
A Novel CDK4/6 and PARP Dual Inhibitor ZC-22 Effectively Suppresses Tumor Growth and Improves the Response to Cisplatin Treatment in Breast and Ovarian Cancer

 , ,
, , 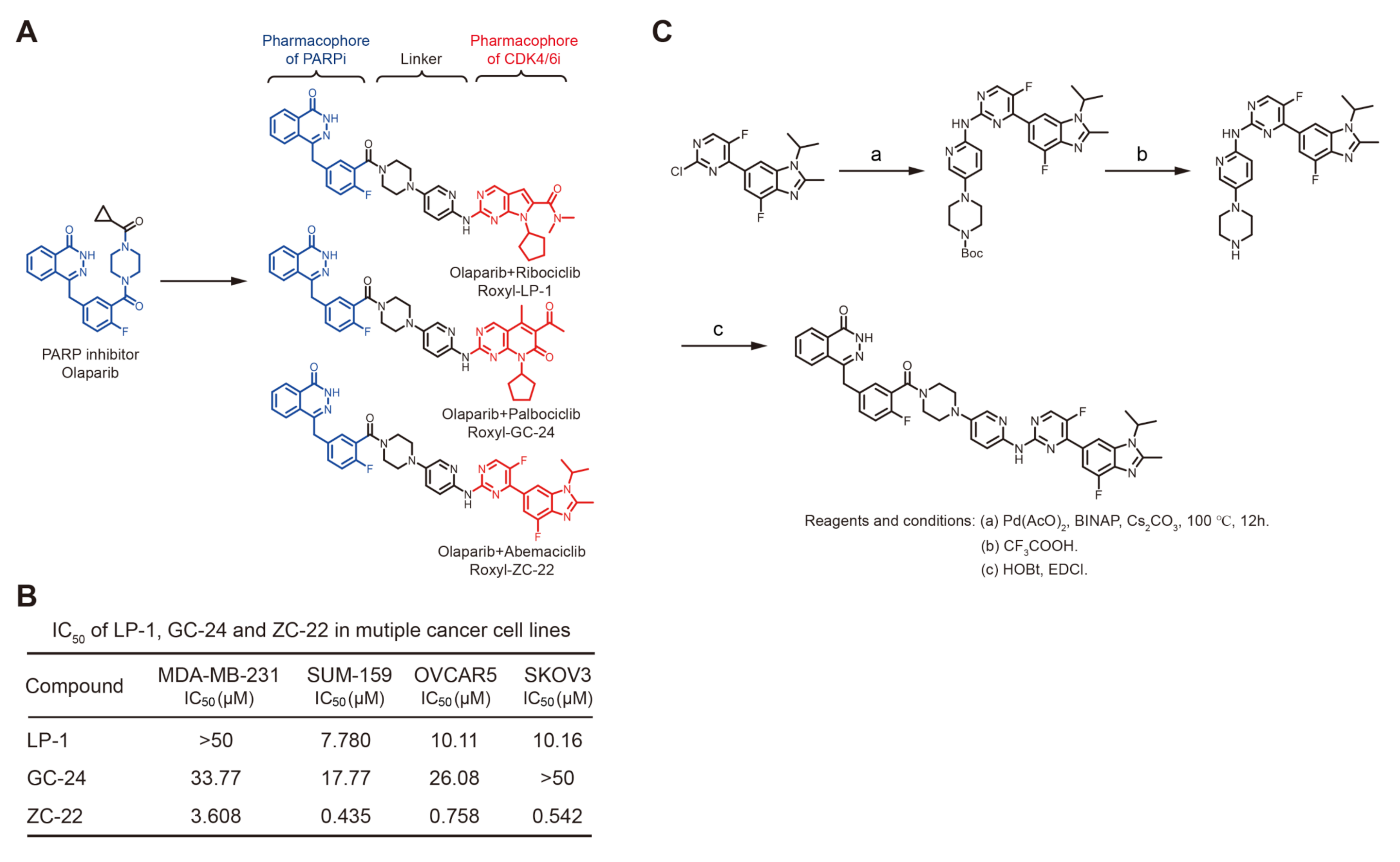{kind=link}
{kind=link}
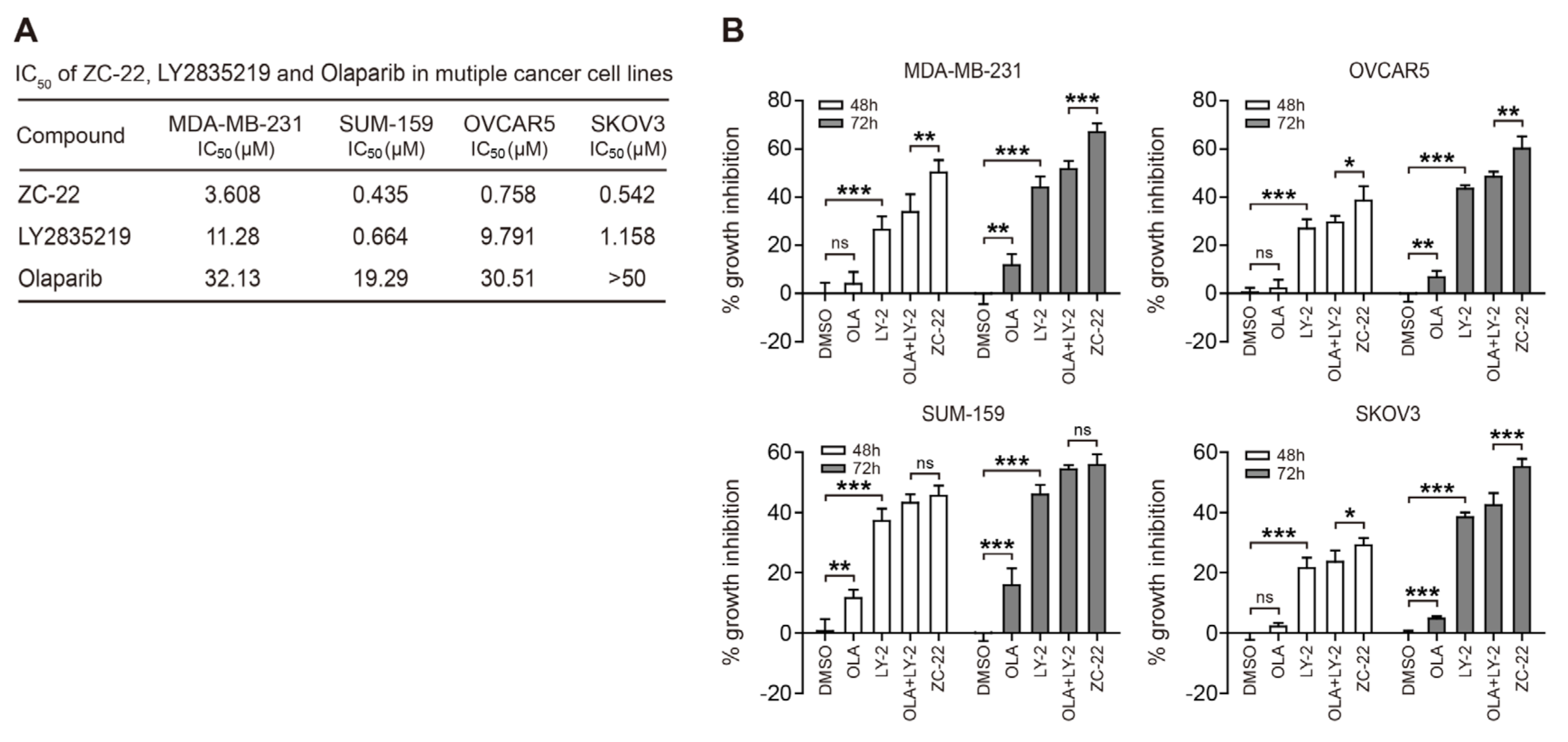
{kind=link}
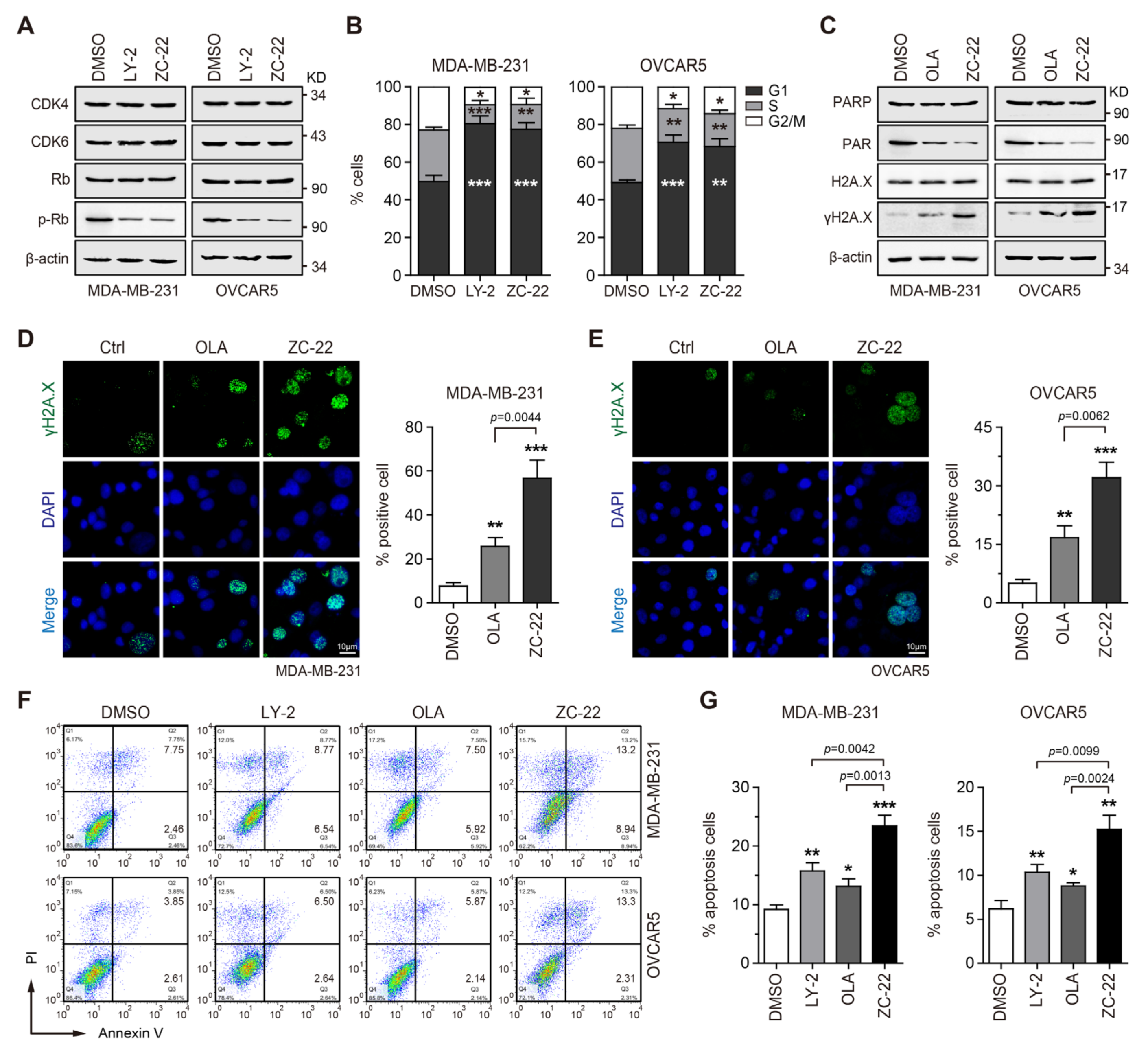
{kind=link}
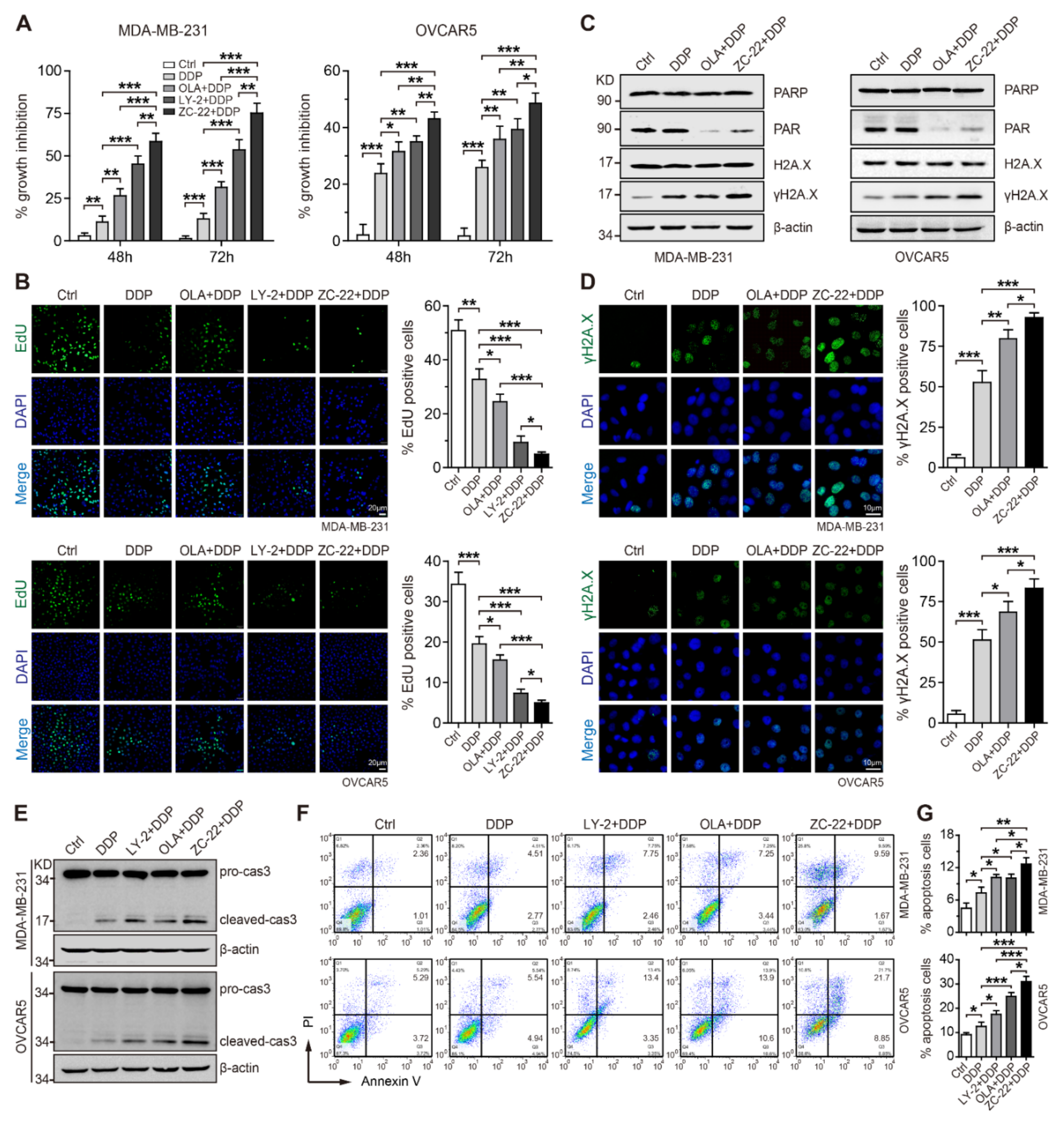{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Antibodies and Reagents
2.3. Cell Viability Assay
2.4. Western Blot Analysis
2.5. Cell Cycle Analysis
2.6. Cell Apoptosis Analysis
2.7. EdU Incorporation Assay
2.8. Immunofluorescence
2.9. Xenograft Mice Models
2.10. Histology
2.11. Statistical Analysis
3. Results
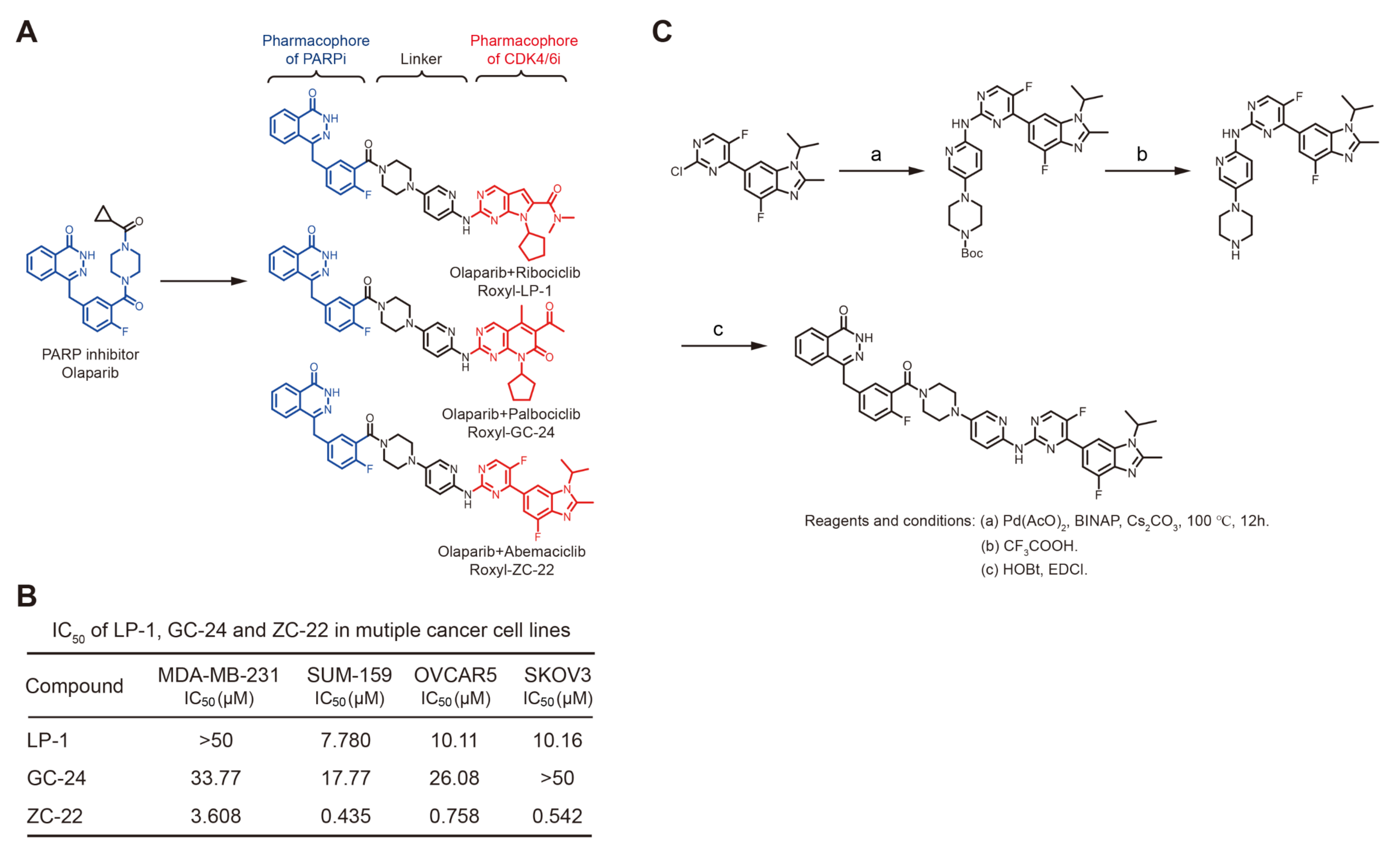
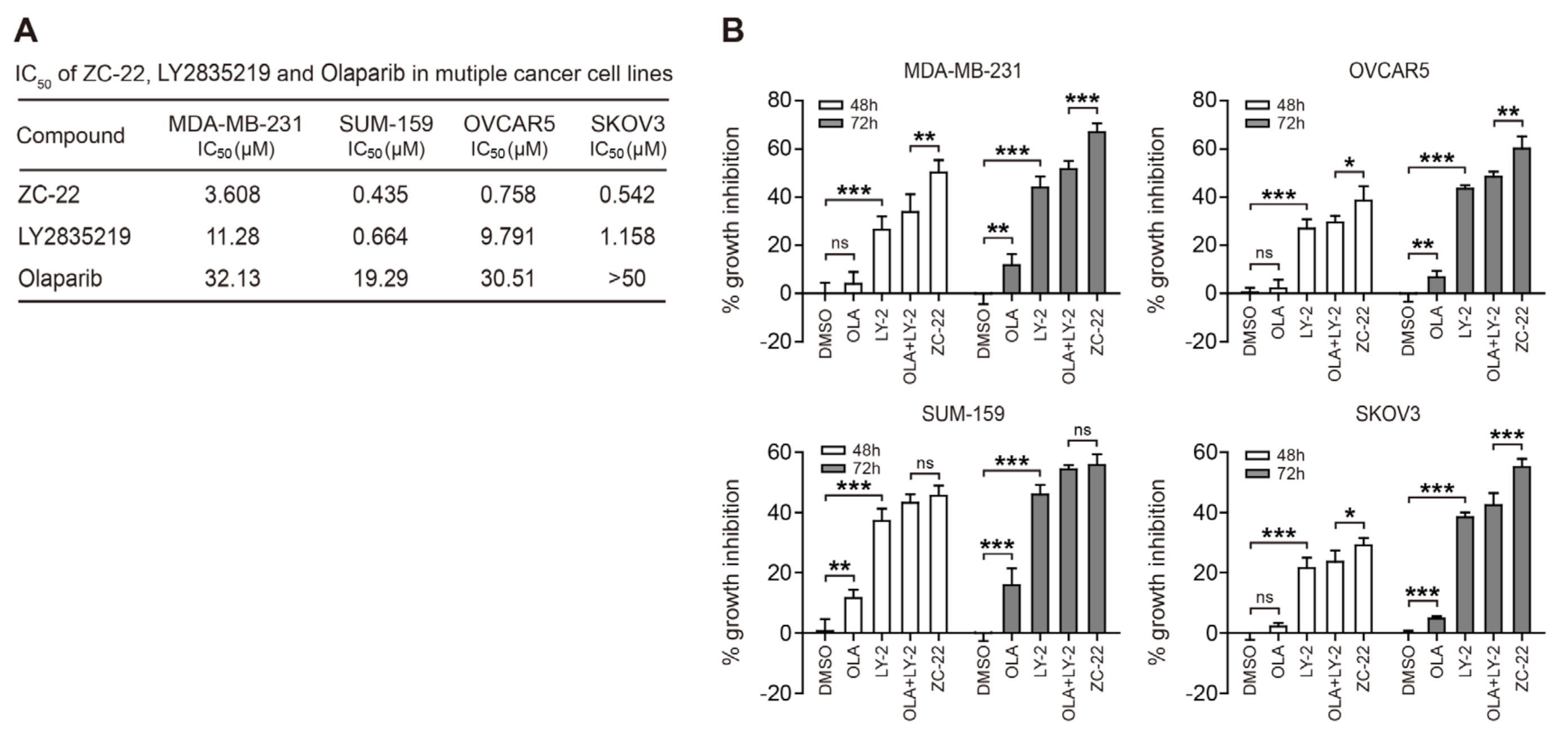
3.1. Synthesis of ZC-22 and Analysis of Its Activity
3.2. ZC-22 Has Betterr Anti-Tumor Efficacy Than Olaparib and Abemaciclib Alone and in Combination
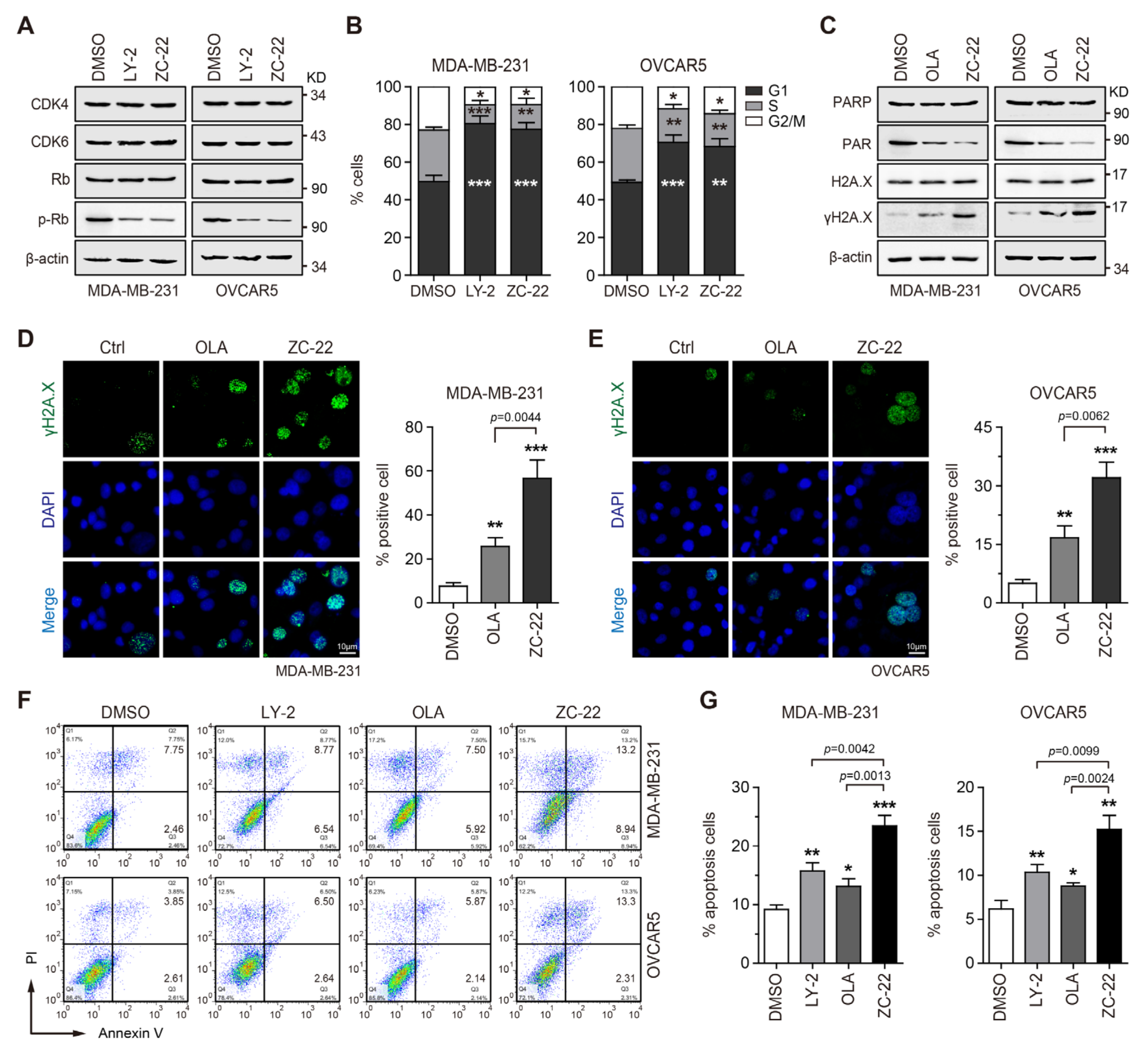
3.3. ZC-22 Effectively Targets CDK4/6 and PARP to Induce Cell Cycle Arrest and Apoptosis
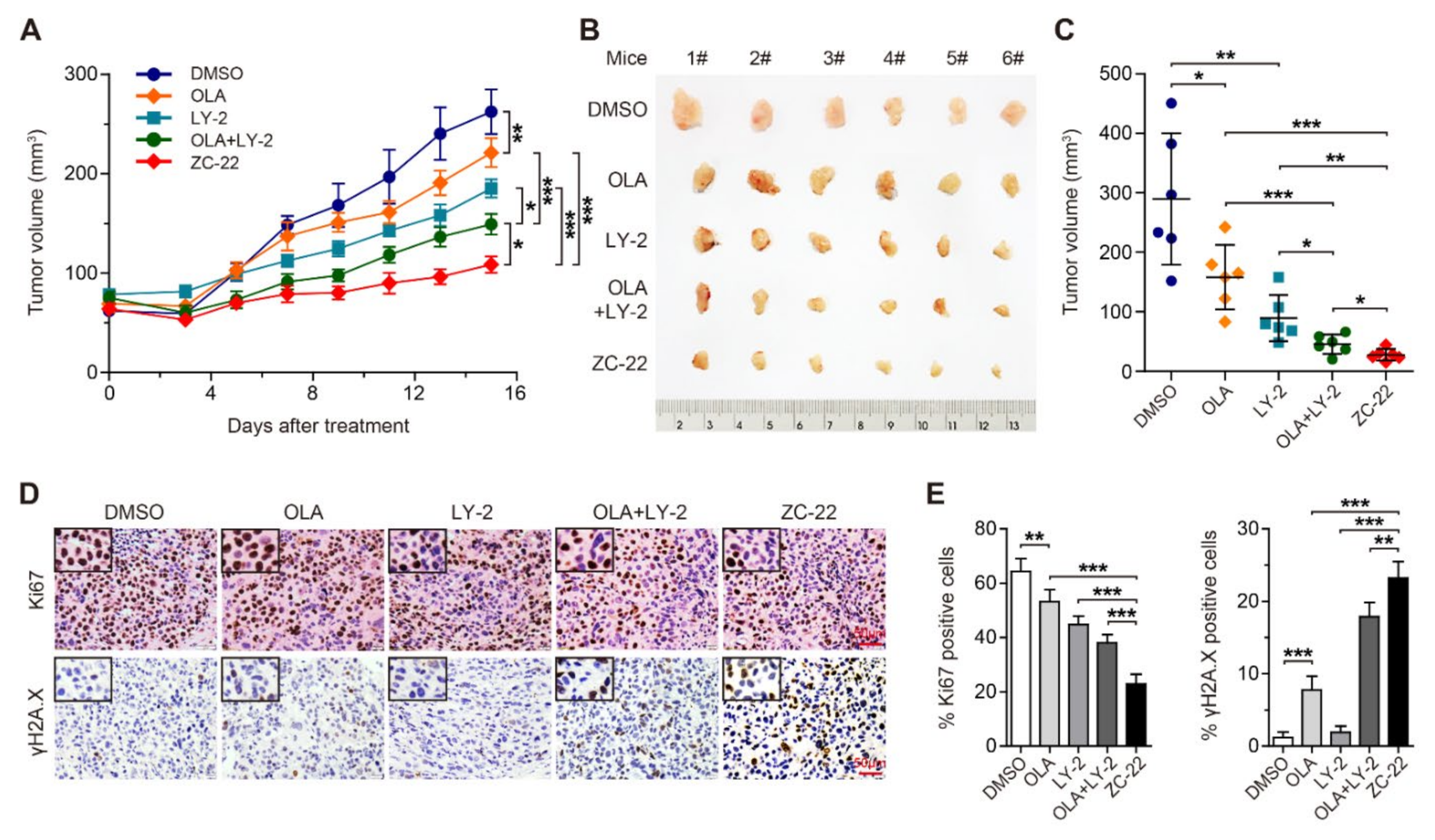
3.4. ZC-22 Monotherapy Displays Better Anti-Tumor Efficacy Than Olaparib and Abemaciclib Combination Therapy in Breast Cancer In Vivo
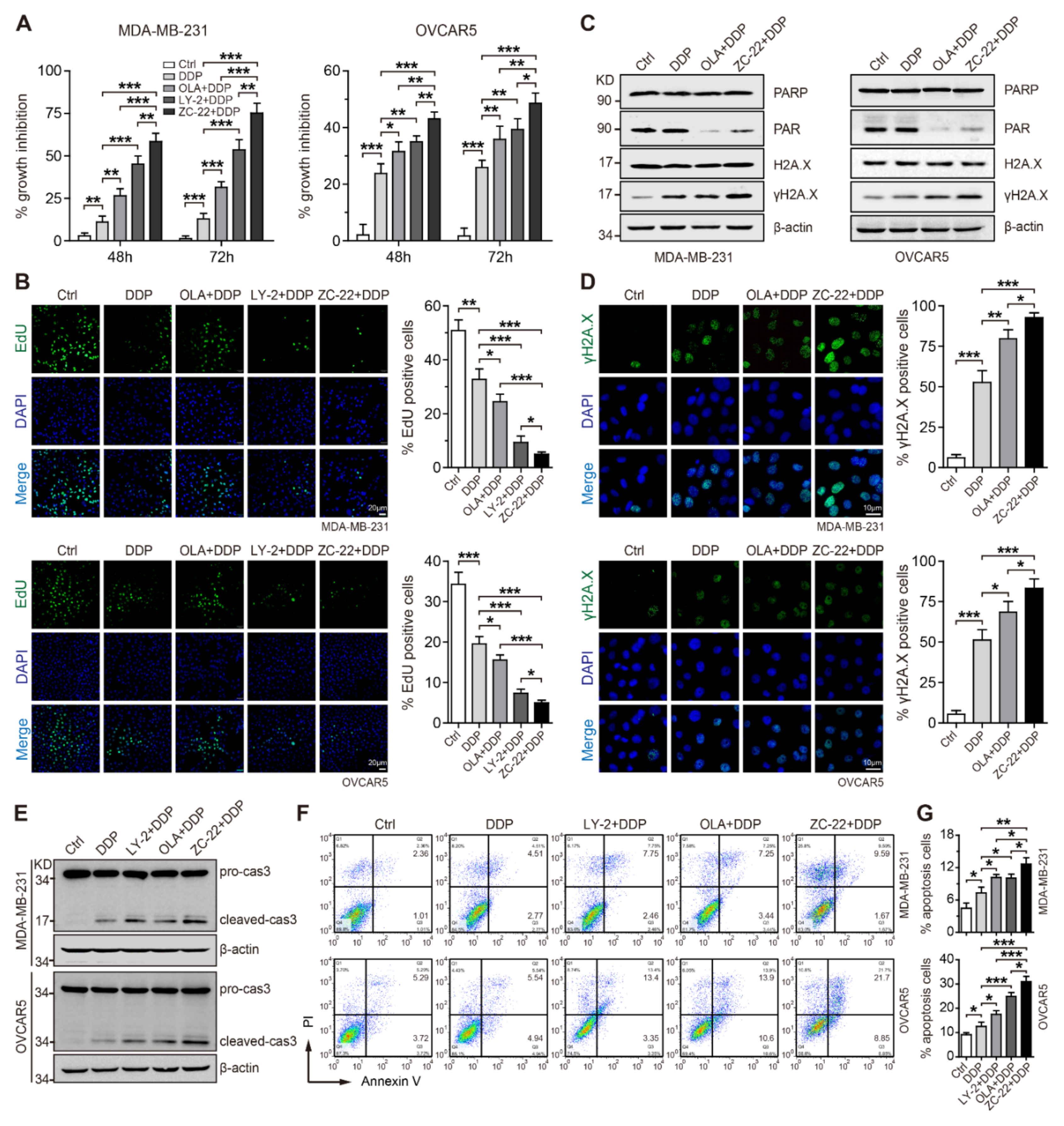
3.5. ZC-22 Sensitizes Breast and Ovarian Cancer Cells to Cisplatin In Vitro
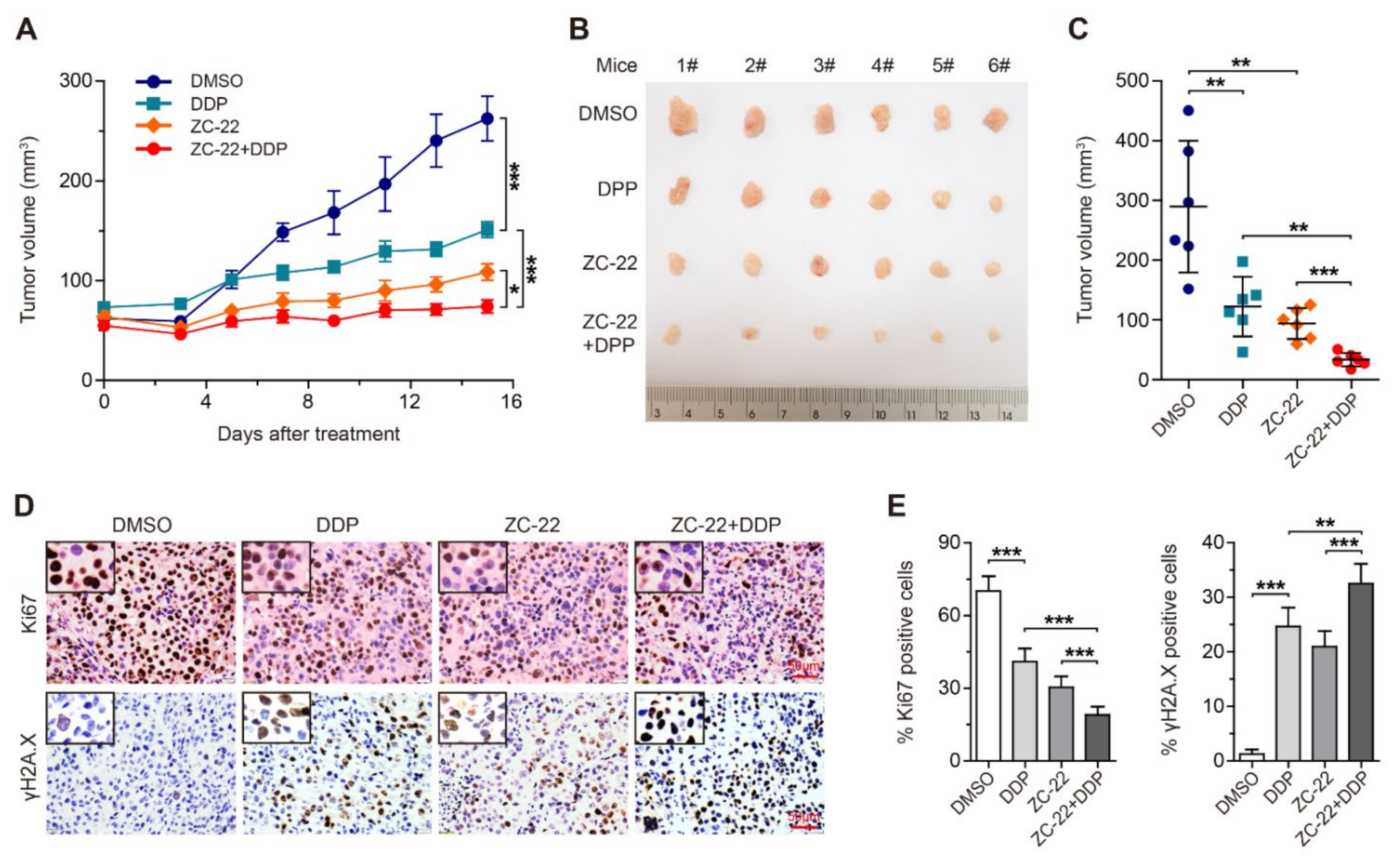
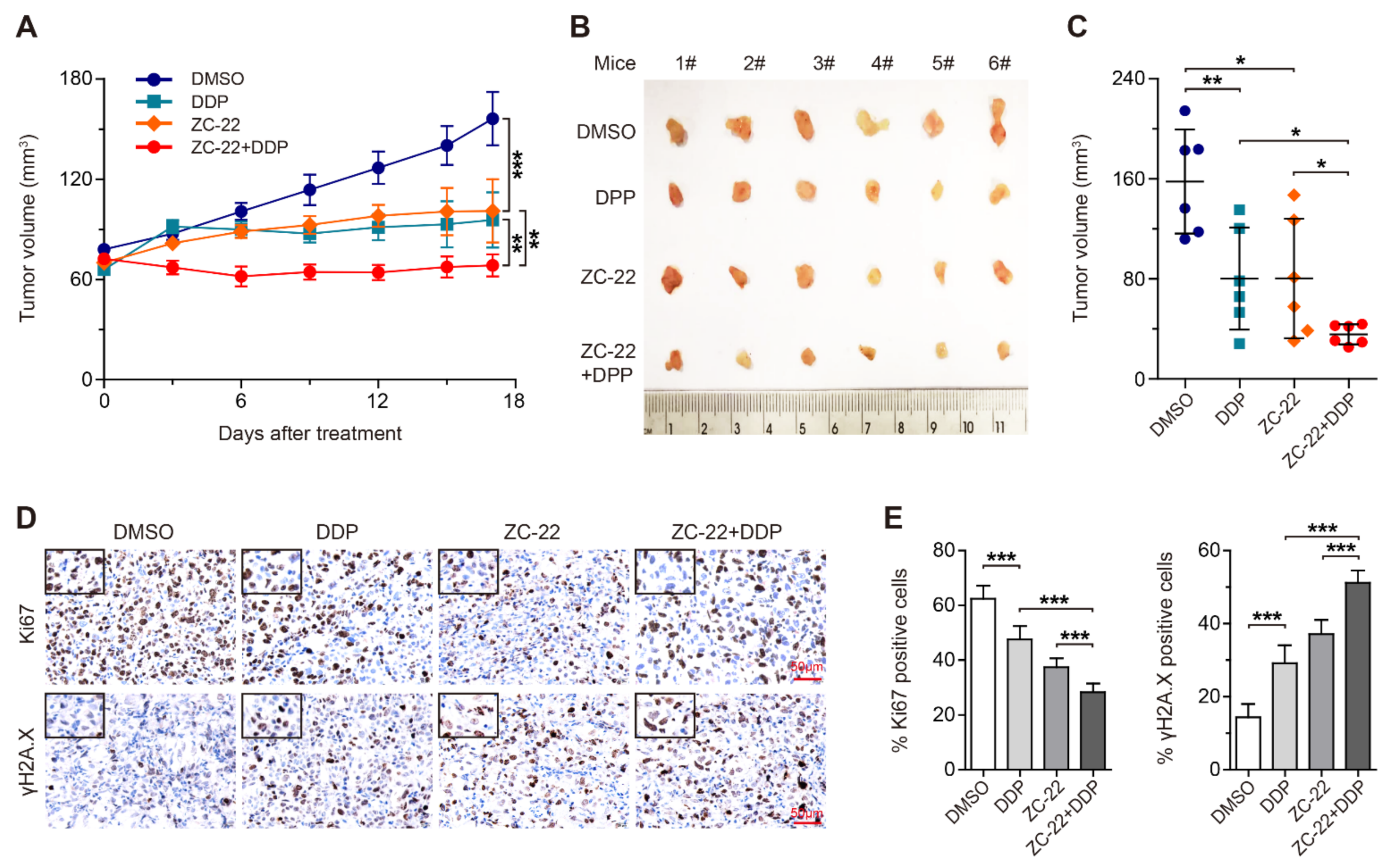
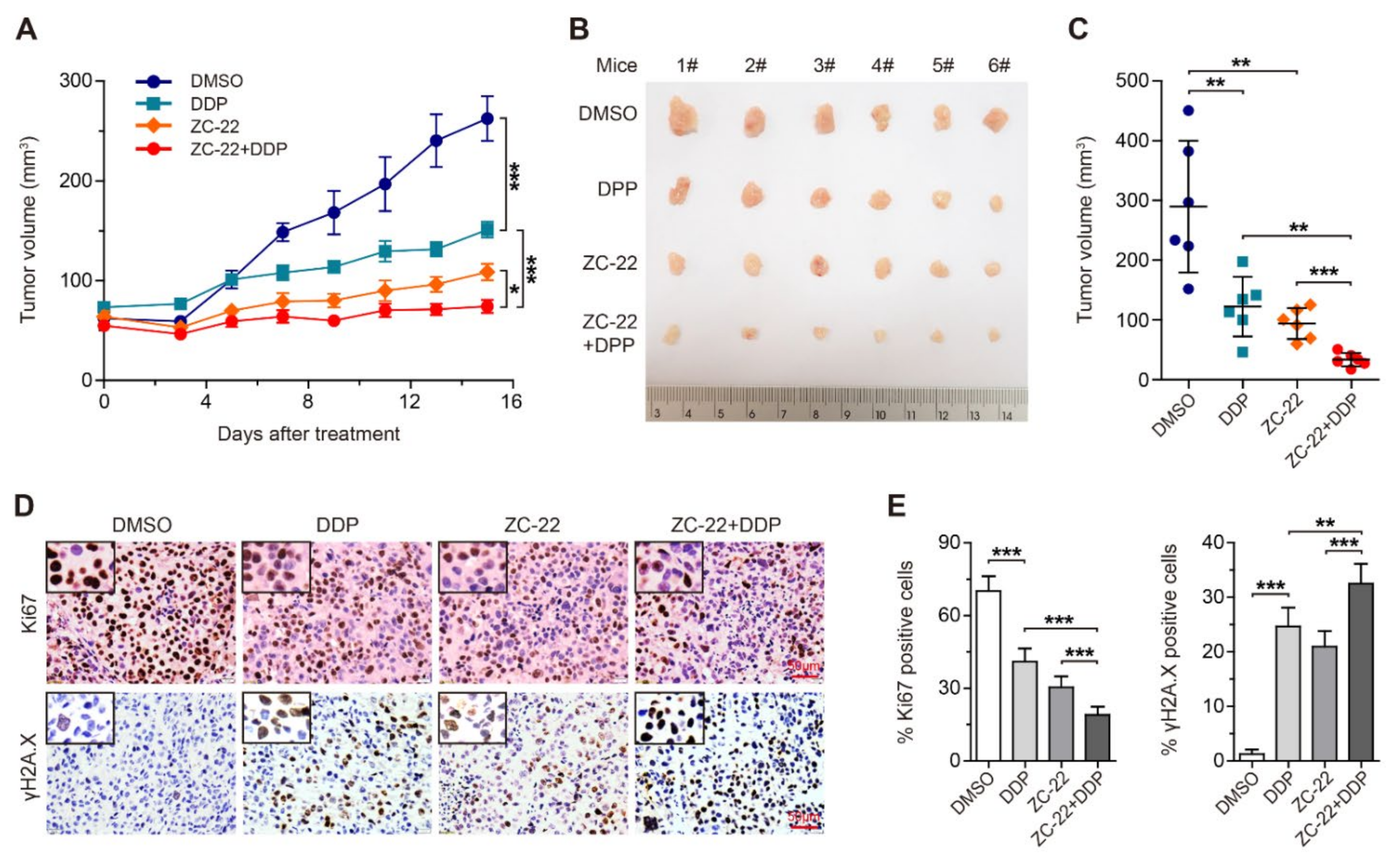
