
Novel Design of Neuropeptide-Based Drugs with β-Sheet Breaking Potential in Amyloid-Beta Cascade: Molecular and Structural Deciphers

 ,
,  ,
,  , and
, and
Abstract
:1. Introduction
2. Results
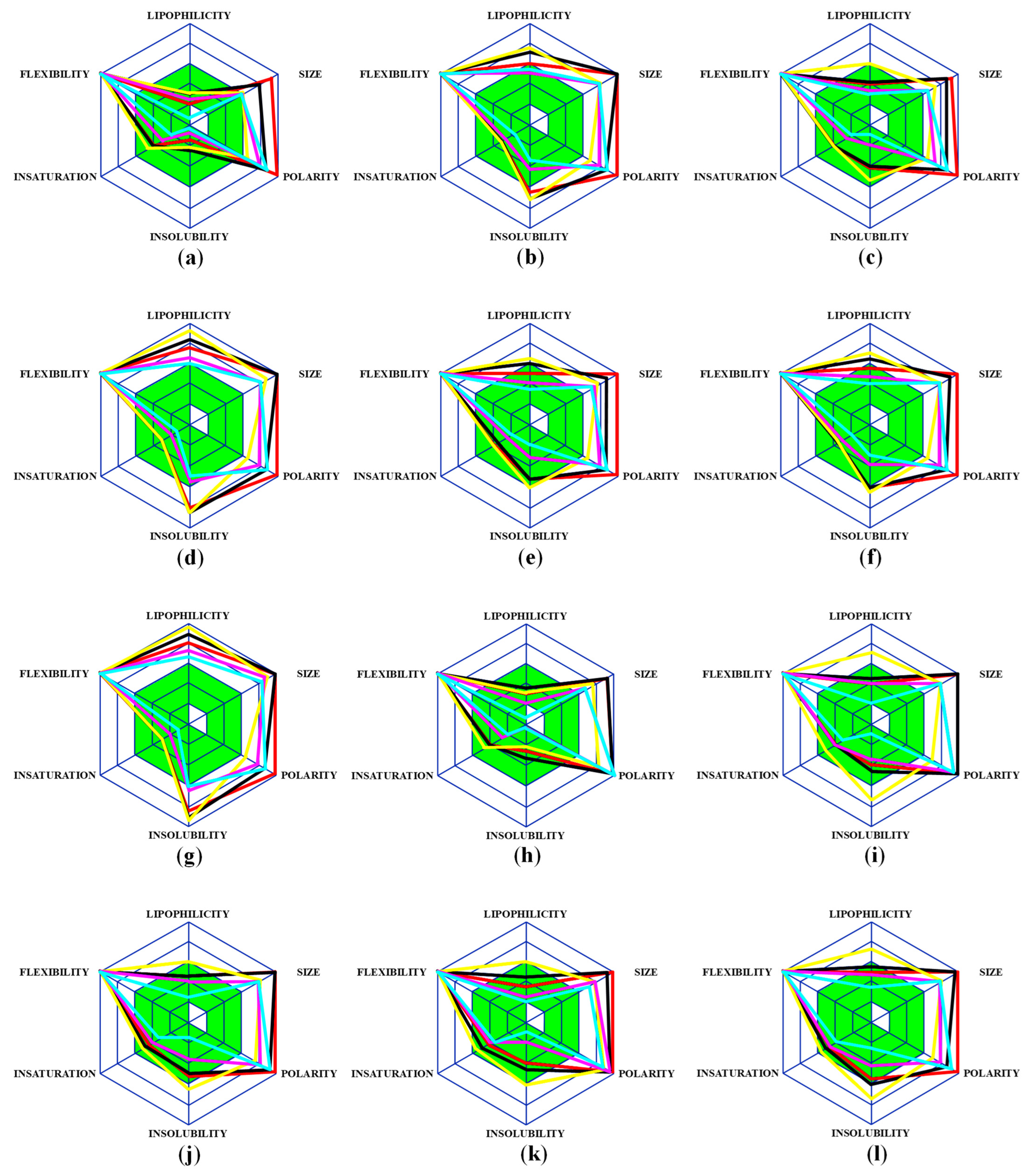
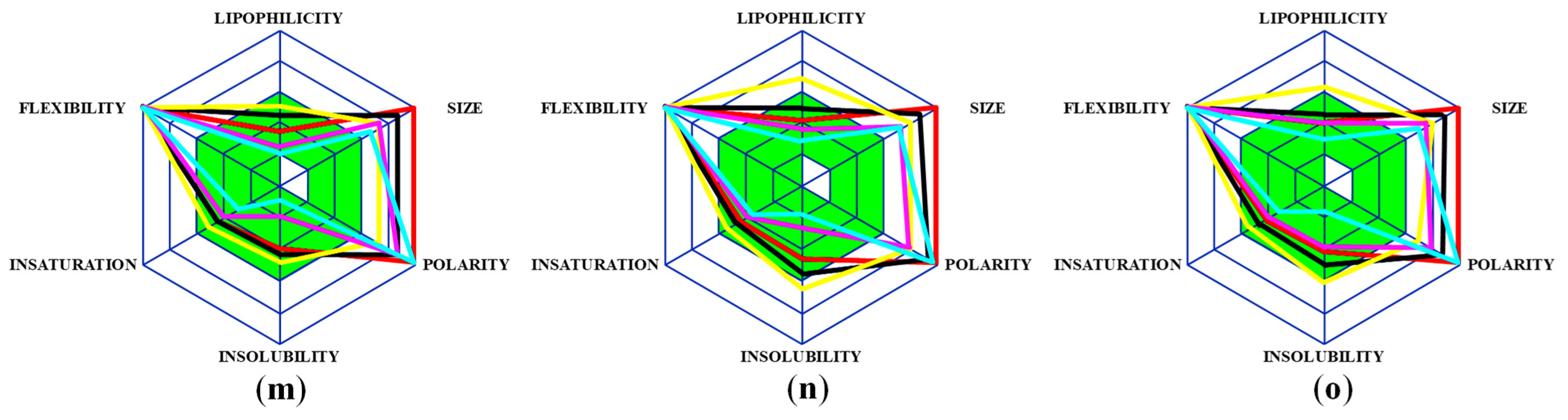
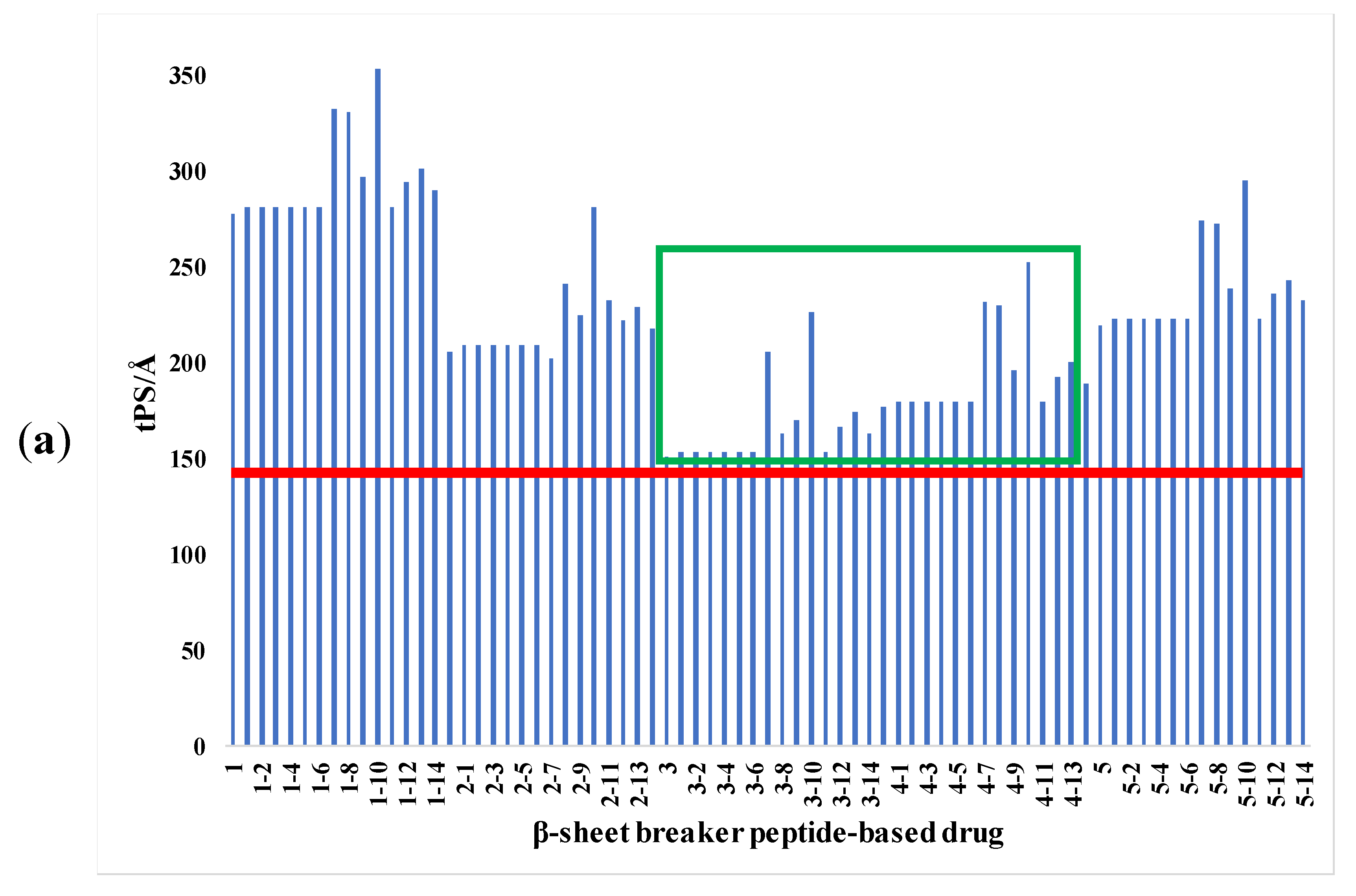
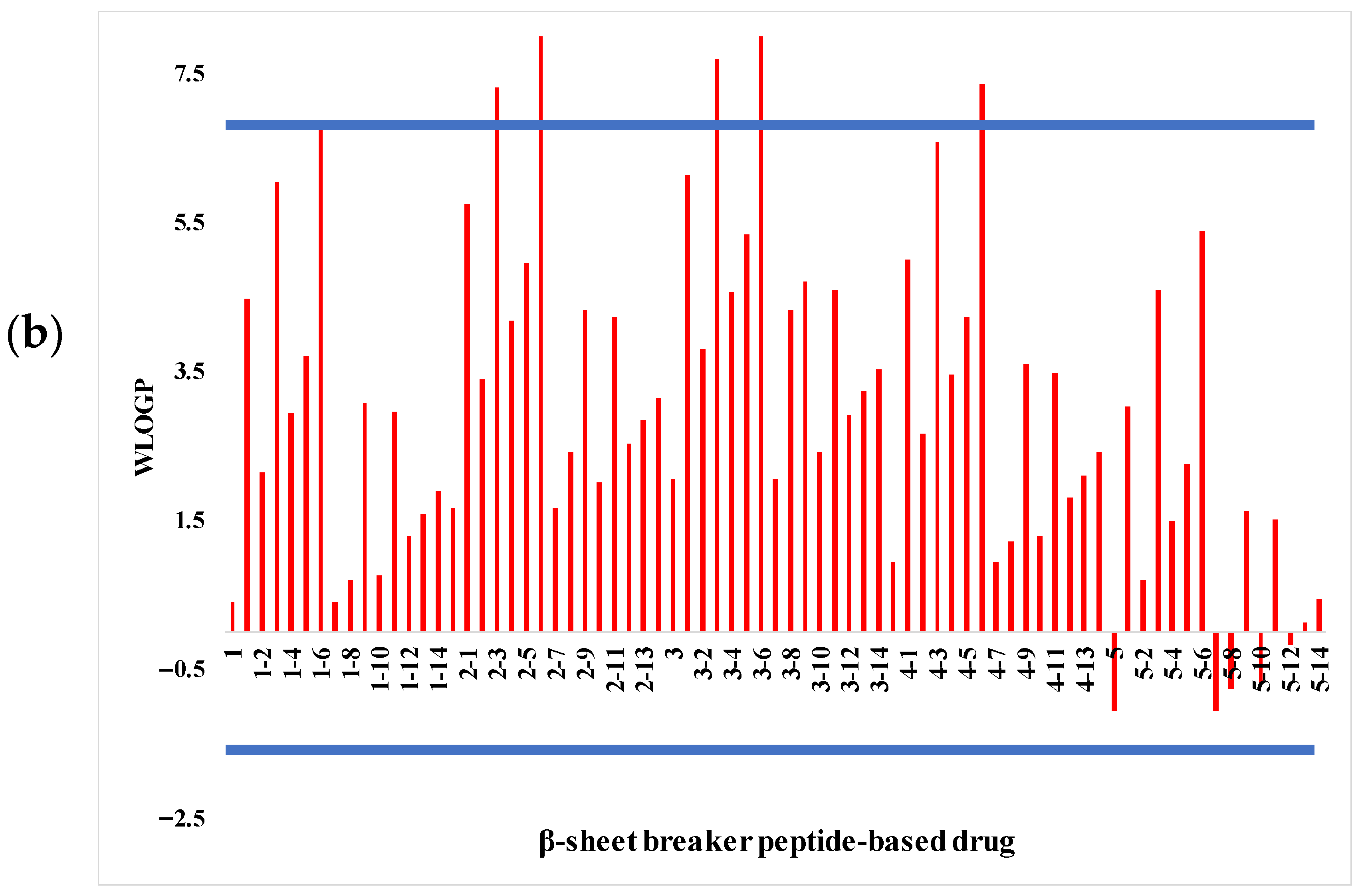
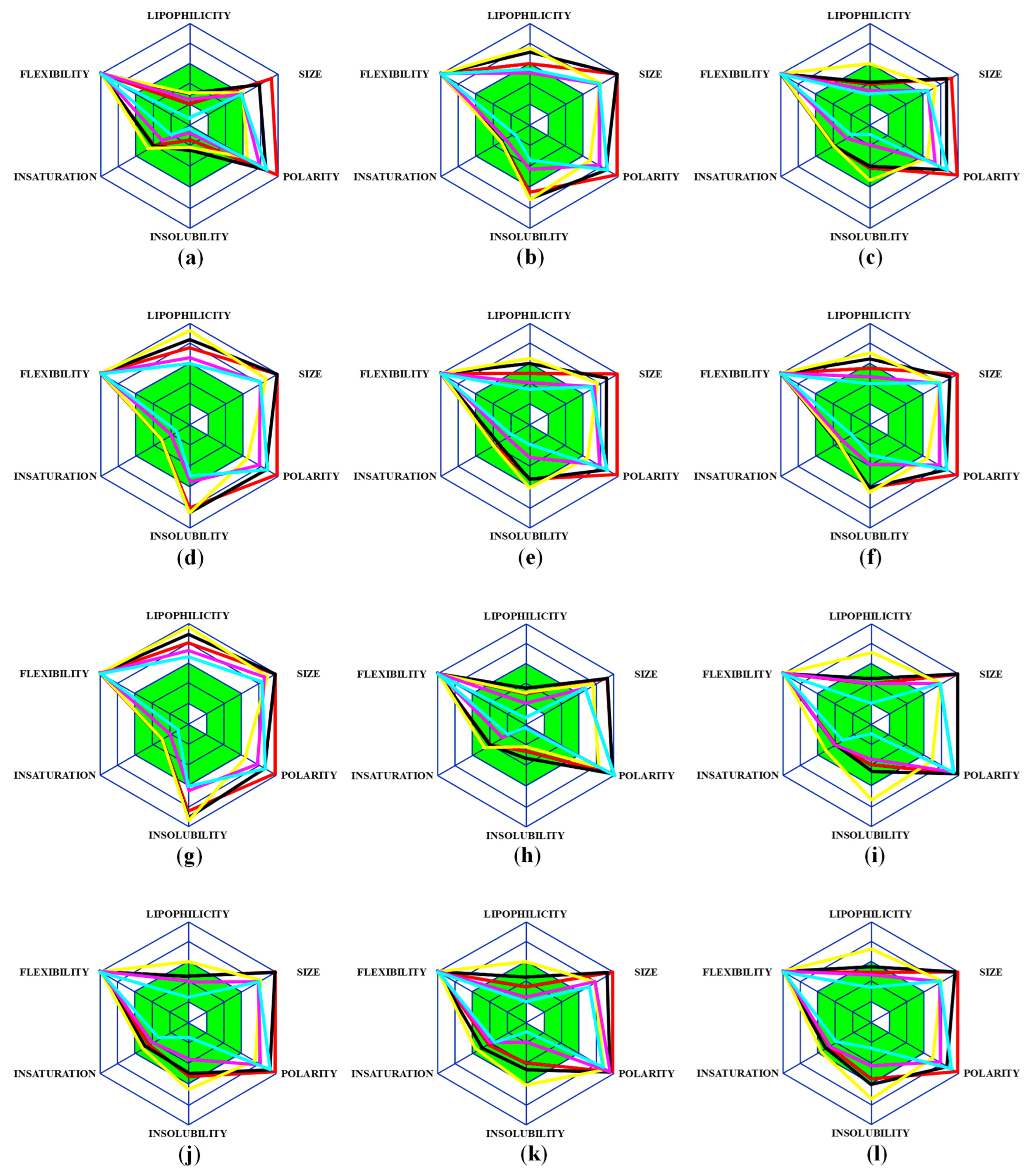
2.1. Analysis of Pharmacological Properties
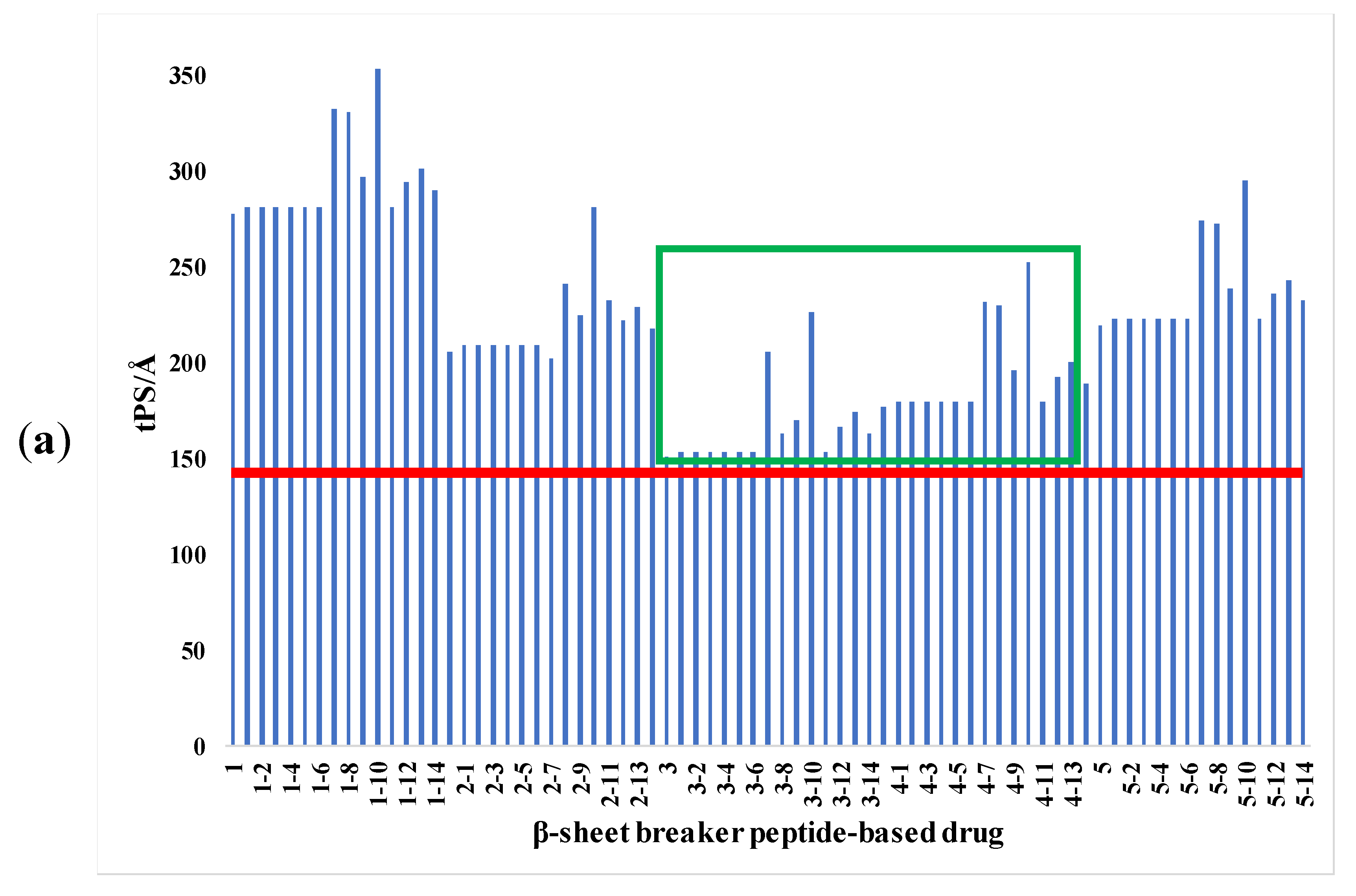
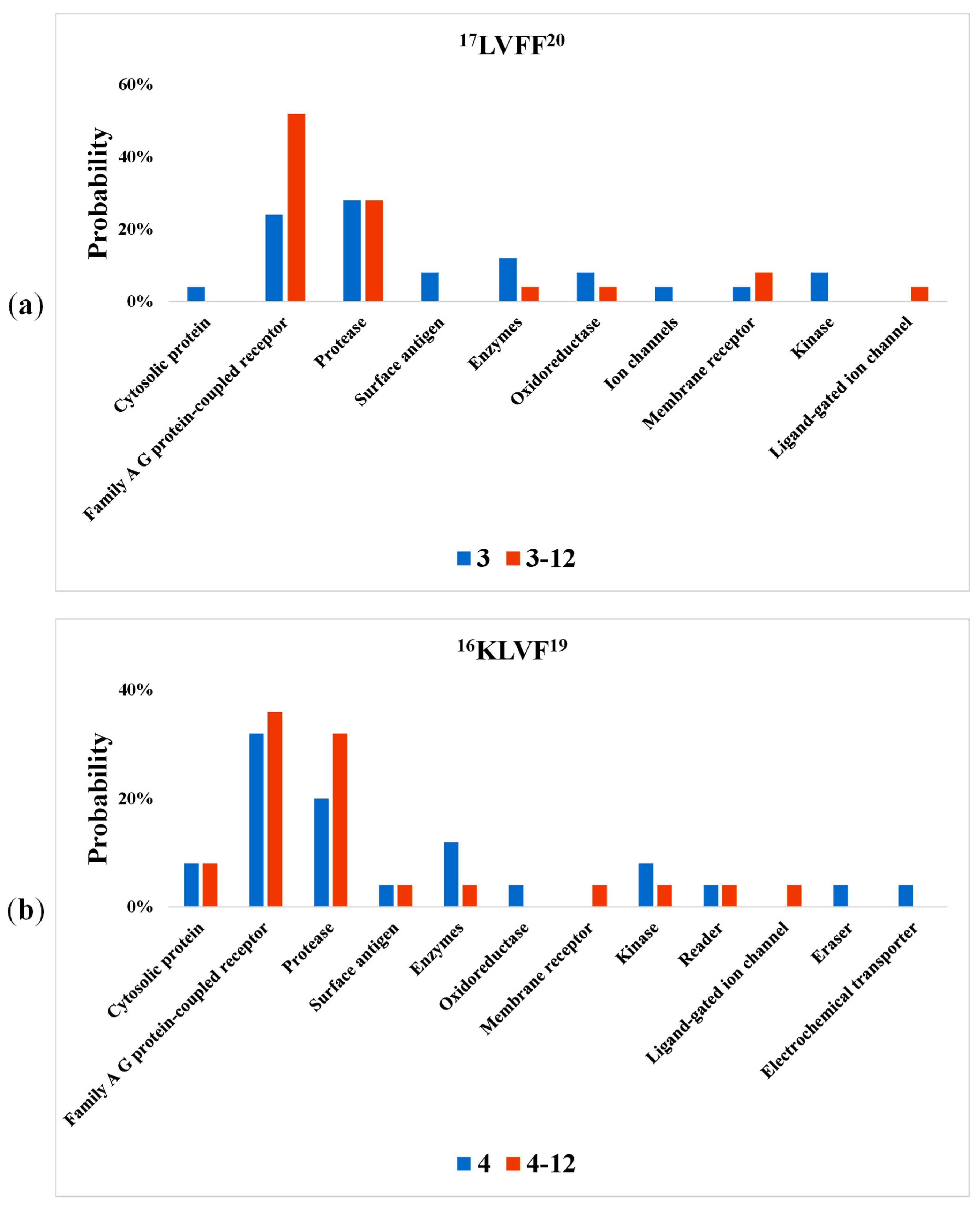
2.2. Bioactivity Screening
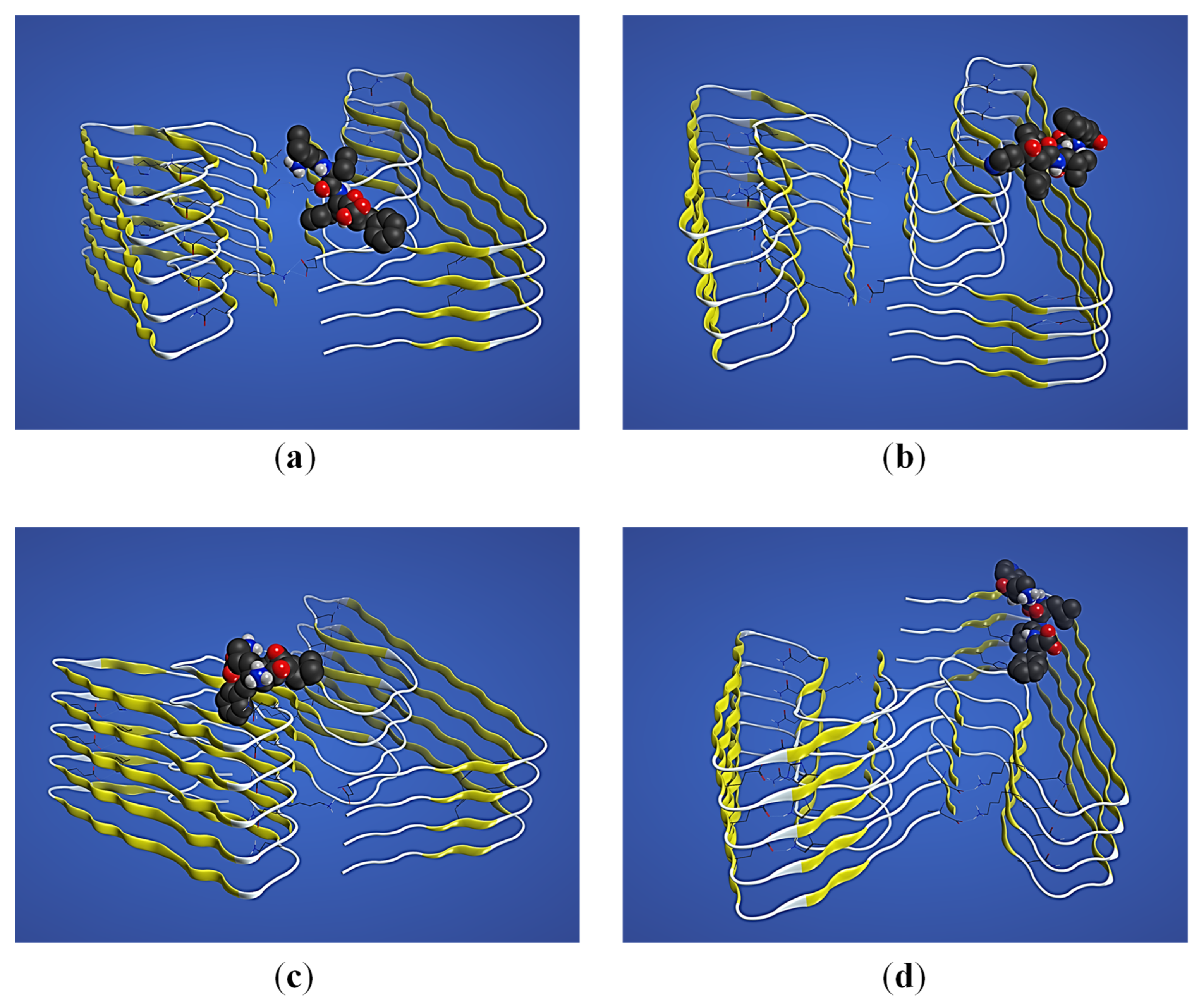
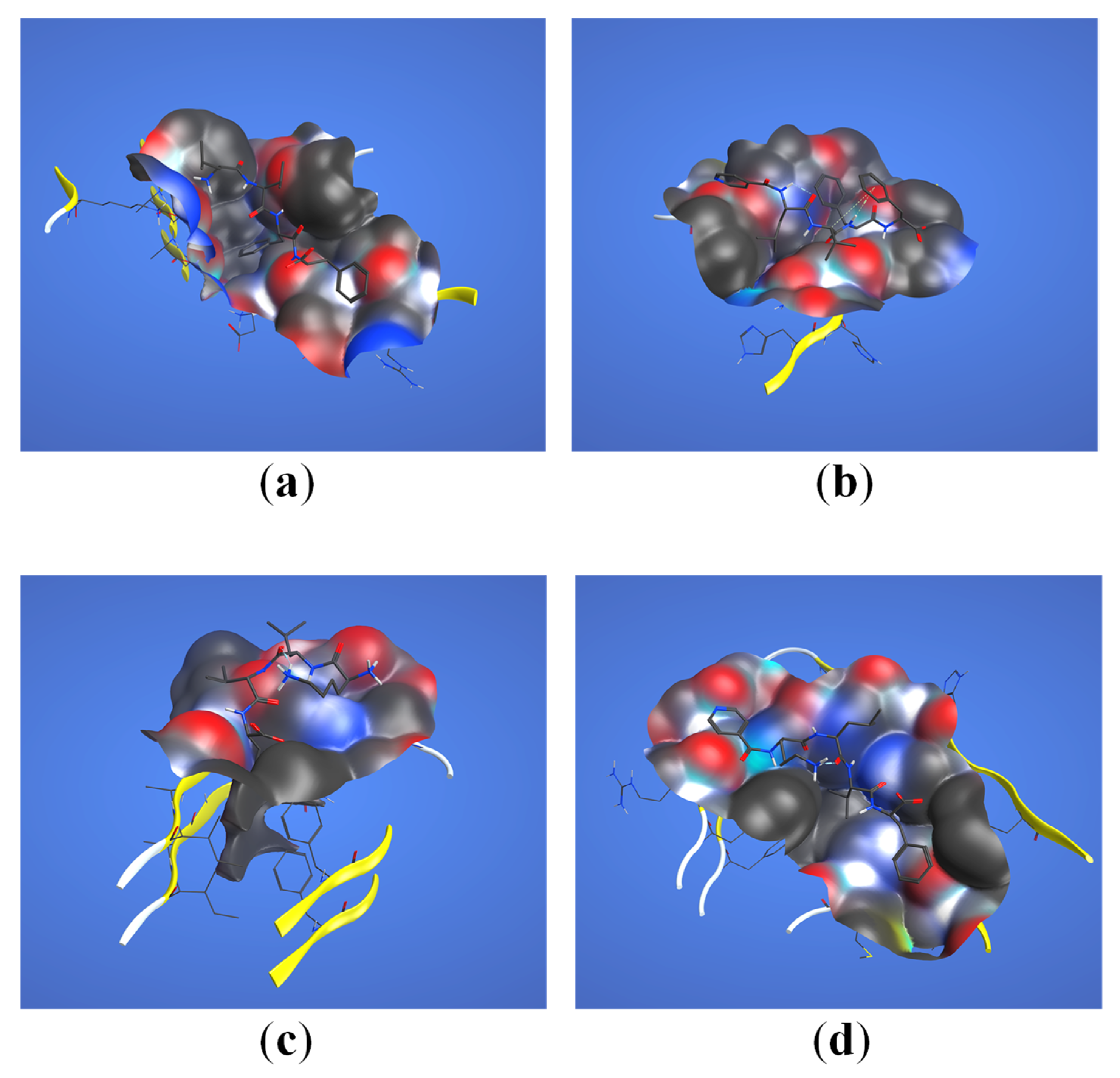
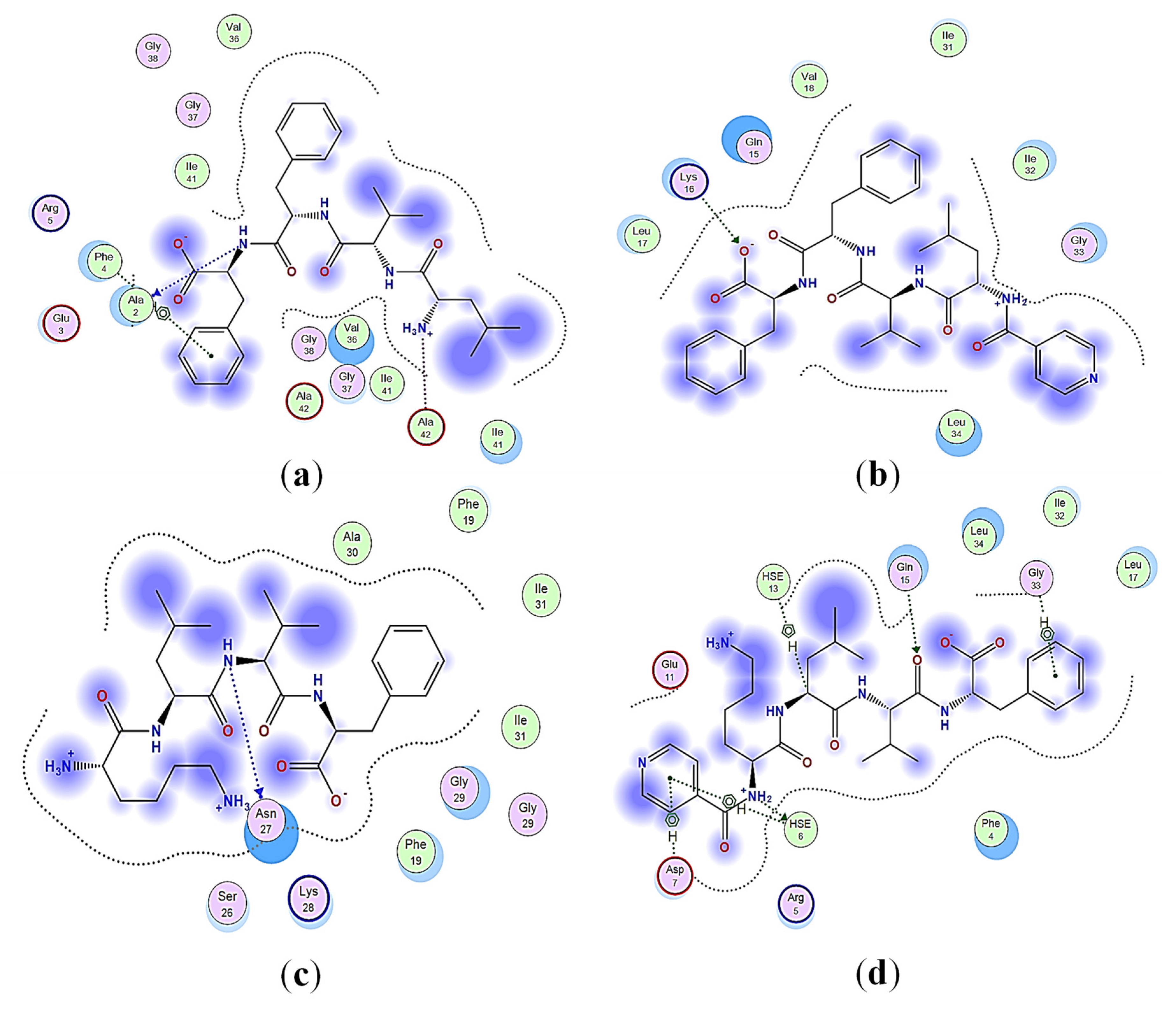
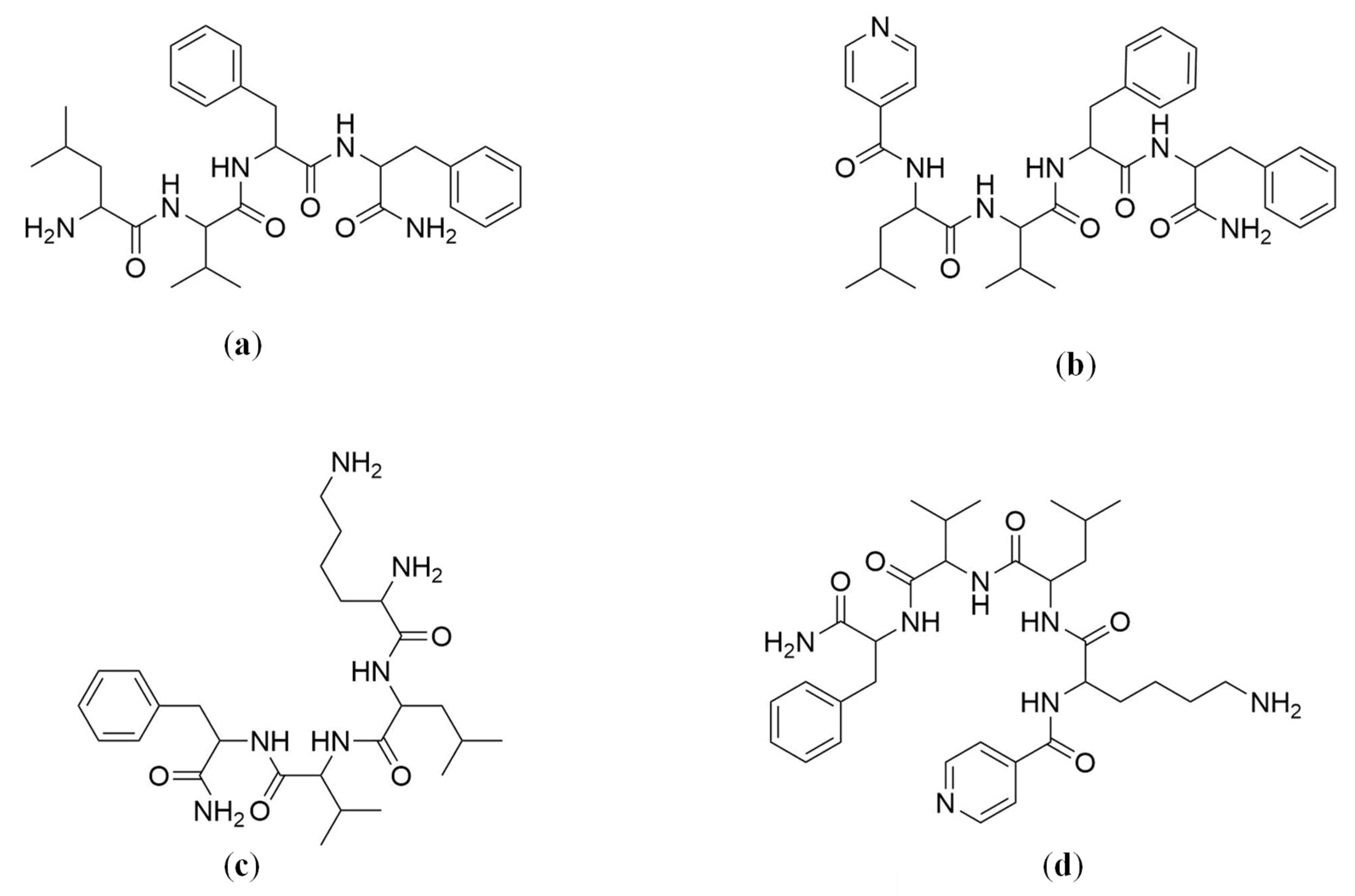
2.3. Structural Interaction and Docking
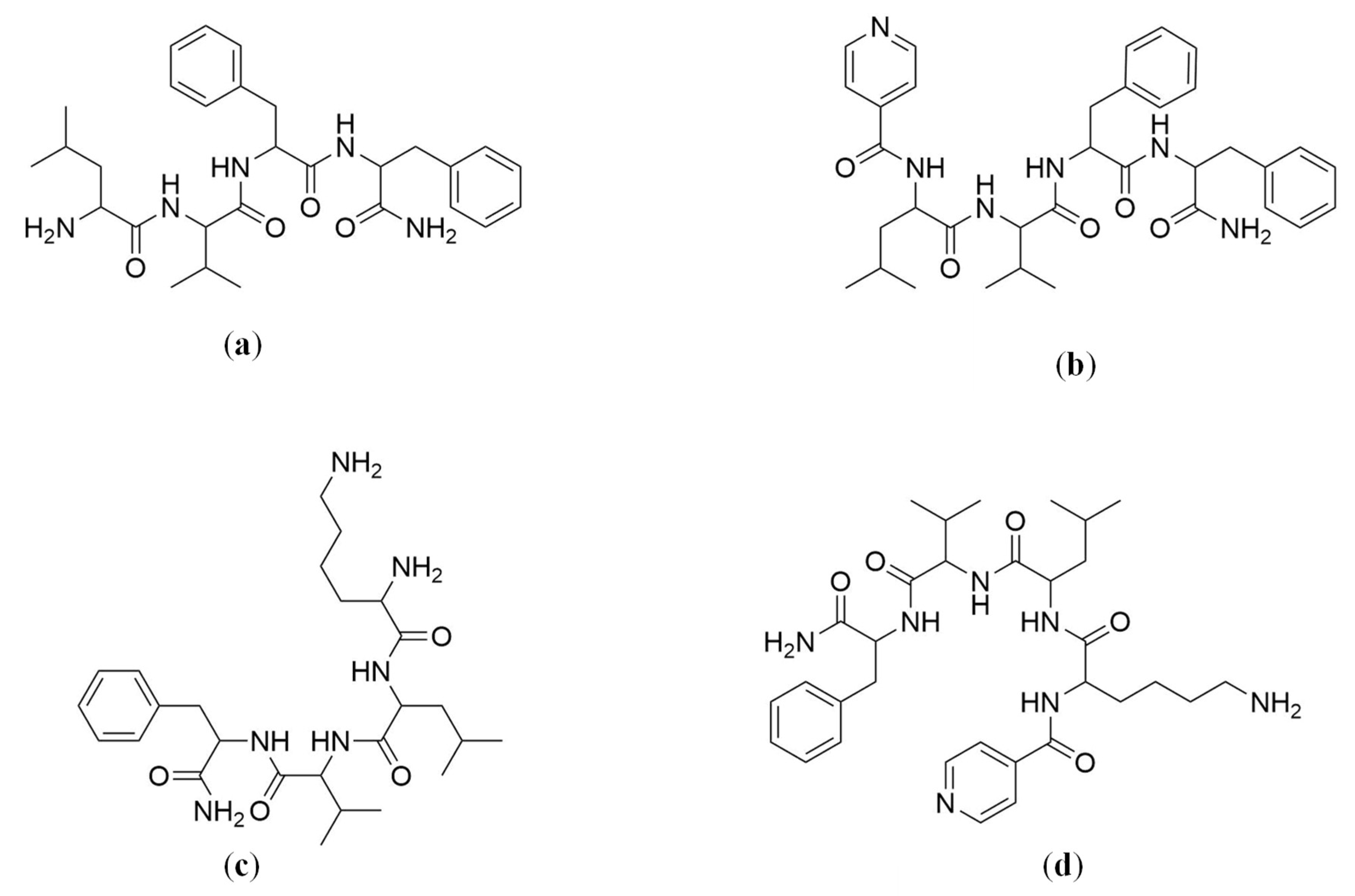
2.4. Solid-Phase Peptide Synthesis (SPPS)
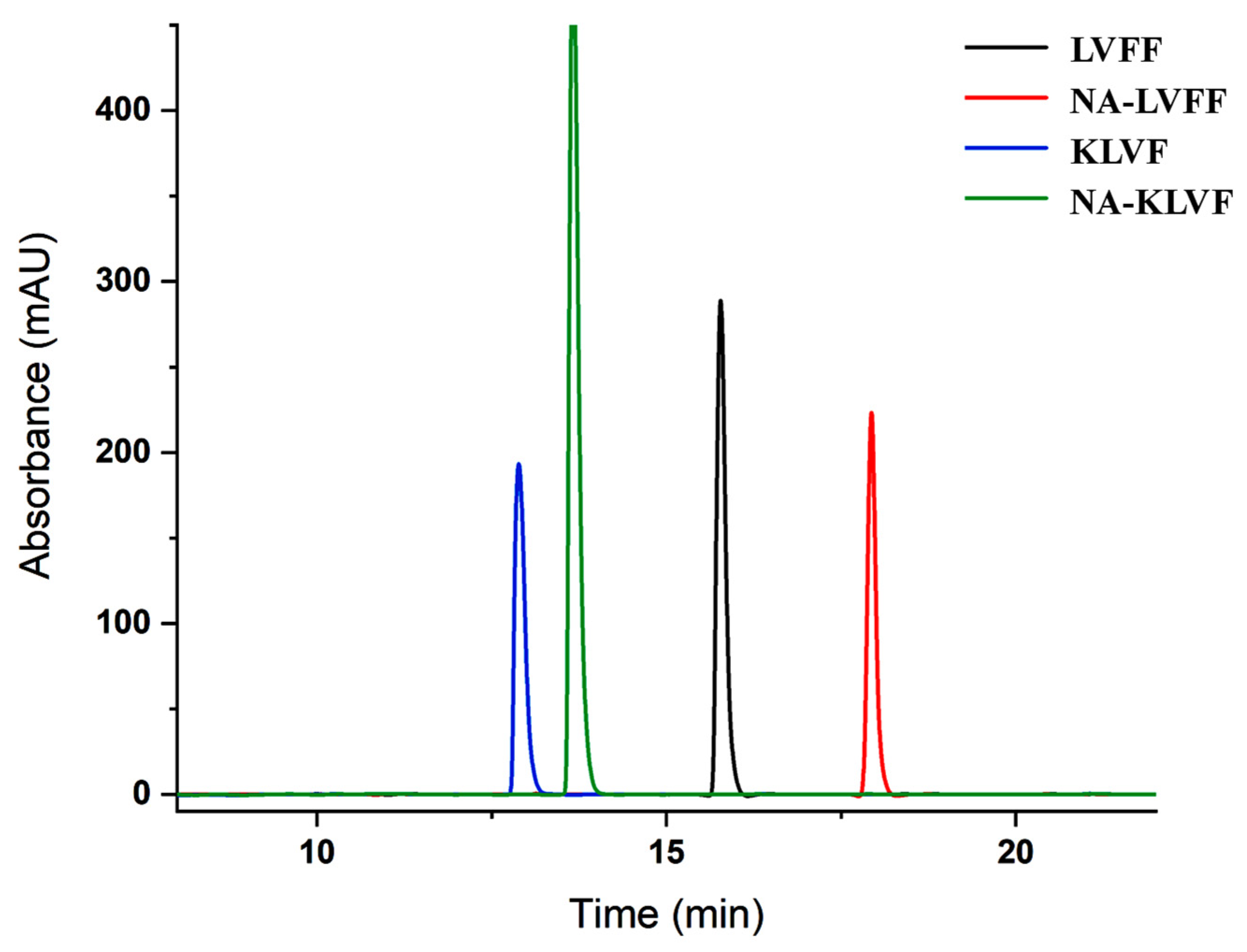
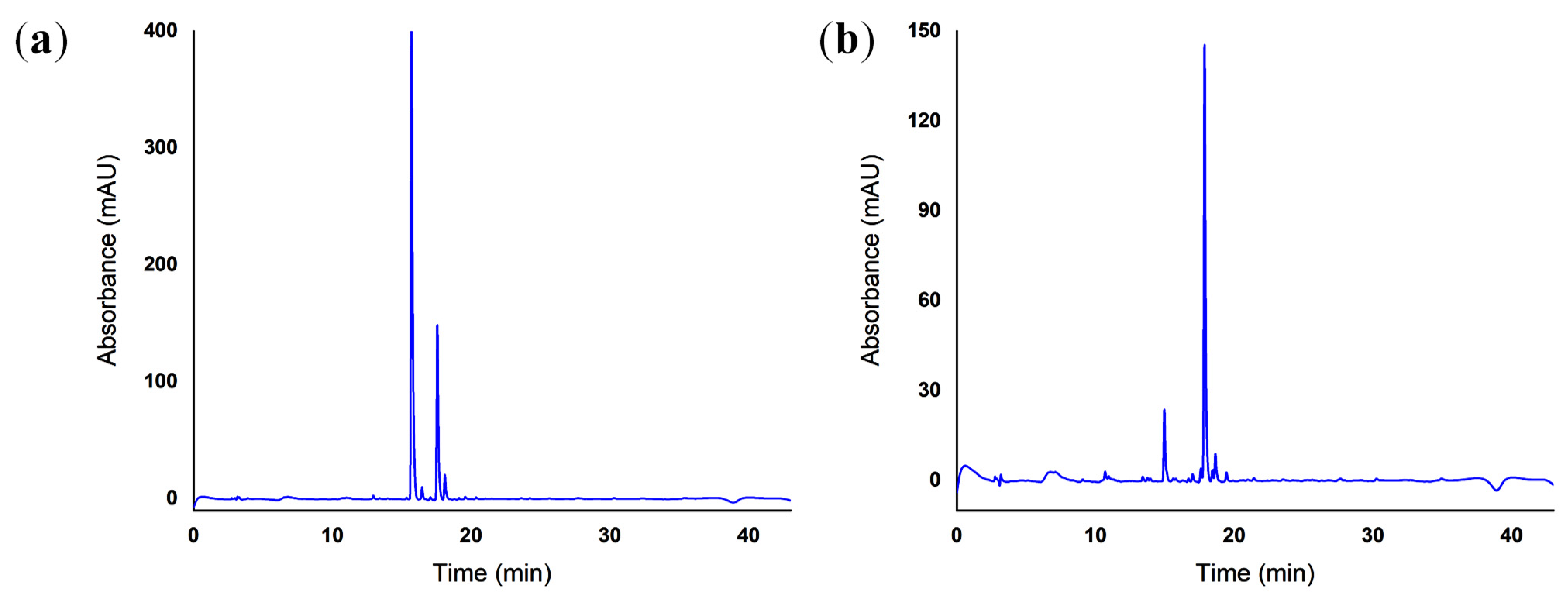
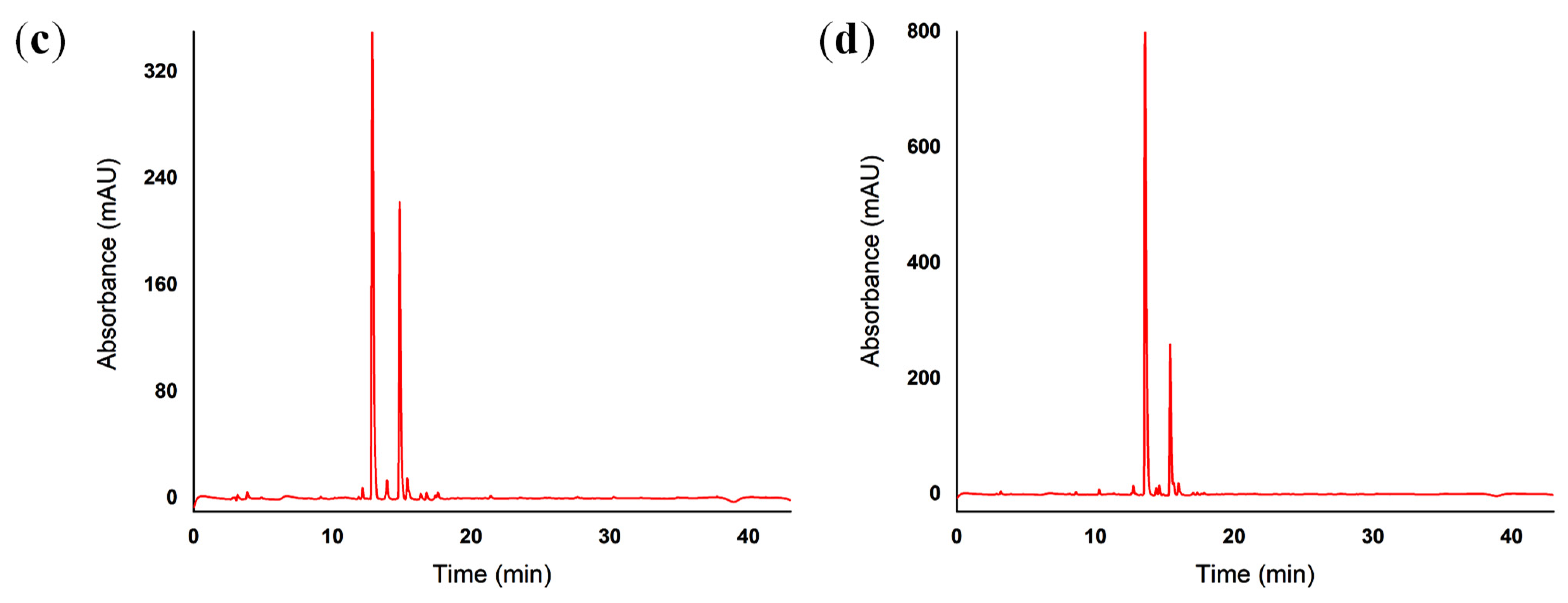
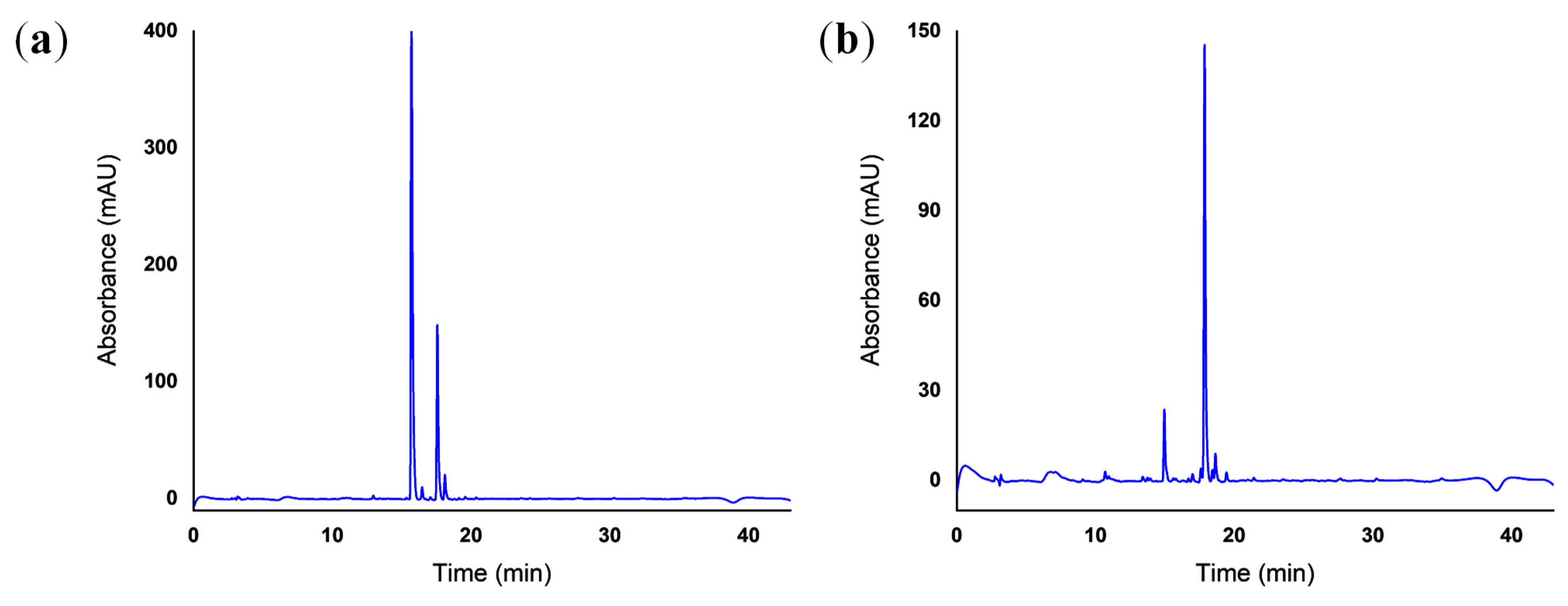
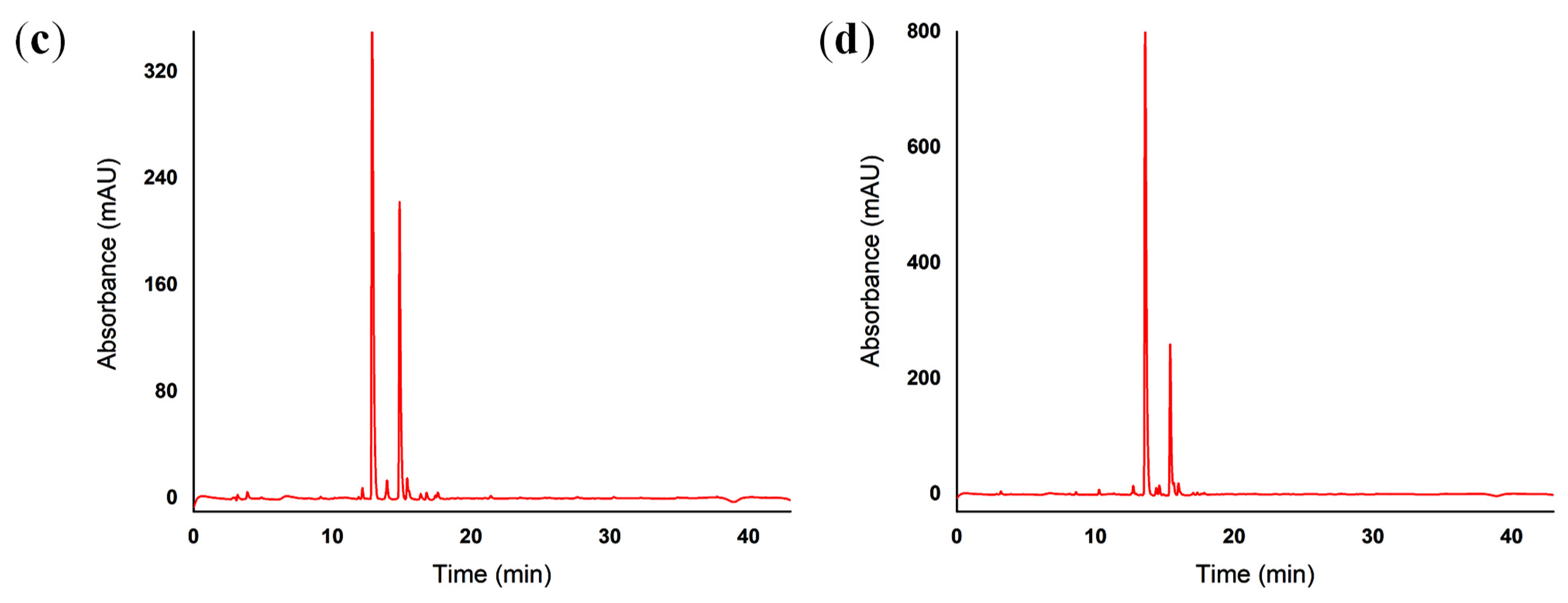
2.5. Reverse-Phase High-Performance Liquid Chromatography (RP-HPLC)
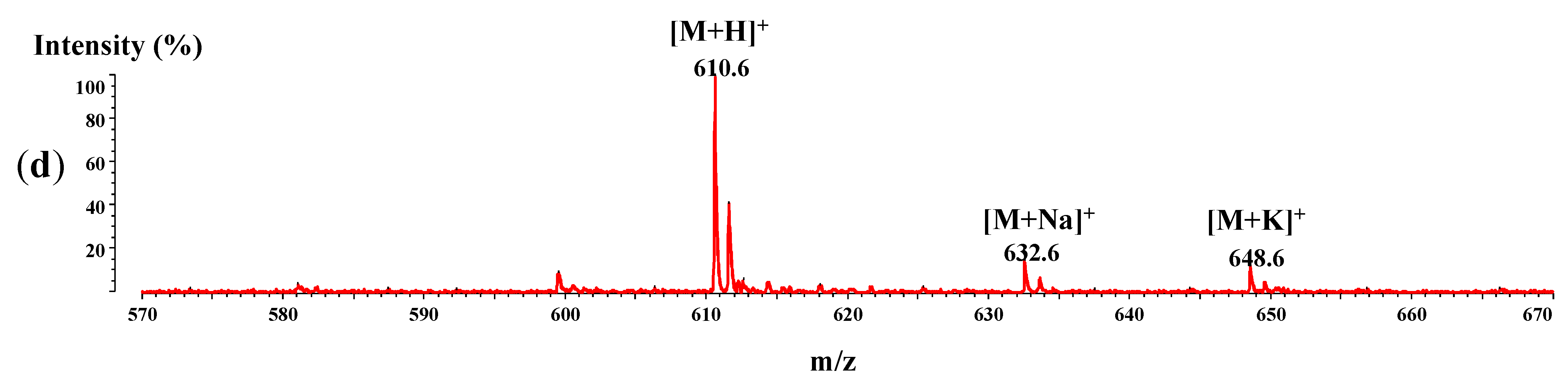
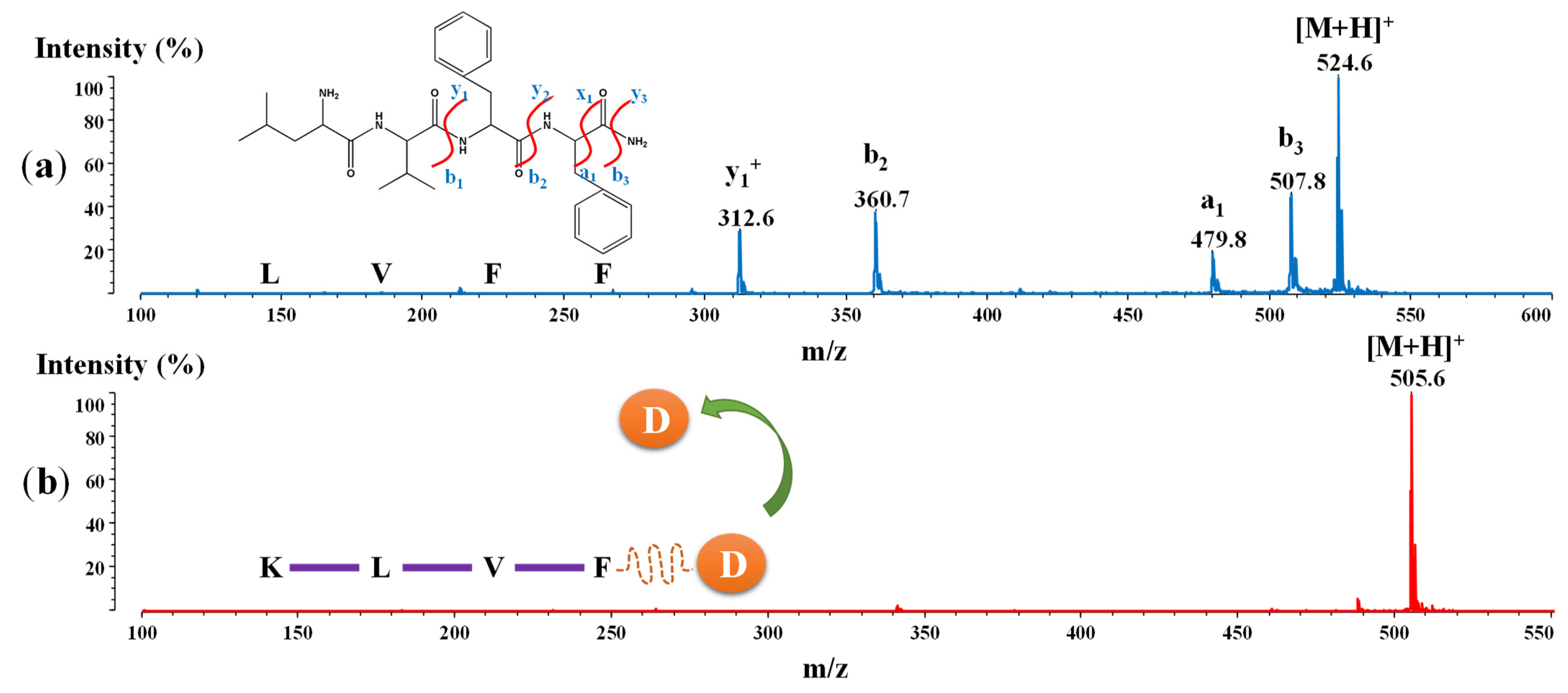
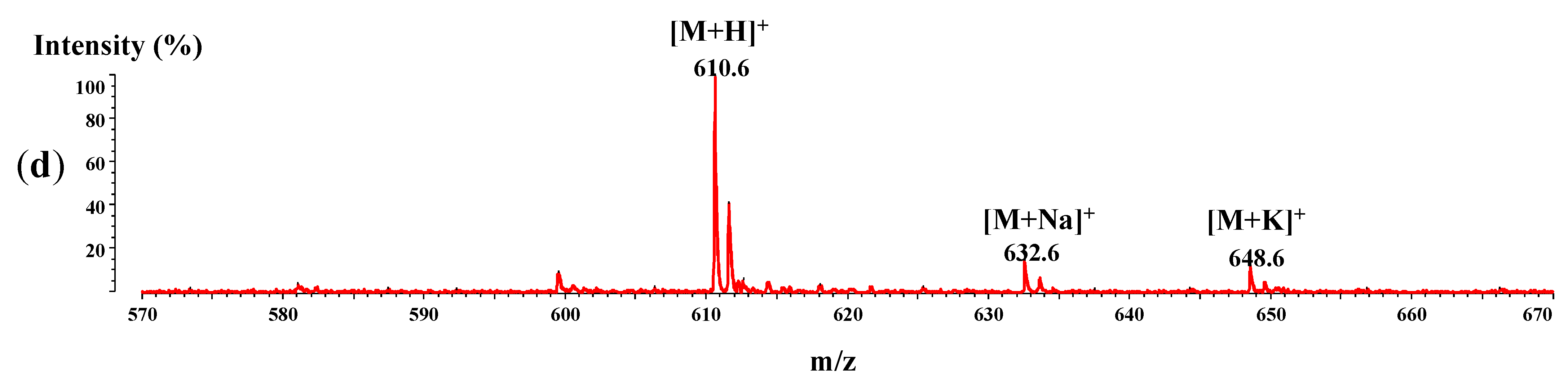
2.6. MALDI-ToF/ToFMass Spectrometry
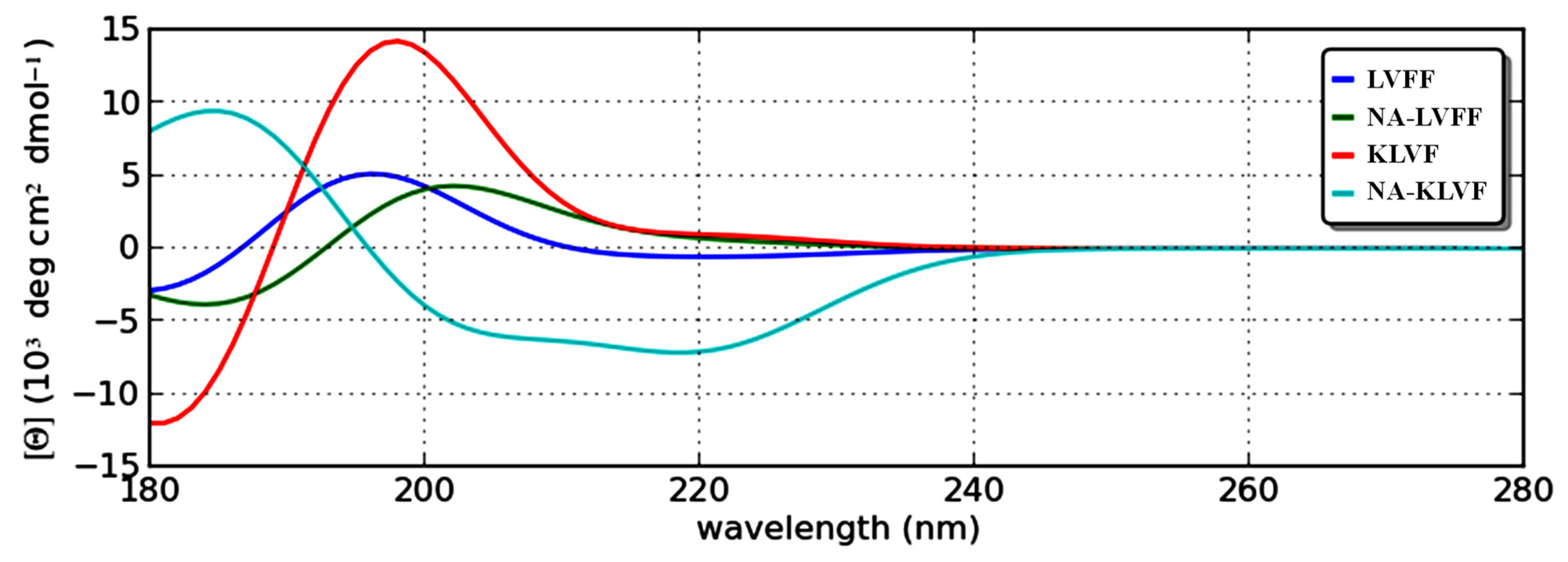
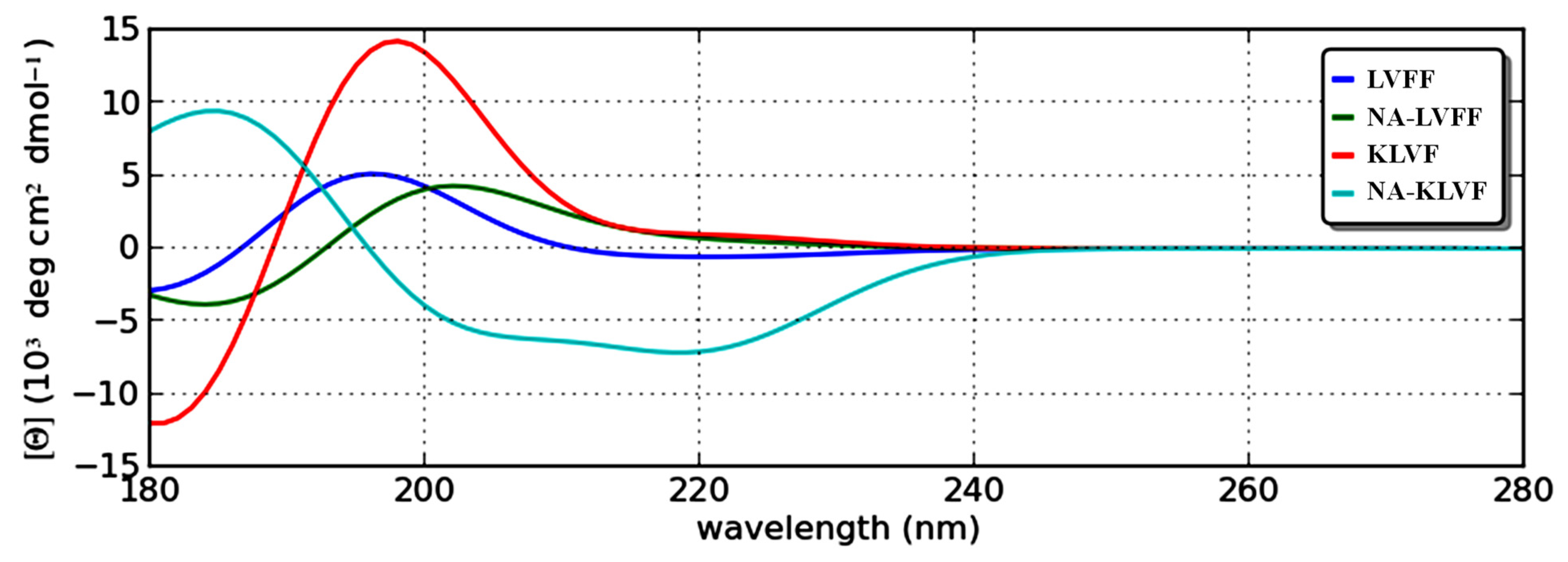
2.7. Theoretical Circular Dichroism
3. Discussion
4. Materials and Methods
4.1. Estimation of the Pharmacological Properties
4.2. Bioactivity Screening
4.3. Structural Interaction and Docking
4.4. Solid-Phase Peptide Synthesis (SPPS)
4.5. Reverse-Phase High-Performance Liquid Chromatography (RP-HPLC)
4.6. MALDI-ToF Mass Spectrometry
4.7. Circular Dichroism
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | % A (0.1% TFA) | % B (80% AcCN in 0.1% TFA, v/v) |
|---|---|---|
| 0 | 95 | 5 |
| 2 | 95 | 5 |
| 30 | 0 | 100 |
| 35 | 0 | 100 |
| 40 | 95 | 5 |
| 43 | 95 | 5 |

| Peptide | Purity (%) |
|---|---|
| 17LVFF20 | 73.94 |
| NA-17LVFF20 | 83.52 |
| 16KLVF19 | 66.02 |
| NA-16KLVF19 | 78.96 |
