
Comparative Analysis of Exo- and Endonuclease Activities of APE1-like Enzymes

 , , , and
, , , and 
Abstract
1. Introduction
2. Results
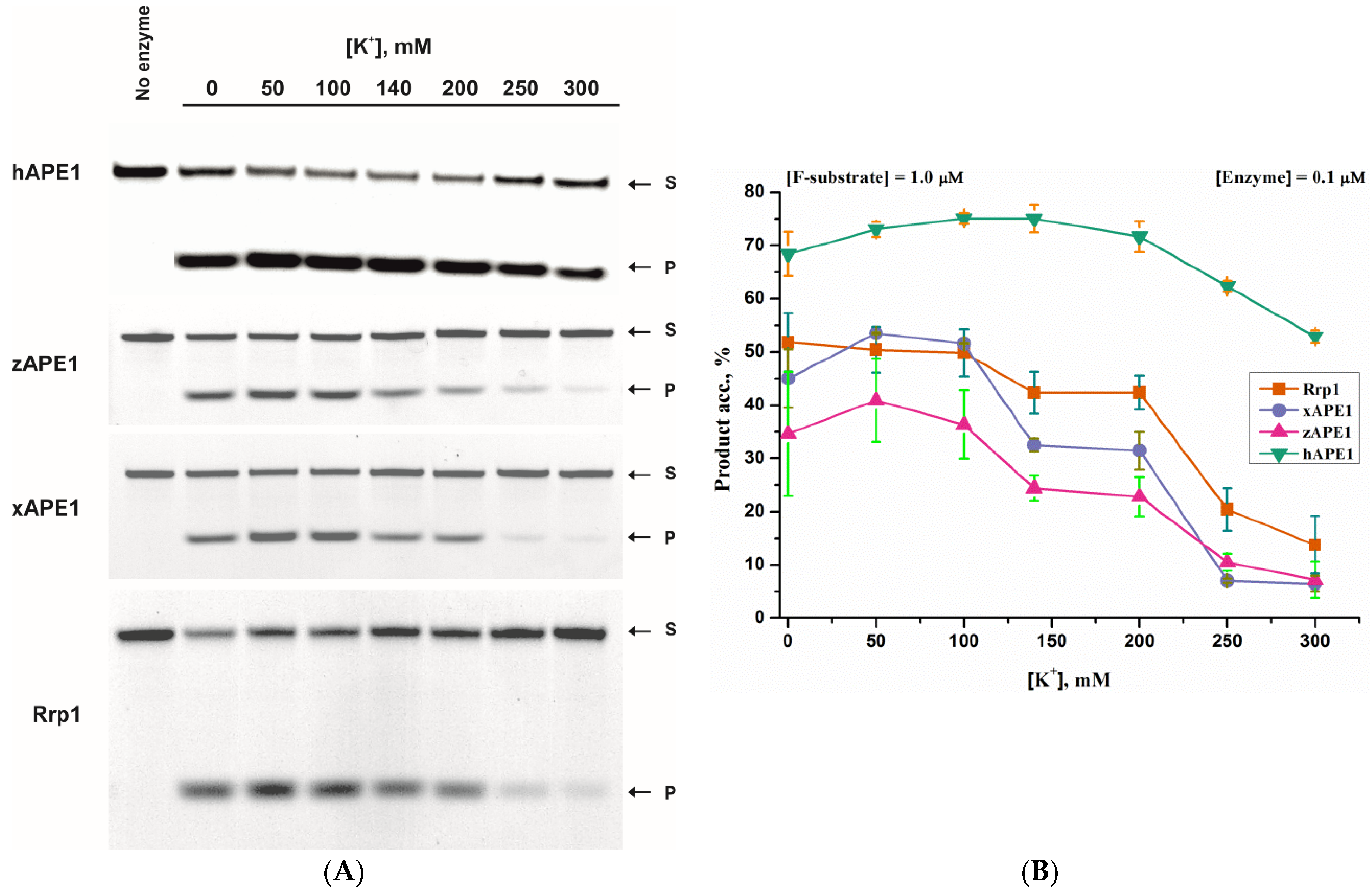
2.1. AP-Endonuclease Activity
2.2. 3′-5′ Exonuclease Activity
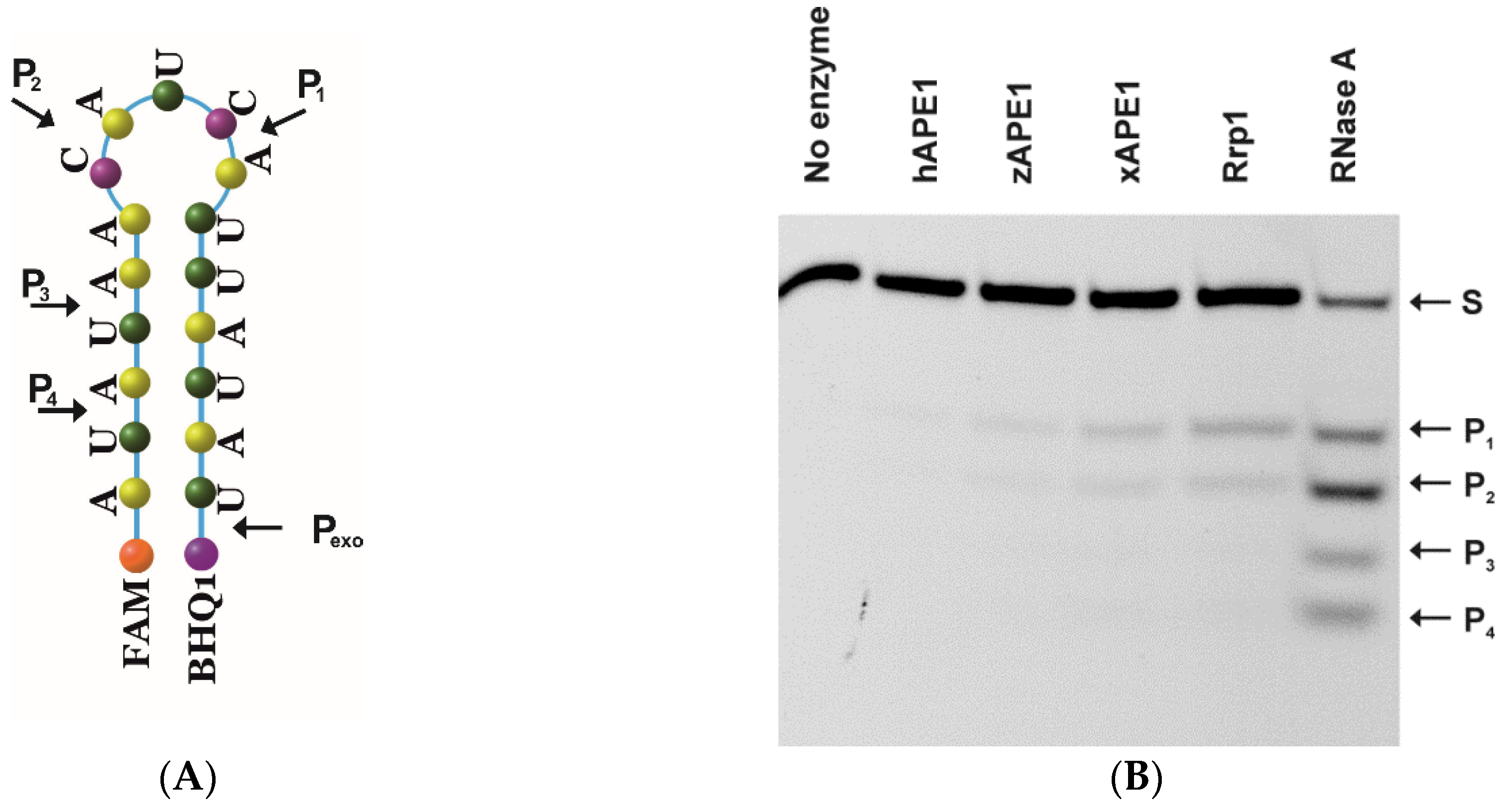
2.3. Endoribonuclease Activity
3. Discussion
4. Materials and Methods
4.1. Cloning of AP-Endonucleases
4.2. Enzyme Purification
4.3. Oligodeoxynucleotides (ODNs)
4.4. PAGE Experiments
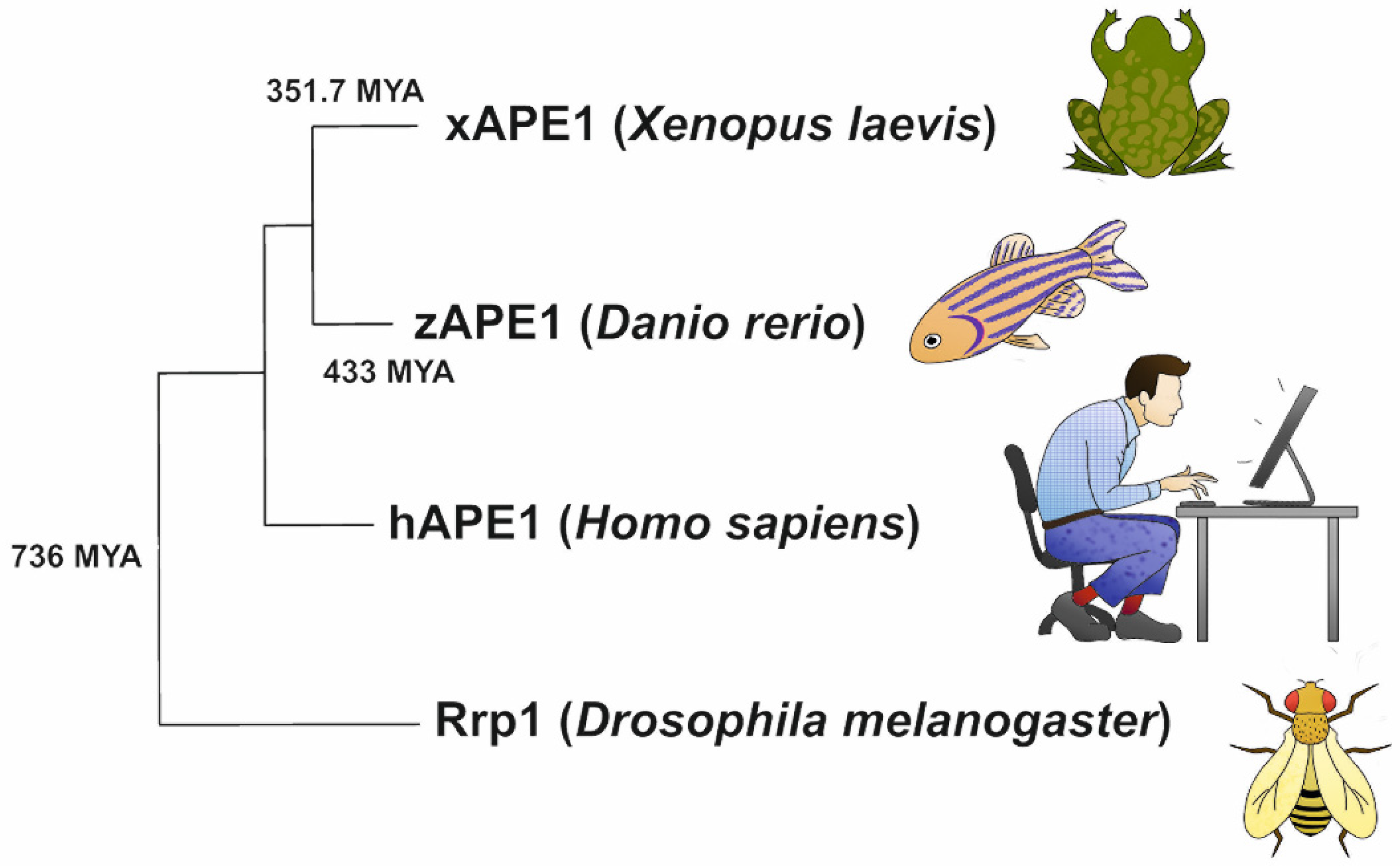
4.5. Phylogenetic Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fromme, J.C.; Banerjee, A.; Verdine, G.L. DNA glycosylase recognition and catalysis. Curr. Opin. Struct. Biol. 2004, 14, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Gros, L.; Saparbaev, M.K.; Laval, J. Enzymology of the repair of free radicals-induced DNA damage. Oncogene 2002, 21, 8905–8925. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C.; Roger, A.S.; Wolfram, S.; Graham, C.W.; Tom, E.; Richard, D.W. DNA Repair and Mutagenesis, 2nd ed.; American Society of Microbiology: Washington, DC, USA, 2006; ISBN 9781555813192. [Google Scholar]
- Li, M.; Wilson, D.M., 3rd. Human apurinic/apyrimidinic endonuclease 1. Antioxid. Redox Signal. 2014, 20, 678–707. [Google Scholar] [CrossRef] [PubMed]
- Demple, B.; Sung, J.-S. Molecular and biological roles of Ape1 protein in mammalian base excision repair. DNA Repair 2005, 4, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Ide, H.; Tedzuka, K.; Shimzu, H.; Kimura, Y.; Purmal, A.A.; Wallace, S.S.; Kow, Y.W. α-Deoxyadenosine, a Major Anoxic Radiolysis Product of Adenine in DNA, Is a Substrate for Escherichia coli Endonuclease IV. Biochemistry 1994, 33, 7842–7847. [Google Scholar] [CrossRef] [PubMed]
- Ischenko, A.A.; Saparbaev, M.K. Alternative nucleotide incision repair pathway for oxidative DNA damage. Nature 2002, 415, 183–187. [Google Scholar] [CrossRef]
- Ishchenko, A.A.; Ide, H.; Ramotar, D.; Nevinsky, G.; Saparbaev, M. Alpha-anomeric deoxynucleotides, anoxic products of ionizing radiation, are substrates for the endonuclease IV-type AP endonucleases. Biochemistry 2004, 43, 15210–15216. [Google Scholar] [CrossRef]
- Ishchenko, A.A.; Deprez, E.; Wlaksimenko, A.; Brochon, J.C.; Tauc, P.; Saparbaev, M.K. Uncoupling of the base excision and nucleotide incision repair pathways reveals thier respective biological roles. Proc. Natl. Acad. Sci. USA 2006, 103, 2564–2569. [Google Scholar] [CrossRef]