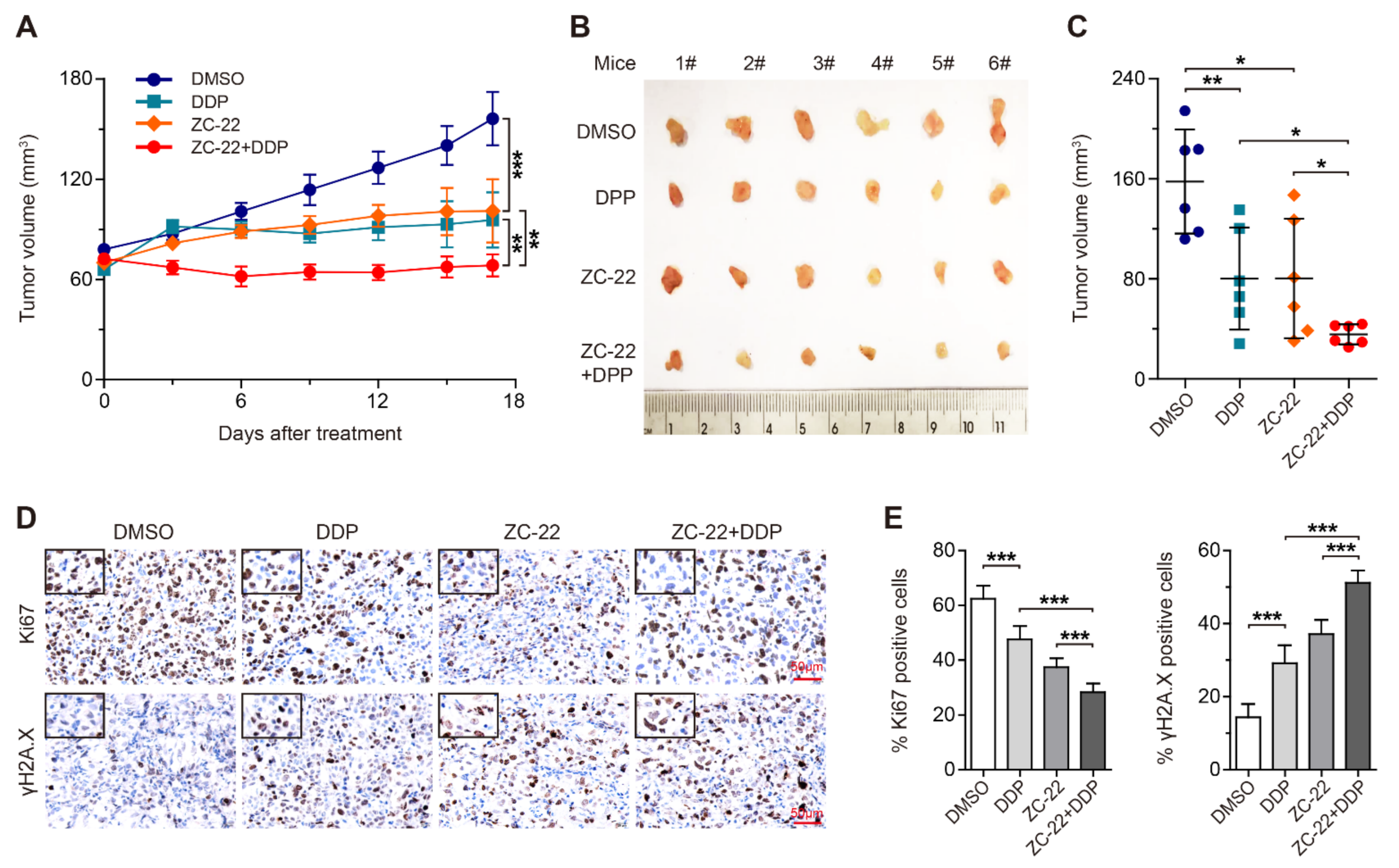
3.6. ZC-22 Greatly Improves the Response of Breast and Ovarian Cancer Cells to Cisplatin Treatment in Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, C.; Ralyea, C.; Lockwood, S. Ovarian Cancer: An Integrated Review. Semin. Oncol. Nurs. 2019, 35, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Pignata, S.; Pisano, C.; Di Napoli, M.; Cecere, S.C.; Tambaro, R.; Attademo, L. Treatment of recurrent epithelial ovarian cancer. Cancer 2019, 125 (Suppl. 24), 4609–4615. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.; Infante, J.R. Targeting CDK4/6 in patients with cancer. Cancer Treat. Rev. 2016, 45, 129–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Eggersmann, T.K.; Degenhardt, T.; Gluz, O.; Wuerstlein, R.; Harbeck, N. CDK4/6 Inhibitors Expand the Therapeutic Options in Breast Cancer: Palbociclib, Ribociclib and Abemaciclib. BioDrugs 2019, 33, 125–135. [Google Scholar] [CrossRef]
- Piezzo, M.; Cocco, S.; Caputo, R.; Cianniello, D.; Di Gioia, G.; Di Lauro, V.; Fusco, G.; Martinelli, C.; Nuzzo, F.; Pensabene, M.; et al. Targeting Cell Cycle in Breast Cancer: CDK4/6 Inhibitors. Int. J. Mol. Sci. 2020, 21, 6479. [Google Scholar] [CrossRef]
- Du, Q.; Guo, X.; Wang, M.; Li, Y.; Sun, X.; Li, Q. The application and prospect of CDK4/6 inhibitors in malignant solid tumors. J. Hematol. Oncol. 2020, 13, 41. [Google Scholar] [CrossRef]