| Peptide | α-Helix Conformation (%) | β-Sheet Conformation (%) | Random Coil Conformation (%) |
|---|---|---|---|
| 17LVFF20 | 2.78 | 52.78 | 44.44 |
| NA-17LVFF20 | 0.00 | 56.14 | 43.86 |
| 16KLVF19 | 0.00 | 67.57 | 32.43 |
| NA-16KLVF19 | 5.05 | 38.38 | 56.57 |
References
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chetelat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Kaur, I.; Fratila, O.; Brata, R.; Bungau, S. Exploring the potential of therapeutic agents targeted towards mitigating the events associated with amyloid-β cascade in Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 7443. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Qin, W.; Zhu, M.; Jia, J. Model-based projection of dementia prevalence in China and worldwide: 2020–2050. J. Alzheimers Dis. 2021, 82, 1823–1831. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, D.; Chandra, R. Amyloid beta hypothesis in Alzheimer’s disease: Major culprits and recent therapeutic strategies. Curr. Drug Targets 2019, 21, 148–166. [Google Scholar] [CrossRef] [PubMed]
- Frisoni, G.B.; Altomare, D.; Thal, D.R.; Ribaldi, F.; van der Kant, R.; Ossenkoppele, R.; Blennow, K.; Cummings, J.; van Dujin, C.; Nilsson, P.M.; et al. The probabilistic model of Alzheimer disease: The amyloid hypothesis revised. Nat. Rev. Neurosci. 2022, 23, 53–66. [Google Scholar] [CrossRef]
- Calabro, M.; Rinaldi, C.; Santoro, G.; Crisafulli, C. The biological pathways of Alzheimer disease: A review. AIMS Neurosci. 2021, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Toward a comprehensive theory for Alzheimer’s disease. Hypothesis: Alzheimer’s disease is caused by the cerebral accumulation and cytotoxicity of amyloid β-protein. Ann. N. Y. Acad. Sci. 2000, 924, 17–25. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer disease: An update on pathobiology and treatment strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Weaver, D.F.; Meek, A. Amyloid-beta is a cytokine. Alzheimers Dement. 2021, 17, e054673. [Google Scholar]
