell. Biol. 2015, 7, 366–382. [Google Scholar] [CrossRef] [PubMed]
- Gaviraghi, M.; Vivori, C.; Tonon, G. How Cancer Exploits Ribosomal RNA Biogenesis: A Journey beyond the Boundaries of rRNA Transcription. Cells 2019, 8, 1098. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Shalaby, N.A.; Buszczak, M. Changes in rRNA transcription influence proliferation and cell fate within a stem cell lineage. Science 2014, 343, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Stepanenko, A.A.; Dmitrenko, V.V. HEK293 in cell biology and cancer research: Phenotype, karyotype, tumorigenicity, and stress-induced genome-phenotype evolution. Gene 2015, 569, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Kojima-Kita, K.; Kuramochi-Miyagawa, S.; Nagamori, I.; Ogonuki, N.; Ogura, A.; Hasuwa, H.; Akazawa, T.; Inoue, N.; Nakano, T. MIWI2 as an effector of DNA methylation and gene silencing in embryonic male germ cells. Cell Rep. 2016, 16, 2819–2828. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Bushnell, B. BBTools: A Suite of Fast, Multithreaded Bioinformatics Tools Designed for Analysis of DNA and RNA Sequence Data; Joint Genome Institute: Walnut Creek, CA, USA, 2018; Available online: https://sourceforge.net/projects/bbmap/ (accessed on 8 March 2022).
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Kent, W.J.; Zweig, A.S.; Barber, G.; Hinrichs, A.S.; Karolchik, D. BigWig and BigBed: Enabling browsing of large distributed datasets. Bioinformatics 2010, 26, 2204–2207. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Storer, J.; Hubley, R.; Rosen, J.; Wheeler, T.J.; Smit, A.F. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob. DNA 2021, 12, 2. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Johnson, N.; Juettemann, T.; Wilder, S.P.; Flicek, P. WiggleTools: Parallel processing of large collections of genome-wide datasets for visualization and statistical analysis. Bioinformatics 2014, 30, 1008–1009. [Google Scholar] [CrossRef] [PubMed]
- Stempor, P.; Ahringer, J. SeqPlots—Interactive software for exploratory data analyses, pattern discovery and visualization in genomics. Wellcome Open Res. 2016, 1, 14. [Google Scholar] [CrossRef] [PubMed]
- NIH Roadmap Epigenomics. 2021. Available online: https://egg2.wustl.edu/roadmap/web_portal (accessed on 8 March 2022).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. Feature counts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Fantom Consortium; The RIKEN PMI and CLST (DGT); Forrest, A.R.; Kawaji, H.; Rehli, M.; Baillie, J.K.; de Hoon, M.J.; Haberle, V.; Lassmann, T.; Kulakovskiy, I.V.; et al. A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Nishumura, T. Mersenne twister: A 623-dimensionally equidistributed uniform pseudo-random number generator. ACM Trans. Modeling Comput. Simul. 1998, 8, 3–30. [Google Scholar] [CrossRef]
- Durstenfeld, R. Algorithm 235: Random permutation. Commun. ACM 1964, 7, 420. [Google Scholar] [CrossRef]
- Kravatsky, Y.V.; Chechetkin, V.R.; Tchurikov, N.A.; Kravatskaya, G.I. Genome-wide study of correlations between genomic features and their relationship with the regulation of gene expression. DNA Res. 2015, 22, 109–119. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, H.M.; Yang, D.; Liu, Z.F.; Hu, S.Z.; Yan, S.H.; He, X.W. Density distribution of gene expression profiles and evaluation of using maximal information coefficient to identify differentially expressed genes. PLoS ONE 2019, 14, e0219551. [Google Scholar] [CrossRef] [PubMed]
- Stavrovskaya, E.D.; Niranjan, T.; Fertig, E.J.; Wheelan, S.J.; Favorov, A.V.; Mironov, A.A. StereoGene: Rapid estimation of genome-wide correlation of continuous or interval feature data. Bioinformatics 2017, 33, 3158–3165. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tchurikov, N.A.; Klushevskaya, E.S.; Alembekov, I.R.; Bukreeva, A.S.; Kretova, A.N.; Chechetkin, V.R.; Kravatskaya, G.I.; Kravatsky, Y.V. Fragments of rDNA Genes Scattered over the Human Genome Are Targets of Small RNAs. Int. J. Mol. Sci. 2022, 23, 3014. https://doi.org/10.3390/ijms23063014
Tchurikov NA, Klushevskaya ES, Alembekov IR, Bukreeva AS, Kretova AN, Chechetkin VR, Kravatskaya GI, Kravatsky YV. Fragments of rDNA Genes Scattered over the Human Genome Are Targets of Small RNAs. International Journal of Molecular Sciences. 2022; 23(6):3014. https://doi.org/10.3390/ijms23063014
Chicago/Turabian StyleTchurikov, Nickolai A., Elena S. Klushevskaya, Ildar R. Alembekov, Anastasiia S. Bukreeva, Antonina N. Kretova, Vladimir R. Chechetkin, Galina I. Kravatskaya, and Yuri V. Kravatsky. 2022. "Fragments of rDNA Genes Scattered over the Human Genome Are Targets of Small RNAs" International Journal of Molecular Sciences 23, no. 6: 3014. https://doi.org/10.3390/ijms23063014
APA StyleTchurikov, N. A., Klushevskaya, E. S., Alembekov, I. R., Bukreeva, A. S., Kretova, A. N., Chechetkin, V. R., Kravatskaya, G. I., & Kravatsky, Y. V. (2022). Fragments of rDNA Genes Scattered over the Human Genome Are Targets of Small RNAs. International Journal of Molecular Sciences, 23(6), 3014. https://doi.org/10.3390/ijms23063014