- Spring, L.M.; Wander, S.A.; Andre, F.; Moy, B.; Turner, N.C.; Bardia, A. Cyclin-dependent kinase 4 and 6 inhibitors for hormone receptor-positive breast cancer: Past, present, and future. Lancet 2020, 395, 817–827. [Google Scholar] [CrossRef]
- Hartkopf, A.D.; Müller, V.; Wöckel, A.; Lux, M.P.; Janni, W.; Ettl, J.; Belleville, E.; Schütz, F.; Fasching, P.A.; Kolberg, H.-C.; et al. Translational Highlights in Breast and Ovarian Cancer 2019—Immunotherapy, DNA Repair, PI3K Inhibition and CDK4/6 Therapy. Geburtshilfe Frauenheilkd 2019, 79, 1309–1319. [Google Scholar] [CrossRef]
- Dall’Acqua, A.; Bartoletti, M.; Masoudi-Khoram, N.; Sorio, R.; Puglisi, F.; Belletti, B.; Baldassarre, G. Inhibition of CDK4/6 as Therapeutic Approach for Ovarian Cancer Patients: Current Evidences and Future Perspectives. Cancers 2021, 13, 35. [Google Scholar] [CrossRef]
- Sobhani, N.; D’Angelo, A.; Pittacolo, M.; Roviello, G.; Miccoli, A.; Corona, S.P.; Bernocchi, O.; Generali, D.; Otto, T. Updates on the CDK4/6 Inhibitory Strategy and Combinations in Breast Cancer. Cells 2019, 8, 321. [Google Scholar] [CrossRef] [Green Version]
- Eustermann, S.; Wu, W.-F.; Langelier, M.-F.; Yang, J.-C.; Easton, L.E.; Riccio, A.A.; Pascal, J.M.; Neuhaus, D. Structural Basis of Detection and Signaling of DNA Single-Strand Breaks by Human PARP-1. Mol. Cell. 2015, 60, 742–754. [Google Scholar] [CrossRef] [Green Version]
- Ronson, G.E.; Piberger, A.L.; Higgs, M.R.; Olsen, A.L.; Stewart, G.S.; McHugh, P.J.; Petermann, E.; Lakin, N.D. PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat. Commun. 2018, 9, 746. [Google Scholar] [CrossRef] [Green Version]