- Sadigh-Eteghad, S.; Sabermarouf, B.; Majdi, A.; Talebi, M.; Farhoudi, M.; Mahmoudi, J. Amyloid-beta: A crucial factor in Alzheimer’s disease. Med. Princ. Pract. 2015, 24, 1–10. [Google Scholar] [CrossRef]
- Broersen, K.; Rousseau, F.; Schymkowitz, J. The culprit behind amyloid beta peptide related neurotoxicity in Alzheimer’s disease: Oligomer size or conformation? Alzheimers Res. Ther. 2010, 2, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Mocanu, C.S.; Jureschi, M.; Drochioiu, G. Aluminium binding to modified amyloid-β peptides: Implications for Alzheimer’s disease. Molecules 2020, 25, 4536. [Google Scholar] [CrossRef] [PubMed]
- Dufort-Gervais, J.; Provost, C.; Charbonneau, L.; Norris, C.M.; Calon, F.; Mongrain, V.; Brouillette, J. Neuroligin-1 is altered in the hippocampus of Alzheimer’s disease patients and mouse models, and modulates the toxicity of amyloid-beta oligomers. Sci. Rep. 2020, 10, 6956. [Google Scholar] [CrossRef] [PubMed]
- Millucci, L.; Ghezzi, L.; Bernardini, G.; Santucci, A. Conformations and biological activities of amyloid beta peptide 25–35. Curr. Protein Pept. Sci. 2010, 11, 54–67. [Google Scholar] [CrossRef]
- Murariu, M.; Ion, L.; Ciobanu, C.I.; Petre, B.A.; Drochioiu, G. MS, CD, and FT-IR characterization of five newly synthesized histidine-containing Ala-and Gly-based peptides. Rev. Roum. Chim. 2017, 62, 277–289. [Google Scholar]
- Rocha, S.; Thunemann, A.F.; do Carmo Pereira, M.; Coelho, M.; Mohwald, H.; Brezesinski, G. Influence of fluorinated and hydrogenated nanoparticles on the structure and fibrillogenesis of amyloid beta-peptide. Biophys. Chem. 2008, 137, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Murariu, M.; Habasescu, L.; Ciobanu, C.I.; Gradinaru, R.V.; Pui, A.; Drochioiu, G.; Mangalagiu, I. Interaction of amyloid Aβ (9–16) peptide fragment with metal ions: CD, FT-IR, and fluorescence spectroscopic studies. Int. J. Pept. Res. Ther. 2019, 25, 897–909. [Google Scholar] [CrossRef]
- Razzokov, J.; Yusupov, M.; Bogaerts, A. Oxidation destabilizes toxic amyloid beta peptide aggregation. Sci. Rep. 2019, 9, 5476. [Google Scholar] [CrossRef]