- Gros, L.; Ishchenko, A.A.; Ide, H.; Elder, R.H.; Saparbaev, M.K. The major human AP endonuclease (Ape1) is involved in the nucleotide incision repair pathway. Nucleic Acids Res. 2004, 32, 73–81. [Google Scholar] [CrossRef]
- Timofeyeva, N.A.; Koval, V.V.; Knorre, D.G.; Zharkov, D.O.; Saparbaev, M.K.; Ishchenko, A.A.; Fedorova, O.S. Conformational Dynamics of Human AP Endonuclease in Base Excision and Nucleotide Incision Repair Pathways. J. Biomol. Struct. Dyn. 2009, 26, 637–652. [Google Scholar] [CrossRef]
- Timofeyeva, N.A.; Fedorova, O.S. A kinetic mechanism of repair of DNA containing alpha-anomeric deoxyadenosine by human apurinic/apyrimidinic endonuclease 1. Mol. Biosyst. 2016, 12, 3435–3446. [Google Scholar] [CrossRef] [PubMed]
- Davletgildeeva, A.T.; Ishchenko, A.A.; Saparbaev, M.; Fedorova, O.S.; Kuznetsov, N.A. The Enigma of Substrate Recognition and Catalytic Efficiency of APE1-Like Enzymes. Front. Cell Dev. Biol. 2021, 9, 532. [Google Scholar] [CrossRef] [PubMed]
- Prorok, P.; Alili, D.; Saint-Pierre, C.; Gasparutto, D.; Zharkov, D.O.; Ishchenko, A.A.; Tudek, B.; Saparbaev, M.K. Uracil in duplex DNA is a substrate for the nucleotide incision repair pathway in human cells. Proc. Natl. Acad. Sci. USA 2013, 110, E3695–E3703. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.-M.; Cheng, Y.-C. The exonuclease activity of human apurinic/apyrimidinic endonuclease (APE1). Biochemical properties and inhibition by the natural dinucleotide Gp4G. J. Biol. Chem. 2003, 278, 18289–18296. [Google Scholar] [CrossRef]
- Wong, D.; DeMott, M.S.; Demple, B. Modulation of the 3′→5′-Exonuclease Activity of Human Apurinic Endonuclease (Ape1) by Its 5′-incised Abasic DNA Product. J. Biol. Chem. 2003, 278, 36242–36249. [Google Scholar] [CrossRef]
- Kuznetsova, A.; Fedorova, O.; Kuznetsov, N. Kinetic Features of 3′-5′ Exonuclease Activity of Human AP-Endonuclease APE1. Molecules 2018, 23, 2101. [Google Scholar] [CrossRef]
- Barzilay, G.; Hickson, I.D. Structure and Function of Apurinic/Apyrimidinic Endonucleases. Bioessays 1995, 17, 713–719. [Google Scholar] [CrossRef]
- Berquist, B.R.; McNeill, D.R.; Wilson, D.M., 3rd. Characterization of abasic endonuclease activity of human Ape1 on alternative substrates, as well as effects of ATP and sequence context on AP site incision. J. Mol. Biol. 2008, 379, 17–27. [Google Scholar] [CrossRef]