- Boussios, S.; Karihtala, P.; Moschetta, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N. Combined Strategies with Poly (ADP-Ribose) Polymerase (PARP) Inhibitors for the Treatment of Ovarian Cancer: A Literature Review. Diagnostics 2019, 9, 87. [Google Scholar] [CrossRef] [Green Version]
- Mullard, A. PARP inhibitor pick-me-up. Nat. Rev. Drug Discov. 2019, 18, 814. [Google Scholar] [CrossRef]
- Slade, D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020, 34, 360–394. [Google Scholar] [CrossRef] [Green Version]
- Konecny, G.E. Combining PARP and CDK4/6 inhibitors in MYC driven ovarian cancer. EBioMedicine 2019, 43, 9–10. [Google Scholar] [CrossRef] [Green Version]
- Geenen, J.J.J.; Linn, S.C.; Beijnen, J.H.; Schellens, J.H.M. PARP Inhibitors in the Treatment of Triple-Negative Breast Cancer. Clin. Pharmacokinet. 2018, 57, 427–437. [Google Scholar] [CrossRef]
- Pilie, P.G.; Gay, C.M.; Byers, L.A.; O’Connor, M.J.; Yap, T.A. PARP Inhibitors: Extending Benefit Beyond BRCA-Mutant Cancers. Clin. Cancer Res. 2019, 25, 3759–3771. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Chen, L.; Huang, B.; Li, X.; Yang, L.; Hu, X.; Jiang, Y.; Shao, Z.; Wang, Z. Efficacy and mechanism of the combination of PARP and CDK4/6 inhibitors in the treatment of triple-negative breast cancer. J. Exp. Clin. Cancer Res. 2021, 40, 122. [Google Scholar] [CrossRef]