- Jobke, B.; McBride, T.; Nevin, L.; Peiperl, L.; Ross, A.; Stone, C.; Turner, R.; PLOS Medicine Editors. Setbacks in Alzheimer research demand new strategies, not surrender. PLoS Med. 2018, 15, e1002518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, C.; Hong, F.; Yang, S. Amyloidosis in Alzheimer’s disease: Pathogeny, etiology, and related therapeutic directions. Molecules 2022, 27, 1210. [Google Scholar] [CrossRef] [PubMed]
- Lalatsa, A.; Schatzlein, A.G.; Uchegbu, I.F. Strategies to deliver peptide drugs to the brain. Mol. Pharm. 2014, 11, 1081–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mocanu, C.S.; Bocec, A.S.; Gradinaru, V.R.; Anton-Paduraru, D.T. A biochemical method for tyrosine determination in phenylketonuria using a colorimetric enzymatic approach. Rev. Chim. 2020, 71, 285–294. [Google Scholar] [CrossRef]
- Drochioiu, G.; Ion, L.; Ciobanu, C.; Habasescu, L.; Mangalagiu, I. Mass spectrometric approach of high pH-and copper-induced glutathione oxidation. Eur. J. Mass Spectrom. 2013, 19, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Hamman, J.H.; Enslin, G.M.; Kotze, A.F. Oral delivery of peptide drugs. BioDrugs 2005, 19, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Calva, C.B.; Fadel, J.R. Intranasal administration of orexin peptides: Mechanisms and therapeutic potential for age-related cognitive dysfunction. Brain Res. 2020, 1731, 145921. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Jin, K. A gold mine for drug discovery: Strategies to develop cyclic peptides into therapies. Med. Res. Rev. 2020, 40, 753–810. [Google Scholar] [CrossRef] [PubMed]
- Mocanu, C.S.; Drochioiu, G. The interaction of possible anti-AD ASA-NAP peptide conjugate with tubulin: A theoretical and experimental insight. Int. J. Pept. Res. Ther. 2021, 27, 2487–2503. [Google Scholar] [CrossRef]
- Ciobanu, C.I.; Stefanescu, R.; Niculaua, M.; Teslaru, T.; Gradinaru, R.; Drochioiu, G. Mass spectrometric evidence for iron binding to the neuroprotective peptide NAP and its Cys5 mutant. Eur. J. Mass Spectrom. 2016, 22, 97–104. [Google Scholar] [CrossRef]
