
The Molecular Basis of FIX Deficiency in Hemophilia B



Abstract
1. Introduction
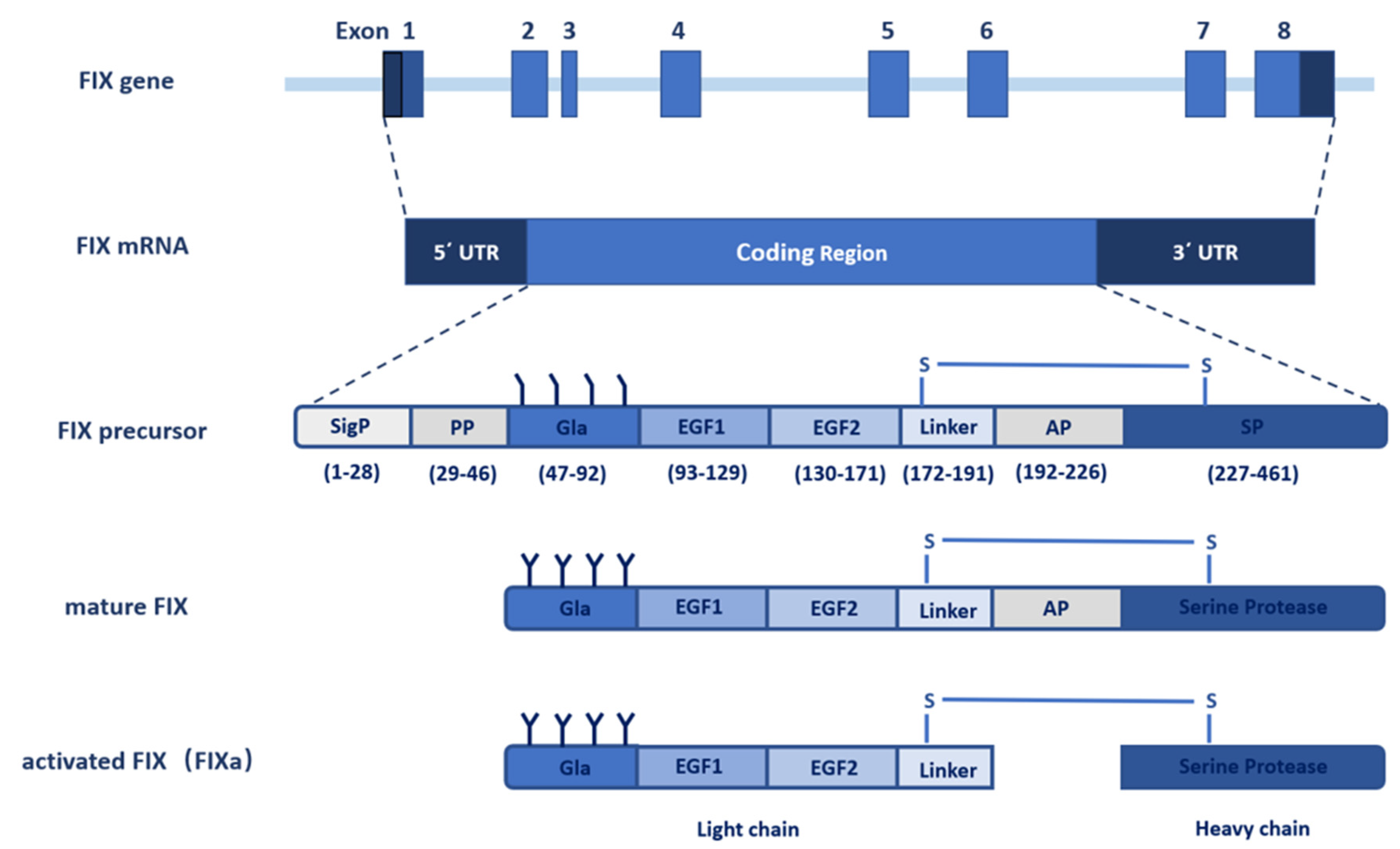
2. The Gene of FIX
3. FIX Protein
3.1. Signal Peptide and Propeptide
3.2. Gla Domain
3.3. EGF-like Domains
3.4. Activation Peptide
3.5. Serine Protease Domain
3.6. Activation of FIX
4. Distribution of F9 Mutations in Hemophilia B
5. Mechanisms of FIX Deficiency
5.1. Point Mutations in Noncoding Region
5.1.1. Mutations in F9 Promoter
5.1.2. Mutations in Introns
5.1.3. Mutations in 3′UTR
5.2. Point Mutations in Coding Region
5.2.1. Silent Mutations
5.2.2. Nonsense Mutations and Ribosome Readthrough
5.2.3. Missense Mutations in Signal Peptide and Propeptide
5.2.4. Missense Mutations in Gla Domain
5.2.5. Missense Mutations in EGF1 and EGF2 Domains
5.2.6. Missense Mutations at Cleavage Site of Activation Peptide
5.2.7. Missense Mutations in SP Domain
6. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Staber, J.; Croteau, S.E.; Davis, J.; Grabowski, E.F.; Kouides, P.; Sidonio, R.F., Jr. The spectrum of bleeding in women and girls with haemophilia B. Haemophilia 2017, 24, 180–185. [Google Scholar] [CrossRef]
- Biggs, R.; Douglas, A.S.; Macfarlane, R.G.; Dacie, J.V.; Pitney, W.R.; Merskey. Christmas disease: A condition previously mistaken for haemophilia. Br. Med. J. 1952, 2, 1378–1382. [Google Scholar] [CrossRef] [PubMed]
- Sidonio, R.F., Jr.; Malec, L. Hemophilia B (Factor IX Deficiency). Hematol. Oncol. Clin. N. Am. 2021, 35, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Peyvandi, F.; Garagiola, I.; Young, G. The past and future of haemophilia: Diagnosis, treatments, and its complications. Lancet 2016, 388, 187–197. [Google Scholar] [CrossRef]
- White, G.C., 2nd; Rosendaal, F.; Aledort, L.M.; Lusher, J.M.; Rothschild, C.; Ingerslev, J. Definitions in hemophilia. Recommendation of the scientific subcommittee on factor VIII and factor IX of the scientific and standardization committee of the International Society on Thrombosis and Haemostasis. Thromb. Haemost. 2001, 85, 560. [Google Scholar] [CrossRef] [PubMed]
- Stafford, D.W. Extravascular FIX and coagulation. Thromb. J. 2016, 14, 35. [Google Scholar] [CrossRef]
- Castaman, G.; Matino, D. Hemophilia A and B: Molecular and clinical similarities and differences. Haematologica 2019, 104, 1702–1709. [Google Scholar] [CrossRef]
- Gao, W.; Xu, Y.; Liu, H.; Gao, M.; Cao, Q.; Wang, Y.; Cui, L.; Huang, R.; Shen, Y.; Li, S.; et al. Characterization of missense mutations in the signal peptide and propeptide of FIX in hemophilia B by a cell-based assay. Blood Adv. 2020, 4, 3659–3667. [Google Scholar] [CrossRef]
- Goodeve, A.C. Hemophilia B: Molecular pathogenesis and mutation analysis. J. Thromb. Haemost. 2015, 13, 1184–1195. [Google Scholar] [CrossRef]
- Choo, K.H.; Gould, K.G.; Rees, D.J.; Brownlee, G.G. Molecular cloning of the gene for human anti-haemophilic factor IX. Nature 1982, 299, 178–180. [Google Scholar] [CrossRef]