- Yi, J.; Liu, C.; Tao, Z.; Wang, M.; Jia, Y.; Sang, X.; Shen, L.; Xue, Y.; Jiang, K.; Luo, F.; et al. MYC status as a determinant of synergistic response to Olaparib and Palbociclib in ovarian cancer. EBioMedicine 2019, 43, 225–237. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.; Liu, L.; Ashby, J.M.; Wu, D.; Chen, Y.; O’Neill, S.S.; Huang, S.; Wang, J.; Wang, G.; Cheng, D.; et al. Recruitment of KMT2C/MLL3 to DNA Damage Sites Mediates DNA Damage Responses and Regulates PARP Inhibitor Sensitivity in Cancer. Cancer Res. 2021, 81, 3358–3373. [Google Scholar] [CrossRef]
- Xu, M.; Huang, S.; Dong, X.; Chen, Y.; Li, M.; Shi, W.; Wang, G.; Huang, C.; Wang, Q.; Liu, Y.; et al. A novel isoform of ATOH8 promotes the metastasis of breast cancer by regulating RhoC. J. Mol. Cell Biol. 2021, 13, 59–71. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [Green Version]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- Matulonis, U.A.; Penson, R.T.; Domchek, S.M.; Kaufman, B.; Shapira-Frommer, R.; Audeh, M.W.; Kaye, S.; Molife, L.R.; Gelmon, K.A.; Robertson, J.D.; et al. Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: A multistudy analysis of response rates and safety. Ann. Oncol. 2016, 27, 1013–1019. [Google Scholar] [CrossRef]
- Dai, M.; Boudreault, J.; Wang, N.; Poulet, S.; Daliah, G.; Yan, G.; Moamer, A.; Burgos, S.A.; Sabri, S.; Ali, S.; et al. Differential Regulation of Cancer Progression by CDK4/6 Plays a Central Role in DNA Replication and Repair Pathways. Cancer Res. 2021, 81, 1332–1346. [Google Scholar] [CrossRef]