- Lupaescu, A.V.; Mocanu, C.S.; Drochioiu, G.; Ciobanu, C.I. Zinc Binding to NAP-type neuroprotective peptides: Nuclear magnetic resonance studies and molecular modeling. Pharmaceuticals 2021, 14, 1011. [Google Scholar] [CrossRef]
- Zhao, S.; Li, J.; Wang, F.; Yu, T.; Zhou, Y.; He, L.; Zhang, Y.; Yang, J. Semi-elastic core-shell nanoparticles enhanced the oral bioavailability of peptide drugs. Chin. Chem. Lett. 2020, 31, 1147–1152. [Google Scholar] [CrossRef]
- Stepensky, D. Pharmacokinetics of toxin-derived peptide drugs. Toxins 2018, 10, 483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, R.; Wong, S.W.; Ge, L.; Shaw, C.; Siu, S.W.; Kwok, H.F. In vitro and MD simulation study to explore physicochemical parameters for antibacterial peptide to become potent anticancer peptide. Mol. Ther.-Oncolytics 2020, 16, 7–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjernberg, L.O.; Naslund, J.; Lindqvist, F.; Johansson, J.; Karlstrom, A.R.; Thyberg, J.; Tereniust, L.; Nordstedt, C. Arrest ofamyloid fibril formation by a pentapeptide ligand (∗). J. Biol. Chem. 1996, 271, 8545–8548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjernberg, L.O.; Lilliehoook, C.; Callaway, D.J.; Naslund, J.; Hahne, S.; Thyberg, J.; Terenius, C.; Nordstedt, C. Controlling amyloid β-peptide fibril formation with protease-stable ligands. J. Biol. Chem. 1997, 272, 12601–12605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benzinger, T.L.; Gregory, D.M.; Burkoth, T.S.; Miller-Auer, H.; Lynn, D.G.; Botto, R.E.; Meredith, S.C. Two-dimensional structure of β-amyloid (10–35) fibrils. Biochemistry 2000, 39, 3491–3499. [Google Scholar] [CrossRef]
- Mulligan, V.K. The emerging role of computational design in peptide macrocycle drug discovery. Expert Opin. Drug Discov. 2020, 15, 833–852. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Wildman, S.A.; Crippen, G.M. Prediction of physicochemical parameters by atomic contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Ludwig-Muller, J. Indole-3-butyric acid in plant growth and development. Plant Growth Regul. 2000, 32, 219–230. [Google Scholar] [CrossRef]