- Barnes, T.; Kim, W.-C.; Mantha, A.K.; Kim, S.-E.; Izumi, T.; Mitra, S.; Lee, C.H. Identification of Apurinic/apyrimidinic endonuclease 1 (APE1) as the endoribonuclease that cleaves c-myc mRNA. Nucleic Acids Res. 2009, 37, 3946–3958. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Novopashina, D.S.; Fedorova, O.S.; Kuznetsov, N.A. Effect of the Substrate Structure and Metal Ions on the Hydrolysis of Undamaged RNA by Human AP Endonuclease APE1. Acta Nat. 2020, 2, 33–44. [Google Scholar] [CrossRef]
- Mol, C.D.; Izumi, T.; Mitra, S.; Talner, J.A. DNA-bound structures and mutants reveal abasic DNA binding by APE1 DNA repair and coordination. Nature 2000, 403, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Mol, C.D.; Hosfield, D.J.; Tainer, J.A. Abasic site recognition by two apurinic/apyrimidinic endonuclease families in DNA base excision repair: The 3′ ends justify the means. Mutat. Res. 2000, 460, 211–229. [Google Scholar] [CrossRef]
- Tsutakawa, S.E.; Shin, D.S.; Mol, C.D.; Izumi, T.; Arvai, A.S.; Mantha, A.K.; Szczesny, B.; Ivanov, I.N.; Hosfield, D.J.; Maiti, B.; et al. Conserved Structural Chemistry for Incision Activity in Structurally Non-homologous Apurinic/Apyrimidinic Endonuclease APE1 and Endonuclease IV DNA Repair Enzymes. J. Biol. Chem. 2013, 288, 8445–8455. [Google Scholar] [CrossRef] [PubMed]
- Freudenthal, B.D.; Beard, W.A.; Cuneo, M.J.; Dyrkheeva, N.S.; Wilson, S.H. Capturing snapshots of APE1 processing DNA damage. Nat. Struct. Mol. Biol. 2015, 22, 924–931. [Google Scholar] [CrossRef]
- Gorman, M.A.; Morera, S.; Rothwell, D.G.; De La Fortelle, E.; Mol, C.D.; Tainer, J.A.; Hickson, I.D.; Freemont, P.S. The crystal structure of the human DNA repair endonuclease HAP1 suggests the recognition of extra-helical deoxyribose at DNA abasic sites. EMBO J. 1997, 16, 6548–6558. [Google Scholar] [CrossRef]
- Alekseeva, I.V.; Kuznetsova, A.A.; Bakman, A.S.; Fedorova, O.S.; Kuznetsov, N.A. The role of active-site amino acid residues in the cleavage of DNA and RNA substrates by human apurinic/apyrimidinic endonuclease APE1. Biochim. Biophys. Acta-Gen. Subj. 2020, 1864, 129718. [Google Scholar] [CrossRef]
- Beernink, P.T.; Segelke, B.W.; Hadi, M.Z.; Erzberger, J.P.; Wilson, D.M., 3rd; Rupp, B. Two divalent metal ions in the active site of a new crystal form of human apurinic/apyrimidinic endonuclease, Ape1: Implications for the catalytic mechanism. J. Mol. Biol. 2001, 307, 1023–1034. [Google Scholar] [CrossRef]