- Kurachi, K.; Davie, E.W. Isolation and characterization of a cDNA coding for human factor IX. Proc. Natl. Acad. Sci. USA 1982, 79, 6461–6464. [Google Scholar] [CrossRef] [PubMed]
- Chance, P.F.; Dyer, K.A.; Kurachi, K.; Yoshitake, S.; Ropers, H.H.; Wieacker, P.; Gartler, S.M. Regional localization of the human factor IX gene by molecular hybridization. Hum. Genet. 1983, 65, 207–208. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.H. The Clinical Genetics of Hemophilia B (Factor IX Deficiency). Appl. Clin. Genet. 2021, 14, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Yoshitake, S.; Schach, B.G.; Foster, D.C.; Davie, E.W.; Kurachi, K. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry 1985, 24, 3736–3750. [Google Scholar] [CrossRef] [PubMed]
- Lillicrap, D. The molecular basis of haemophilia B. Haemophilia 1998, 4, 350–357. [Google Scholar] [CrossRef]
- Arruda, V.R.; Hagstrom, J.N.; Deitch, J.; Heiman-Patterson, T.; Camire, R.M.; Chu, K.; Fields, P.A.; Herzog, R.W.; Couto, L.B.; Larson, P.J.; et al. Posttranslational modifications of recombinant myotube-synthesized human factor IX. Blood 2001, 97, 130–138. [Google Scholar] [CrossRef]
- Kaufman, R.J. Post-translational modifications required for coagulation factor secretion and function. Thromb. Haemost. 1998, 79, 1068–1079. [Google Scholar] [CrossRef]
- Higgins-Gruber, S.L.; Mutucumarana, V.P.; Lin, P.J.; Jorgenson, J.W.; Stafford, D.W.; Straight, D.L. Effect of vitamin K-dependent protein precursor propeptide, vitamin K hydroquinone, and glutamate substrate binding on the structure and function of {gamma}-glutamyl carboxylase. J. Biol. Chem. 2010, 285, 31502–31508. [Google Scholar] [CrossRef]
- Tjarnlund-Wolf, A.; Lassila, R. Phenotypic characterization of haemophilia B—Understanding the underlying biology of coagulation factor IX. Haemophilia 2019, 25, 567–574. [Google Scholar] [CrossRef]
- Hao, Z.; Jin, D.Y.; Stafford, D.W.; Tie, J.K. Vitamin K-dependent carboxylation of coagulation factors: Insights from a cell-based functional study. Haematologica 2020, 105, 2164–2173. [Google Scholar] [CrossRef]
- Bentley, A.K.; Rees, D.J.; Rizza, C.; Brownlee, G.G. Defective propeptide processing of blood clotting factor IX caused by mutation of arginine to glutamine at position-4. Cell 1986, 45, 343–348. [Google Scholar] [CrossRef]
- Ware, J.; Diuguid, D.L.; Liebman, H.A.; Rabiet, M.J.; Kasper, C.K.; Furie, B.C.; Furie, B.; Stafford, D.W. Factor IX San Dimas. Substitution of glutamine for Arg-4 in the propeptide leads to incomplete gamma-carboxylation and altered phospholipid binding properties. J. Biol. Chem. 1989, 264, 11401–11406. [Google Scholar] [CrossRef]
- Dahms, S.O.; Arciniega, M.; Steinmetzer, T.; Huber, R.; Than, M.E. Structure of the unliganded form of the proprotein convertase furin suggests activation by a substrate-induced mechanism. Proc. Natl. Acad. Sci. USA 2016, 113, 11196–11201. [Google Scholar] [CrossRef]