- Kwon, W.Y.; Suh, G.J.; Kim, K.S.; Lee, H.J.; Jeong, K.Y.; Kwak, Y.H.; Kim, K. Niacin suppresses the mitogen-activated protein kinase pathway and attenuates brain injury after cardiac arrest in rats. Crit. Care Med. 2013, 41, e223–e232. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.B.; Han, J.; Zhang, N.; Tian, Z.; Li, X.B.; Zhao, M.G. Neuroprotective effects of oestrogen against oxidative toxicity through activation of G-protein-coupled receptor 30 receptor. Clin. Exp. Pharmacol. Physiol. 2011, 38, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Ying, G.; Iribarren, P.; Zhou, Y.; Gong, W.; Zhang, N.; Yu, Z.X.; Le, Y.; Cui, Y.; Wang, J.M. Humanin, a newly identified neuroprotective factor, uses the G protein-coupled formylpeptide receptor-like-1 as a functional receptor. J. Immunol. 2004, 172, 7078–7085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Waterschoot, R.A.; Schinkel, A.H. A critical analysis of the interplay between cytochrome P450 3A and P-glycoprotein: Recent insights from knockout and transgenic mice. Pharmacol. Rev. 2011, 63, 390–410. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Shou, M.; Grogan, J.; Mancewicz, J.A.; Krausz, K.W.; Gonzalez, F.J.; Gelboin, H.V.; Korzekwa, K.R. Activation of CYP3A4: Evidence for the simultaneous binding of two substrates in a cytochrome P450 active site. Biochemistry 1994, 33, 6450–6455. [Google Scholar] [CrossRef]
- Mocanu, C.S.; Petre, B.A.; Ion, L.D.; Drochioiu, G.; Niculaua, M.; Stoica, I.; Homocianu, M.; Nita, L.E.; Gradinaru, V.R. Structural characterization of a new collagen biomimetic octapeptide with nanoscale self-assembly potential: Experimental and theoretical approaches. ChemPlusChem 2021, 87, e202100462. [Google Scholar] [CrossRef]
- Hamilton, J.A.; Brunaldi, K. A model for fatty acid transport into the brain. J. Mol. Neurosci. 2007, 33, 12–17. [Google Scholar] [CrossRef]