- Manvilla, B.A.; Pozharski, E.; Toth, E.A.; Drohat, A.C. Structure of human apurinic/apyrimidinic endonuclease 1 with the essential Mg2+ cofactor. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 2555–2562. [Google Scholar] [CrossRef]
- Lipton, A.S.; Heck, R.W.; Primak, S.; McNeill, D.R.; Wilson, D.M., 3rd; Ellis, P.D. Characterization of Mg2+ binding to the DNA repair protein apurinic/apyrimidic endonuclease 1 via solid-state 25Mg NMR spectroscopy. J. Am. Chem. Soc. 2008, 130, 9332–9341. [Google Scholar] [CrossRef]
- Oezguen, N.; Schein, C.H.; Peddi, S.R.; Power, T.D.; Izumi, T.; Braun, W. A “moving metal mechanism” for substrate cleavage by the DNA repair endonuclease APE-1. Proteins 2007, 68, 313–323. [Google Scholar] [CrossRef]
- Masuda, Y.; Bennett, R.A.; Demple, B. Rapid dissociation of human apurinic endonuclease (Ape1) from incised DNA induced by magnesium. J. Biol. Chem. 1998, 273, 30360–30365. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Erzberger, J.P.; Wilson, D.M., 3rd. The role of Mg2+ and specific amino acid residues in the catalytic reaction of the major human abasic endonuclease: New insights from EDTA-resistant incision of acyclic abasic site analogs and site-directed mutagenesis. J. Mol. Biol. 1999, 290, 447–457. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Chen, Q.; Georgiadis, M.M. High-resolution crystal structures reveal plasticity in the metal binding site of apurinic/apyrimidinic endonuclease I. Biochemistry 2014, 53, 6520–6529. [Google Scholar] [CrossRef] [PubMed]
- Miroshnikova, A.D.; Kuznetsova, A.A.; Vorobjev, Y.N.; Kuznetsov, N.A.; Fedorova, O.S. Effects of mono- and divalent metal ions on DNA binding and catalysis of human apurinic/apyrimidinic endonuclease 1. Mol. BioSyst. 2016, 12, 1527–1539. [Google Scholar] [CrossRef] [PubMed]
- Aboelnga, M.M.; Wetmore, S.D. Unveiling a Single-Metal-Mediated Phosphodiester Bond Cleavage Mechanism for Nucleic Acids: A Multiscale Computational Investigation of a Human DNA Repair Enzyme. J. Am. Chem. Soc. 2019, 141, 8646–8656. [Google Scholar] [CrossRef] [PubMed]
- Mundle, S.T.; Delaney, J.C.; Essigmann, J.M.; Strauss, P.R. Enzymatic mechanism of human apurinic/apyrimidinic endonuclease against a THF AP site model substrate. Biochemistry 2009, 48, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Batebi, H.; Imhof, P. Phosphodiester hydrolysis computed for cluster models of enzymatic active sites. Theor. Chem. Acc. 2016, 135, 262. [Google Scholar] [CrossRef]