- Huang, M.; Rigby, A.C.; Morelli, X.; Grant, M.A.; Huang, G.; Furie, B.; Seaton, B.; Furie, B.C. Structural basis of membrane binding by Gla domains of vitamin K-dependent proteins. Nat. Struct. Biol. 2003, 10, 751–756. [Google Scholar] [CrossRef]
- Huang, M.; Furie, B.C.; Furie, B. Crystal structure of the calcium-stabilized human factor IX Gla domain bound to a conformation-specific anti-factor IX antibody. J. Biol. Chem. 2004, 279, 14338–14346. [Google Scholar] [CrossRef]
- Zogg, T.; Brandstetter, H. Activation mechanisms of coagulation factor IX. Biol. Chem. 2009, 390, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Freedman, S.J.; Furie, B.C.; Furie, B.; Baleja, J.D. Structure of the calcium ion-bound gamma-carboxyglutamic acid-rich domain of factor IX. Biochemistry 1995, 34, 12126–12137. [Google Scholar] [CrossRef] [PubMed]
- Freedman, S.J.; Furie, B.C.; Furie, B.; Baleja, J.D. Structure of the metal-free gamma-carboxyglutamic acid-rich membrane binding region of factor IX by two-dimensional NMR spectroscopy. J. Biol. Chem. 1995, 270, 7980–7987. [Google Scholar] [CrossRef] [PubMed]
- Feuerstein, G.Z.; Patel, A.; Toomey, J.R.; Bugelski, P.; Nichols, A.J.; Church, W.R.; Valocik, R.; Koster, P.; Baker, A.; Blackburn, M.N. Antithrombotic efficacy of a novel murine antihuman factor IX antibody in rats. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2554–2562. [Google Scholar] [CrossRef][Green Version]
- Feuerstein, G.Z.; Toomey, J.R.; Valocik, R.; Koster, P.; Patel, A.; Blackburn, M.N. An inhibitory anti-factor IX antibody effectively reduces thrombus formation in a rat model of venous thrombosis. Thromb. Haemost. 1999, 82, 1443–1445. [Google Scholar] [CrossRef] [PubMed]
- Frazier, D.; Smith, K.J.; Cheung, W.F.; Ware, J.; Lin, S.W.; Thompson, A.R.; Reisner, H.; Bajaj, S.P.; Stafford, D.W. Mapping of monoclonal antibodies to human factor IX. Blood 1989, 74, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Kirchhofer, D.; Lipari, M.T.; Moran, P.; Eigenbrot, C.; Kelley, R.F. The tissue factor region that interacts with substrates factor IX and Factor, X. Biochemistry 2000, 39, 7380–7387. [Google Scholar] [CrossRef] [PubMed]
- Soeda, T.; Nogami, K.; Nishiya, K.; Takeyama, M.; Ogiwara, K.; Sakata, Y.; Yoshioka, A.; Shima, M. The factor VIIIa C2 domain (residues 2228-2240) interacts with the factor IXa Gla domain in the factor Xase complex. J. Biol. Chem. 2009, 284, 3379–3388. [Google Scholar] [CrossRef] [PubMed]
- Venkateswarlu, D. Structural insights into the interaction of blood coagulation co-factor VIIIa with factor IXa: A computational protein-protein docking and molecular dynamics refinement study. Biochem. Biophys. Res. Commun. 2014, 452, 408–414. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mann, D.M.; Stafford, K.A.; Poon, M.C.; Matino, D.; Stafford, D.W. The Function of extravascular coagulation factor IX in haemostasis. Haemophilia 2021, 27, 332–339. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Rao, Z.; Handford, P.; Mayhew, M.; Knott, V.; Brownlee, G.G.; Stuart, D. The structure of a Ca(2+)-binding epidermal growth factor-like domain: Its role in protein-protein interactions. Cell 1995, 82, 131–141. [Google Scholar] [CrossRef]