- Jang, H.; Lee, S.; Choi, S.L.; Kim, H.Y.; Baek, S.; Kim, Y. Taurine directly binds to oligomeric amyloid-β and recovers cognitive deficits in Alzheimer model mice. In Taurine; Springer: Dordrecht, The Netherlands, 2017; Volume 10, pp. 233–241. [Google Scholar]
- Moutinho, M.; Tsai, A.P.; Puntambekar, S.S.; Patel, J.; Lin, P.B.; Jadhav, V.; Williams, R.Y.; Shekhar, A.; Landreth, G.E. Therapeutic potential of niacin in Alzheimer’s disease: Nonhuman/target identification and validation studies: Other. Alzheimers Dement. 2020, 16, e040679. [Google Scholar] [CrossRef]
- De La Cruz, J.P.; Guerrero, A.; Gonzalez-Correa, J.A.; Arrebola, M.M.; Sanchez De La Cuesta, F. Antioxidant effect of acetylsalicylic and salicylic acid in rat brain slices subjected to hypoxia. J. Neurosci. Res. 2004, 75, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Tortosa, E.; Avila, J.; Perez, M. Acetylsalicylic acid decreases tau phosphorylation at serine 422. Neurosci. Lett. 2006, 396, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R.; Lai, L. Computation of octanol—water partition coefficients by guiding an additive model with knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.S. ESOL: Estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef]
- Tian, S.; Wang, J.; Li, Y.; Li, D.; Xu, L.; Hou, T. The application of in silico drug-likeness predictions in pharmaceutical research. Adv. Drug Deliv. Rev. 2015, 86, 2–10. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [Green Version]
- Gfeller, D.; Michielin, O.; Zoete, V. Shaping the interaction landscape of bioactive molecules. Bioinformatics 2013, 29, 3073–3079. [Google Scholar] [CrossRef]
- Crescenzi, O.; Tomaselli, S.; Guerrini, R.; Salvadori, S.; D’Ursi, A.M.; Temussi, P.A.; Picone, D. Solution structure of the Alzheimer amyloid β-peptide (1–42) in an apolar microenvironment: Similarity with a virus fusion domain. Eur. J. Biochem. 2002, 269, 5642–5648. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE) 2016.02, Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montr. QC, Canada, H3A 2R7. 2018. Available online: https://www.chemcomp.com/Products.htm (accessed on 12 February 2022).