- Lowry, D.F.; Hoyt, D.W.; Khazi, F.A.; Bagu, J.; Lindsey, A.G.; Wilson, D.M. Investigation of the Role of the Histidine–Aspartate Pair in the Human Exonuclease III-like Abasic Endonuclease, Ape1. J. Mol. Biol. 2003, 329, 311–322. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Matveeva, A.G.; Milov, A.D.; Vorobjev, Y.N.; Dzuba, S.A.; Fedorova, O.S.; Kuznetsov, N.A. Substrate specificity of human apurinic/apyrimidinic endonuclease APE1 in the nucleotide incision repair pathway. Nucleic Acids Res. 2018, 46, 11454–11465. [Google Scholar] [CrossRef]
- Miroshnikova, A.D.; Kuznetsova, A.A.; Kuznetsov, N.A.; Fedorova, O.S. Thermodynamics of Damaged DNA Binding and Catalysis by Human AP Endonuclease 1. Acta Nat. 2016, 8, 103–110. [Google Scholar] [CrossRef]
- Bulygin, A.A.; Kuznetsova, A.A.; Vorobjev, Y.N.; Fedorova, O.S.; Kuznetsov, N.A. The Role of Active-Site Plasticity in Damaged-Nucleotide Recognition by Human Apurinic/Apyrimidinic Endonuclease APE1. Molecules 2020, 25, 3940. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, A.A.; Senchurova, S.I.; Ishchenko, A.A.; Saparbaev, M.; Fedorova, O.S.; Kuznetsov, N.A. Common Kinetic Mechanism of Abasic Site Recognition by Structurally Different Apurinic/Apyrimidinic Endonucleases. Int. J. Mol. Sci. 2021, 22, 8874. [Google Scholar] [CrossRef] [PubMed]
- Mega 11. Available online: https://www.megasoftware.net/download_form (accessed on 1 February 2022).
- Wang, Y.; Shupenko, C.C.; Melo, L.F.; Strauss, P.R. DNA Repair Protein Involved in Heart and Blood Development. Mol. Cell. Biol. 2006, 26, 9083–9093. [Google Scholar] [CrossRef] [PubMed]
- Sander, M.; Lowenhaupt, K.; Rich, A. Drosophila Rrp1 protein: An apurinic endonuclease with homologous recombination activities. Proc. Natl. Acad. Sci. USA 1991, 88, 6780–6784. [Google Scholar] [CrossRef]
- Sander, M. Drosophila Rrp1 3’-exonuclease: Demonstration of DNA sequence dependence and DNA strand specificity. Nucleic Acids Res. 1996, 24, 3926–3933. [Google Scholar] [CrossRef][Green Version]
- Reardon, B.J.; Lombardo, C.R.; Sander, M. DrosophilaRrp1 Domain Structure as Defined by Limited Proteolysis and Biophysical Analyses. J. Biol. Chem. 1998, 273, 33991–33999. [Google Scholar] [CrossRef]
- Kim, W.-C.; Berquist, B.R.; Chohan, M.; Uy, C.; Wilson, D.M.; Lee, C.H. Characterization of the Endoribonuclease Active Site of Human Apurinic/Apyrimidinic Endonuclease 1. J. Mol. Biol. 2011, 411, 960–971. [Google Scholar] [CrossRef]