- Dean, J.L.; McClendon, A.K.; Knudsen, E.S. Modification of the DNA damage response by therapeutic CDK4/6 inhibition. J. Biol. Chem. 2012, 287, 29075–29087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Luo, J.; Chen, X.; Yang, Z.; Mei, X.; Ma, J.; Zhang, Z.; Guo, X.; Yu, X. CDK4/6 inhibitors: A novel strategy for tumor radiosensitization. J. Exp. Clin. Cancer Res. 2020, 39, 188. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, M.; Kastan, M.B. The DNA damage response: Implications for tumor responses to radiation and chemotherapy. Annu. Rev. Med. 2015, 66, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Sikov, W.M. Assessing the role of platinum agents in aggressive breast cancers. Curr. Oncol. Rep. 2015, 17, 3. [Google Scholar] [CrossRef]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef]
- Tchounwou, P.B.; Dasari, S.; Noubissi, F.K.; Ray, P.; Kumar, S. Advances in Our Understanding of the Molecular Mechanisms of Action of Cisplatin in Cancer Therapy. J. Exp. Pharmacol. 2021, 13, 303–328. [Google Scholar] [CrossRef]
- Kiss, R.C.; Xia, F.; Acklin, S. Targeting DNA Damage Response and Repair to Enhance Therapeutic Index in Cisplatin-Based Cancer Treatment. Int. J. Mol. Sci. 2021, 22, 8199. [Google Scholar] [CrossRef]