- Zogg, T.; Brandstetter, H. Structural basis of the cofactor- and substrate-assisted activation of human coagulation factor IXa. Structure 2009, 17, 1669–1678. [Google Scholar] [CrossRef][Green Version]
- Brandstetter, H.; Bauer, M.; Huber, R.; Lollar, P.; Bode, W. X-ray structure of clotting factor IXa: Active site and module structure related to Xase activity and hemophilia B. Proc. Natl. Acad. Sci. USA 1995, 92, 9796–9800. [Google Scholar] [CrossRef]
- McNicholas, S.; Potterton, E.; Wilson, K.S.; Noble, M.E.M. Presenting your structures: The CCP4mg molecular-graphics software. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 386–394. [Google Scholar] [CrossRef]
- Rallapalli, P.M.; Kemball-Cook, G.; Tuddenham, E.G.; Gomez, K.; Perkins, S.J. An interactive mutation database for human coagulation factor IX provides novel insights into the phenotypes and genetics of hemophilia B. J. Thromb. Haemost. 2013, 11, 1329–1340. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, B.M.; Matafonov, A.; Ivanov, I.; Sun, M.F.; Cheng, Q.; Dickeson, S.K.; Li, C.; Sun, D.; Verhamme, I.M.; Emsley, J.; et al. An update on factor XI structure and function. Thromb. Res. 2018, 161, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Zhong, D.; Bajaj, M.S.; Schmidt, A.E.; Bajaj, S.P. The N-terminal epidermal growth factor-like domain in factor IX and factor X represents an important recognition motif for binding to tissue factor. J. Biol. Chem. 2002, 277, 3622–3631. [Google Scholar] [CrossRef]
- Schmidt, A.E.; Bajaj, S.P. Structure-function relationships in factor IX and factor IXa. Trends Cardiovasc. Med. 2003, 13, 39–45. [Google Scholar] [CrossRef]
- Wilkinson, F.H.; London, F.S.; Walsh, P.N. Residues 88-109 of factor IXa are important for assembly of the factor X activating complex. J. Biol. Chem. 2002, 277, 5725–5733. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, F.H.; Ahmad, S.S.; Walsh, P.N. The factor IXa second epidermal growth factor (EGF2) domain mediates platelet binding and assembly of the factor X activating complex. J. Biol. Chem. 2002, 277, 5734–5741. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chang, Y.J.; Lin, S.W.; Walsh, P.N. Identification of residues Asn89, Ile90, and Val107 of the factor IXa second epidermal growth factor domain that are essential for the assembly of the factor X-activating complex on activated platelets. J. Biol. Chem. 2004, 279, 46400–46405. [Google Scholar] [CrossRef] [PubMed]
- Gailani, D.; Geng, Y.; Verhamme, I.; Sun, M.F.; Bajaj, S.P.; Messer, A.; Emsley, J. The mechanism underlying activation of factor IX by factor XIa. Thromb. Res. 2014, 133 (Suppl. 1), S48–S51. [Google Scholar] [CrossRef]
- Agarwala, K.L.; Kawabata, S.; Takao, T.; Murata, H.; Shimonishi, Y.; Nishimura, H.; Iwanaga, S. Activation peptide of human factor IX has oligosaccharides O-glycosidically linked to threonine residues at 159 and 169. Biochemistry 1994, 33, 5167–5171. [Google Scholar] [CrossRef]