- Kim, W.-C.C.; King, D.; Lee, C.H. RNA-cleaving properties of human apurinic/apyrimidinic endonuclease 1 (APE1). Int. J. Biochem. Mol. Biol. 2010, 1, 12–25. [Google Scholar]
- Kuznetsova, A.A.; Akhmetgalieva, A.A.; Ulyanova, V.V.; Ilinskaya, O.N.; Fedorova, O.S.; Kuznetsov, N.A. Efficiency of RNA Hydrolysis by Binase from Bacillus pumilus: The Impact of Substrate Structure, Metal Ions, and Low Molecular Weight Nucleotide Compounds. Mol. Biol. 2020, 54, 769–776. [Google Scholar] [CrossRef]
- Davletgildeeva, A.T.; Kuznetsova, A.A.; Fedorova, O.S.; Kuznetsov, N.A. Activity of Human Apurinic/Apyrimidinic Endonuclease APE1 Toward Damaged DNA and Native RNA With Non-canonical Structures. Front. Cell Dev. Biol. 2020, 8, 590848. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Gavrilova, A.A.; Novopashina, D.S.; Fedorova, O.S.; Kuznetsov, N.A. Mutational and Kinetic Analysis of APE1 Endoribonuclease Activity. Mol. Biol. 2021, 55, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.E.; Gorrell, A.; Rader, S.D.; Lee, C.H. Endoribonuclease activity of human apurinic/apyrimidinic endonuclease 1 revealed by a real-time fluorometric assay. Anal. Biochem. 2010, 398, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.M., III; Barsky, D. The major human abasic endonuclease: Formation, consequences and repair of abasic lesions in DNA. Mutat. Res. 2001, 485, 283–307. [Google Scholar] [CrossRef]
- Chou, K.M.; Cheng, Y.C. An exonucleolytic activity of human apurinic/apyrimidinic endonuclease on 3′ mispaired DNA. Nature 2002, 415, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Demple, B.; Harrison, L. Repair of oxidative damage to DNA: Enzymology and biology. Annu. Rev. Biochem. 1994, 63, 915–948. [Google Scholar] [CrossRef]
- Burkovics, P.; Szukacsov, V.; Unk, I.; Haracska, L. Human Ape2 protein has a 3′-5′ exonuclease activity that acts preferentially on mismatched base pairs. Nucleic Acids Res. 2006, 34, 2508–2515. [Google Scholar] [CrossRef] [PubMed]
- Joldybayeva, B.; Prorok, P.; Grin, I.R.; Zharkov, D.O.; Ishenko, A.A.; Tudek, B.; Bissenbaev, A.K.; Saparbaev, M. Cloning and Characterization of a Wheat Homologue of Apurinic/Apyrimidinic Endonuclease Ape1L. PLoS ONE 2014, 9, e92963. [Google Scholar] [CrossRef]
- Abeldenov, S.; Talhaoui, I.; Zharkov, D.O.; Ishchenko, A.A.; Ramanculov, E.; Saparbaev, M.; Khassenov, B. Characterization of DNA substrate specificities of apurinic/apyrimidinic endonucleases from Mycobacterium tuberculosis. DNA Repair 2015, 33, 1–16. [Google Scholar] [CrossRef]