- Garutti, M.; Pelizzari, G.; Bartoletti, M.; Malfatti, M.C.; Gerratana, L.; Tell, G.; Puglisi, F. Platinum Salts in Patients with Breast Cancer: A Focus on Predictive Factors. Int. J. Mol. Sci. 2019, 20, 3390. [Google Scholar] [CrossRef] [Green Version]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Drew, Y.; Kristeleit, R.S. Homologous recombination deficiency and ovarian cancer. Eur. J. Cancer 2016, 60, 49–58. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.-Y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Sugimura, K.; Takebayashi, S.; Taguchi, H.; Takeda, S.; Okumura, K. PARP-1 ensures regulation of replication fork progression by homologous recombination on damaged DNA. J. Cell Biol. 2008, 183, 1203–1212. [Google Scholar] [CrossRef] [Green Version]
- Ström, C.E.; Johansson, F.; Uhlen, M.; Szigyarto, C.A.-K.; Erixon, K.; Helleday, T. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011, 39, 3166–3175. [Google Scholar] [CrossRef]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.-S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef] [Green Version]
- Maya-Mendoza, A.; Moudry, P.; Merchut-Maya, J.M.; Lee, M.; Strauss, R.; Bartek, J. High speed of fork progression induces DNA replication stress and genomic instability. Nature 2018, 559, 279–284. [Google Scholar] [CrossRef]
- Salvador-Barbero, B.; Alvarez-Fernández, M.; Zapatero-Solana, E.; El Bakkali, A.; Menéndez, M.D.C.; López-Casas, P.P.; Di Domenico, T.; Xie, T.; VanArsdale, T.; Shields, D.J.; et al. CDK4/6 Inhibitors Impair Recovery from Cytotoxic Chemotherapy in Pancreatic Adenocarcinoma. Cancer Cell 2020, 38, 584. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, C.; Wei, Y.; Li, J.; Huang, Z.; Wang, Q.; Lin, Y.; Lv, X.; Chen, Y.; Fan, Y.; Sun, P.; et al. A Novel CDK4/6 and PARP Dual Inhibitor ZC-22 Effectively Suppresses Tumor Growth and Improves the Response to Cisplatin Treatment in Breast and Ovarian Cancer. Int. J. Mol. Sci. 2022, 23, 2892. https://doi.org/10.3390/ijms23052892
Tian C, Wei Y, Li J, Huang Z, Wang Q, Lin Y, Lv X, Chen Y, Fan Y, Sun P, et al. A Novel CDK4/6 and PARP Dual Inhibitor ZC-22 Effectively Suppresses Tumor Growth and Improves the Response to Cisplatin Treatment in Breast and Ovarian Cancer. International Journal of Molecular Sciences. 2022; 23(5):2892. https://doi.org/10.3390/ijms23052892
Chicago/Turabian StyleTian, Chenchen, Yufan Wei, Jianjun Li, Zhi Huang, Qiong Wang, Yingxue Lin, Xingping Lv, Yanan Chen, Yan Fan, Peiqing Sun, and et al. 2022. "A Novel CDK4/6 and PARP Dual Inhibitor ZC-22 Effectively Suppresses Tumor Growth and Improves the Response to Cisplatin Treatment in Breast and Ovarian Cancer" International Journal of Molecular Sciences 23, no. 5: 2892. https://doi.org/10.3390/ijms23052892
APA StyleTian, C., Wei, Y., Li, J., Huang, Z., Wang, Q., Lin, Y., Lv, X., Chen, Y., Fan, Y., Sun, P., Xiang, R., Chang, A., & Yang, S. (2022). A Novel CDK4/6 and PARP Dual Inhibitor ZC-22 Effectively Suppresses Tumor Growth and Improves the Response to Cisplatin Treatment in Breast and Ovarian Cancer. International Journal of Molecular Sciences, 23(5), 2892. https://doi.org/10.3390/ijms23052892