- Makino, Y.; Omichi, K.; Kuraya, N.; Ogawa, H.; Nishimura, H.; Iwanaga, S.; Hase, S. Structural analysis of N-linked sugar chains of human blood clotting factor IX. J. Biochem. 2000, 128, 175–180. [Google Scholar] [CrossRef]
- White, G.C., 2nd; Beebe, A.; Nielsen, B. Recombinant factor IX. Thromb. Haemost. 1997, 78, 261–265. [Google Scholar] [CrossRef]
- Vysotchin, A.; Medved, L.V.; Ingham, K.C. Domain structure and domain-domain interactions in human coagulation factor IX. J. Biol. Chem. 1993, 268, 8436–8446. [Google Scholar] [CrossRef]
- Kolkman, J.A.; Mertens, K. Insertion loop 256-268 in coagulation factor IX restricts enzymatic activity in the absence but not in the presence of factor VIII. Biochemistry 2000, 39, 7398–7405. [Google Scholar] [CrossRef] [PubMed]
- Kolkman, J.A.; Lenting, P.J.; Mertens, K. Regions 301–303 and 333–339 in the catalytic domain of blood coagulation factor IX are factor VIII-interactive sites involved in stimulation of enzyme activity. Biochem. J. 1999, 339 Pt 2, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, J.B.; Nuss, R.; Haver, T.; Jacobson, L.; Thompson, A.R.; Manco-Johnson, M. Factor IX Denver, ASN 346-->ASP mutation resulting in a dysfunctional protein with defective factor VIIIa interaction. Thromb. Haemost. 2001, 86, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Mathur, A.; Bajaj, S.P. Protease and EGF1 domains of factor IXa play distinct roles in binding to factor VIIIa. Importance of helix 330 (helix 162 in chymotrypsin) of protease domain of factor IXa in its interaction with factor VIIIa. J. Biol. Chem. 1999, 274, 18477–18486. [Google Scholar] [CrossRef]
- Yang, L.; Manithody, C.; Rezaie, A.R. Localization of the heparin binding exosite of factor IXa. J. Biol. Chem. 2002, 277, 50756–50760. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.P.; Lang, A.; Karcher, A.; Sichler, K.; Kopetzki, E.; Brandstetter, H.; Huber, R.; Bode, W.; Engh, R.A. Coagulation factor IXa: The relaxed conformation of Tyr99 blocks substrate binding. Structure 1999, 7, 989–996. [Google Scholar] [CrossRef]
- Dang, Q.D.; Di Cera, E. Residue 225 determines the Na(+)-induced allosteric regulation of catalytic activity in serine proteases. Proc. Natl. Acad. Sci. USA 1996, 93, 10653–10656. [Google Scholar] [CrossRef]
- Harris, V.A.; Lin, W.; Perkins, S.J. Analysis of 272 Genetic Variants in the Upgraded Interactive FXI Web Database Reveals New Insights into FXI Deficiency. TH Open 2021, 5, e543–e556. [Google Scholar] [CrossRef]
- Byrne, R.; Link, R.P.; Castellino, F.J. A kinetic evaluation of activated bovine blood coagulation factor IX toward synthetic substrates. J. Biol. Chem. 1980, 255, 5336–5341. [Google Scholar] [CrossRef]
- van Dieijen, G.; Tans, G.; Rosing, J.; Hemker, H.C. The role of phospholipid and factor VIIIa in the activation of bovine factor X. J. Biol. Chem. 1981, 256, 3433–3442. [Google Scholar] [CrossRef]