- Redrejo-Rodriguez, M.; Vigouroux, A.; Mursalimov, A.; Grin, I.; Alili, D.; Koshenov, Z.; Akishev, Z.; Maksimenko, A.; Bissenbaev, A.K.; Matkarimov, B.T.; et al. Structural comparison of AP endonucleases from the exonuclease III family reveals new amino acid residues in human AP endonuclease 1 that are involved in incision of damaged DNA. Biochimie 2016, 128, 20–33. [Google Scholar] [CrossRef]
- Daviet, S.; Couvé-Privat, S.; Gros, L.; Shinozuka, K.; Ide, H.; Saparbaev, M.; Ishchenko, A.A.; Couve-Privat, S.; Gros, L.; Shinozuka, K.; et al. Major oxidative products of cytosine are substrates for the nucleotide incision repair pathway. DNA Repair 2007, 6, 8–18. [Google Scholar] [CrossRef]
- Fantini, D.; Vascotto, C.; Marasco, D.; D’Ambrosio, C.; Romanello, M.; Vitagliano, L.; Pedone, C.; Poletto, M.; Cesaratto, L.; Quadrifoglio, F.; et al. Critical lysine residues within the overlooked N-terminal domain of human APE1 regulate its biological functions. Nucleic Acids Res. 2010, 38, 8239–8256. [Google Scholar] [CrossRef] [PubMed]
- Poletto, M.; Vascotto, C.; Scognamiglio, P.L.; Lirussi, L.; Marasco, D.; Tell, G. Role of the unstructured N-terminal domain of the hAPE1 (human apurinic/apyrimidinic endonuclease 1) in the modulation of its interaction with nucleic acids and NPM1 (nucleophosmin). Biochem. J. 2013, 452, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, R.; Wiederhold, L.; Szczesny, B.; Boldogh, I.; Hazra, T.K.; Izumi, T.; Mitra, S. Identification and characterization of mitochondrial abasic (AP)-endonuclease in mammalian cells. Nucleic Acids Res. 2006, 34, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Izumi, T.; Mitra, S. Deletion analysis of human AP-endonuclease: Minimum sequence required for the endonuclease activity. Carcinogenesis 1998, 19, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, A.A.; Kuznetsov, N.A.; Ishchenko, A.A.; Saparbaev, M.K.; Fedorova, O.S. Pre-steady-state fluorescence analysis of damaged DNA transfer from human DNA glycosylases to AP endonuclease APE1. Biochim. Biophys. Acta 2014, 1840, 3042–3051. [Google Scholar] [CrossRef]
- Mello, B. Estimating TimeTrees with MEGA and the TimeTree Resource. Mol. Biol. Evol. 2018, 35, 2334–2342. [Google Scholar] [CrossRef]