- Chan, S.L.; Labute, P. Training a scoring function for the alignment of small molecules. J. Chem. Inf. Model. 2010, 50, 1724–1735. [Google Scholar] [CrossRef]
- Kearsley, S.K.; Smith, G.M. An alternative method for the alignment of molecular structures: Maximizing electrostatic and steric overlap. Tetrahedron Comput. Methodol. 1990, 3, 615–633. [Google Scholar] [CrossRef]
- Labute, P.; Williams, C.; Feher, M.; Sourial, E.; Schmidt, J.M. Flexible alignment of small molecules. J. Med. Chem. 2001, 44, 1483–1490. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.M.; Labute, P. 2D depiction of protein− ligand complexes. J. Chem. Inf. Model. 2007, 47, 1933–1944. [Google Scholar] [CrossRef] [PubMed]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Clark, A.M.; Labute, P.; Santavy, M. 2D structure depiction. J. Chem. Inf. Model. 2006, 46, 1107–1123. [Google Scholar] [CrossRef]
- Gremer, L.; Scholzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.; Tusche, M.; Lopez-iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril structure of amyloid-β (1–42) by cryo–electron microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosdidier, A.; Zoete, V.; Michielin, O. Fast docking using the CHARMM force field with EADock DSS. J. Comput. Chem. 2011, 32, 2149–2159. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, H.; Takaishi, H.; Ikeda, H.; Ariumi, H.; Hamada, Y.; Yamashita, K.; Usui, K. Synthesis of peptide-immobilized magnetic beads, and peptide reactivity assay for assessing skin sensitization utilizing chromophore. Processes 2020, 8, 1257. [Google Scholar] [CrossRef]
- Bulheller, B.M.; Hirst, J.D. DichroCalc—Circular and linear dichroism online. Bioinformatics 2009, 25, 539–540. [Google Scholar] [CrossRef]
- Wiedemann, C.; Bellstedt, P.; Gorlach, M. CAPITO—A web server-based analysis and plotting tool for circular dichroism data. Bioinformatics 2013, 29, 1750–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]

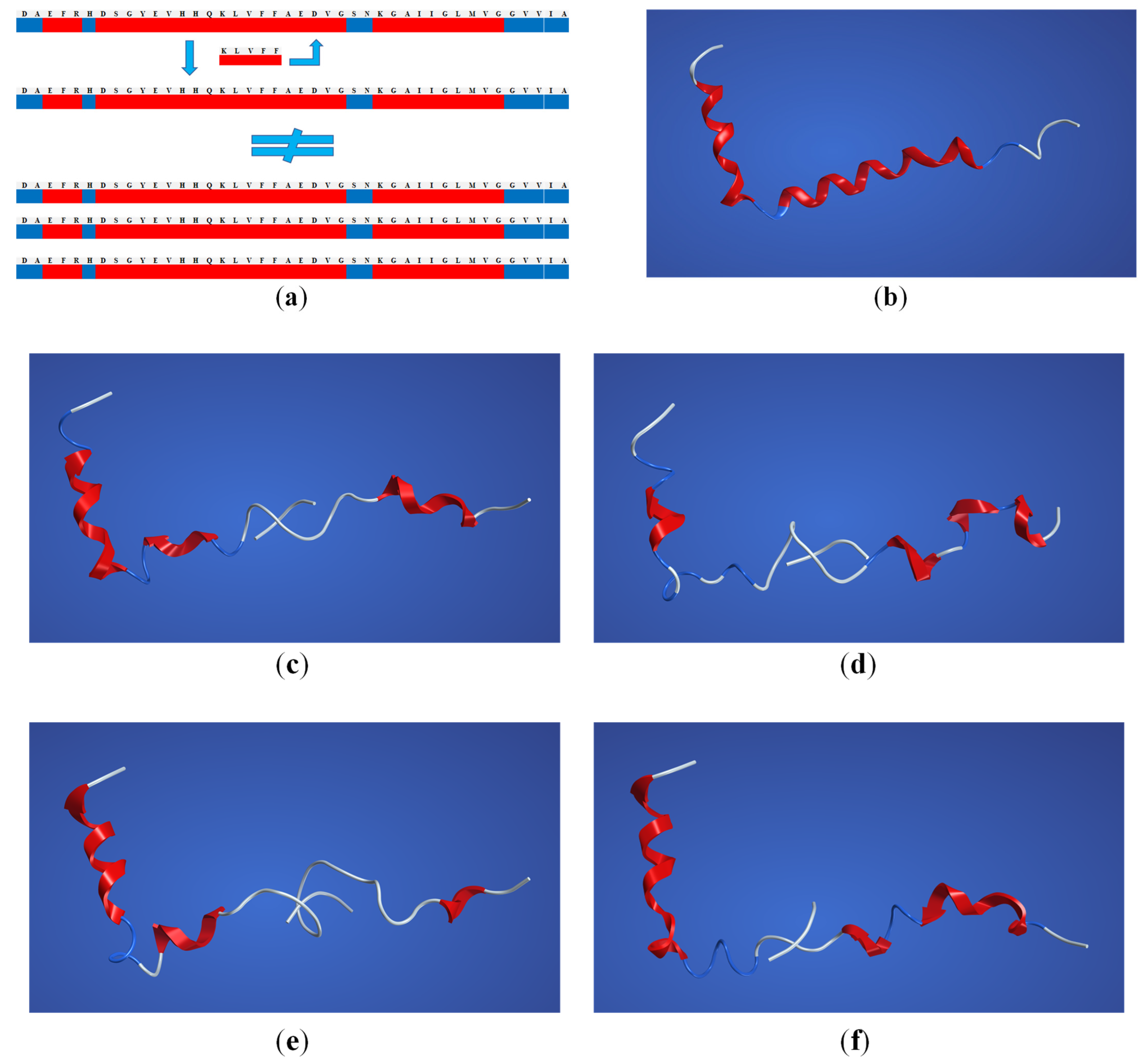
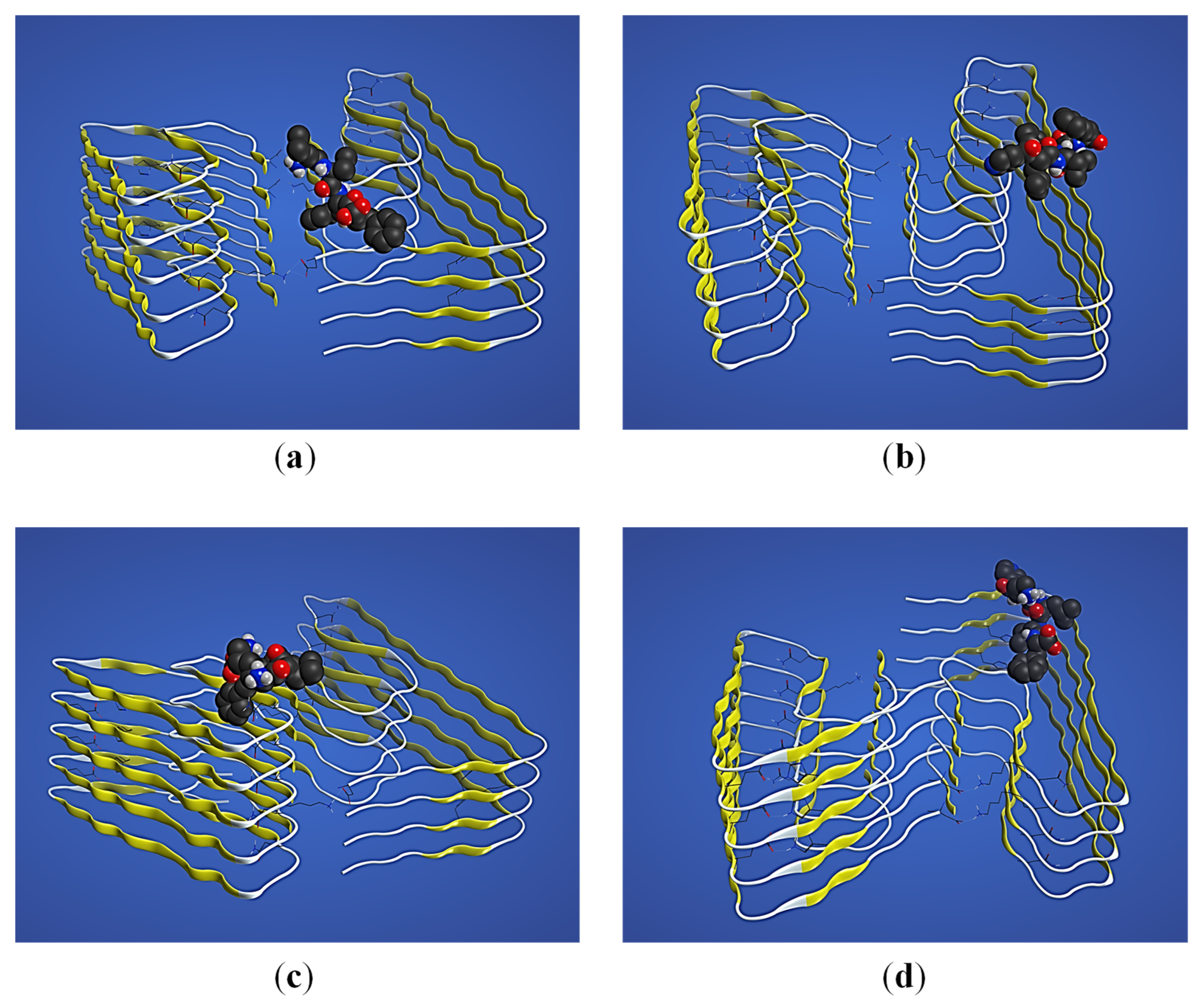



| Code | OC | Code | OC | Code | OC | Code | OC | Code | OC | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Peptide ↓ | Peptide ↓ | Peptide ↓ | Peptide ↓ | Peptide ↓ | ||||||||||
| 1-1 | 1 | 1 | 2-1 | 2 | 1 | 3-1 | 3 | 1 | 4-1 | 4 | 1 | 5-1 | 5 | 1 |
| 1-2 | 1 | 2 | 2-2 | 2 | 2 | 3-2 | 3 | 2 | 4-2 | 4 | 2 | 5-2 | 5 | 2 |
| 1-3 | 1 | 3 | 2-3 | 2 | 3 | 3-3 | 3 | 3 | 4-3 | 4 | 3 | 5-3 | 5 | 3 |
| 1-4 | 1 | 4 | 2-4 | 2 | 4 | 3-4 | 3 | 4 | 4-4 | 4 | 4 | 5-4 | 5 | 4 |
| 1-5 | 1 | 5 | 2-5 | 2 | 5 | 3-5 | 3 | 5 | 4-5 | 4 | 5 | 5-5 | 5 | 5 |
| 1-6 | 1 | 6 | 2-6 | 2 | 6 | 3-6 | 3 | 6 | 4-6 | 4 | 6 | 5-6 | 5 | 6 |
| 1-7 | 1 | 7 | 2-7 | 2 | 7 | 3-7 | 3 | 7 | 4-7 | 4 | 7 | 5-7 | 5 | 7 |
| 1-8 | 1 | 8 | 2-8 | 2 | 8 | 3-8 | 3 | 8 | 4-8 | 4 | 8 | 5-8 | 5 | 8 |
| 1-9 | 1 | 9 | 2-9 | 2 | 9 | 3-9 | 3 | 9 | 4-9 | 4 | 9 | 5-9 | 5 | 9 |
| 1-10 | 1 | 10 | 2-10 | 2 | 10 | 3-10 | 3 | 10 | 4-10 | 4 | 10 | 5-10 | 5 | 10 |
| 1-11 | 1 | 11 | 2-11 | 2 | 11 | 3-11 | 3 | 11 | 4-11 | 4 | 11 | 5-11 | 5 | 11 |
| 1-12 | 1 | 12 | 2-12 | 2 | 12 | 3-12 | 3 | 12 | 4-12 | 4 | 12 | 5-12 | 5 | 12 |
| 1-13 | 1 | 13 | 2-13 | 2 | 13 | 3-13 | 3 | 13 | 4-13 | 4 | 13 | 5-13 | 5 | 13 |
| 1-14 | 1 | 14 | 2-14 | 2 | 14 | 3-14 | 3 | 14 | 4-14 | 4 | 14 | 5-14 | 5 | 14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mocanu, C.S.; Niculaua, M.; Zbancioc, G.; Mangalagiu, V.; Drochioiu, G. Novel Design of Neuropeptide-Based Drugs with β-Sheet Breaking Potential in Amyloid-Beta Cascade: Molecular and Structural Deciphers. Int. J. Mol. Sci. 2022, 23, 2857. https://doi.org/10.3390/ijms23052857
Mocanu CS, Niculaua M, Zbancioc G, Mangalagiu V, Drochioiu G. Novel Design of Neuropeptide-Based Drugs with β-Sheet Breaking Potential in Amyloid-Beta Cascade: Molecular and Structural Deciphers. International Journal of Molecular Sciences. 2022; 23(5):2857. https://doi.org/10.3390/ijms23052857
Chicago/Turabian StyleMocanu, Cosmin Stefan, Marius Niculaua, Gheorghita Zbancioc, Violeta Mangalagiu, and Gabi Drochioiu. 2022. "Novel Design of Neuropeptide-Based Drugs with β-Sheet Breaking Potential in Amyloid-Beta Cascade: Molecular and Structural Deciphers" International Journal of Molecular Sciences 23, no. 5: 2857. https://doi.org/10.3390/ijms23052857
APA StyleMocanu, C. S., Niculaua, M., Zbancioc, G., Mangalagiu, V., & Drochioiu, G. (2022). Novel Design of Neuropeptide-Based Drugs with β-Sheet Breaking Potential in Amyloid-Beta Cascade: Molecular and Structural Deciphers. International Journal of Molecular Sciences, 23(5), 2857. https://doi.org/10.3390/ijms23052857