- Hedges, S.B.; Marin, J.; Suleski, M.; Paymer, M.; Kumar, S. Tree of life reveals clock-like speciation and diversification. Mol. Biol. Evol. 2015, 32, 835–845. [Google Scholar] [CrossRef]
- Tamura, K.; Battistuzzi, F.U.; Billing-Ross, P.; Murillo, O.; Filipski, A.; Kumar, S. Estimating divergence times in large molecular phylogenies. Proc. Natl. Acad. Sci. USA 2012, 109, 19333–19338. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| F-substrate | 5′–FAM–GCTCAFGTACAGAGCTG–3′ 3′–CGAGTGCATGTCTCGAC–BHQ1–5′ |
| Exo-substrate | 5′–FAM–CAGCTCTGTACGTGAGC–3′ 3′–GTCGAGACATGCACTCGTCACCACTGTG–5′ |
| RNA substrate | 5′–FAM–r(AUAUAACAUCAUUAUAU)–BHQ1–3′ |
| Substrate | Reactant Concentrations | Reaction Conditions |
|---|---|---|
| F-substrate | [enzyme] = 1.0 μM, [substrate] = 1.0 μM | (1) [MgCl2] = 5.0 mM, [KCl] = 0–300 mM, 25 °C, 20 s (2) [MgCl2] = 0–10 mM, [KCl] = 50 mM, 25 °C, 20 s |
| Exo-substrate | [enzyme] = 1.0 μM, [substrate] = 1.0 μM | (1) [MgCl2] = 5.0 mM, [KCl] = 50 mM, 25 °C, 30 min (2) [MgCl2] = 5.0 mM, [KCl] = 0–300 mM, 25 °C, 30 min (3) [MgCl2] = 0–10 mM, [KCl] = 50 mM, 25 °C, 30 min |
| RNA substrate | [enzyme] = 2.0 μM, [substrate] = 1.0 μM | [MgCl2] = 0.0 or 5.0 mM, [KCl] = 50 mM, 25 °C, 1 h |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davletgildeeva, A.T.; Kuznetsova, A.A.; Novopashina, D.S.; Ishchenko, A.A.; Saparbaev, M.; Fedorova, O.S.; Kuznetsov, N.A. Comparative Analysis of Exo- and Endonuclease Activities of APE1-like Enzymes. Int. J. Mol. Sci. 2022, 23, 2869. https://doi.org/10.3390/ijms23052869
Davletgildeeva AT, Kuznetsova AA, Novopashina DS, Ishchenko AA, Saparbaev M, Fedorova OS, Kuznetsov NA. Comparative Analysis of Exo- and Endonuclease Activities of APE1-like Enzymes. International Journal of Molecular Sciences. 2022; 23(5):2869. https://doi.org/10.3390/ijms23052869
Chicago/Turabian StyleDavletgildeeva, Anastasiia T., Alexandra A. Kuznetsova, Darya S. Novopashina, Alexander A. Ishchenko, Murat Saparbaev, Olga S. Fedorova, and Nikita A. Kuznetsov. 2022. "Comparative Analysis of Exo- and Endonuclease Activities of APE1-like Enzymes" International Journal of Molecular Sciences 23, no. 5: 2869. https://doi.org/10.3390/ijms23052869
APA StyleDavletgildeeva, A. T., Kuznetsova, A. A., Novopashina, D. S., Ishchenko, A. A., Saparbaev, M., Fedorova, O. S., & Kuznetsov, N. A. (2022). Comparative Analysis of Exo- and Endonuclease Activities of APE1-like Enzymes. International Journal of Molecular Sciences, 23(5), 2869. https://doi.org/10.3390/ijms23052869