- Veltkamp, J.J.; Meilof, J.; Remmelts, H.G.; van der Vlerk, D.; Loeliger, E.A. Another genetic variant of haemophilia B: Haemophilia B Leyden. Scand. J. Haematol. 1970, 7, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Odaira, K.; Tamura, S.; Suzuki, N.; Kakihara, M.; Hattori, Y.; Tokoro, M.; Suzuki, S.; Takagi, A.; Katsumi, A.; Hayakawa, F.; et al. Apparent synonymous mutation F9 c.87A>G causes secretion failure by in-frame mutation with aberrant splicing. Thromb. Res. 2019, 179, 95–103. [Google Scholar] [CrossRef]
- Green, P.M.; Bentley, D.R.; Mibashan, R.S.; Nilsson, I.M.; Giannelli, F. Molecular pathology of haemophilia B. EMBO J. 1989, 8, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Melendez-Aranda, L.; Jaloma-Cruz, A.R.; Pastor, N.; Romero-Prado, M.M.J. In silico analysis of missense mutations in exons 1-5 of the F9 gene that cause hemophilia B. BMC Bioinform. 2019, 20, 363. [Google Scholar] [CrossRef]
- Meireles, M.R.; Bragatte, M.A.S.; Bandinelli, E.; Salzano, F.M.; Vieira, G.F. A new in silico approach to investigate molecular aspects of factor IX missense causative mutations and their impact on the hemophilia B severity. Hum. Mutat. 2019, 40, 706–715. [Google Scholar] [CrossRef]
- Ahmed, S.Z.; O’Rourke, M.; Jenkins, V.; Regan, I.; Nolan, B. Progressive increase in FIX level in males with haemophilia B Leyden and c.35G > A mutation in early childhood not related to androgen effect. Br. J. Haematol. 2020, 189, e262–e265. [Google Scholar] [CrossRef]
- Crossley, M.; Winship, P.R.; Austen, D.E.; Rizza, C.R.; Brownlee, G.G. A less severe form of Haemophilia B Leyden. Nucleic Acids Res. 1990, 18, 4633. [Google Scholar] [CrossRef][Green Version]
- Crossley, M.; Ludwig, M.; Stowell, K.M.; De Vos, P.; Olek, K.; Brownlee, G.G. Recovery from hemophilia B Leyden: An androgen-responsive element in the factor IX promoter. Science 1992, 257, 377–379. [Google Scholar] [CrossRef]
- Funnell, A.P.; Crossley, M. Hemophilia B Leyden and once mysterious cis-regulatory mutations. Trends Genet. 2014, 30, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Funnell, A.P.; Wilson, M.D.; Ballester, B.; Mak, K.S.; Burdach, J.; Magan, N.; Pearson, R.C.; Lemaigre, F.P.; Stowell, K.M.; Odom, D.T.; et al. A CpG mutational hotspot in a ONECUT binding site accounts for the prevalent variant of hemophilia B Leyden. Am. J. Hum. Genet. 2013, 92, 460–467. [Google Scholar] [CrossRef]
- Kurachi, S.; Huo, J.S.; Ameri, A.; Zhang, K.; Yoshizawa, A.C.; Kurachi, K. An age-related homeostasis mechanism is essential for spontaneous amelioration of hemophilia B Leyden. Proc. Natl. Acad. Sci. USA 2009, 106, 7921–7926. [Google Scholar] [CrossRef] [PubMed]
- Heit, J.A.; Ketterling, R.P.; Zapata, R.E.; Ordonez, S.M.; Kasper, C.K.; Sommer, S.S. Haemophilia B Brandenberg-type promoter mutation. Haemophilia 1999, 5, 73–75. [Google Scholar] [CrossRef]
- Vielhaber, E.; Jacobson, D.P.; Ketterling, R.P.; Liu, J.Z.; Sommer, S.S. A mutation in the 3′ untranslated region of the factor IX gene in four families with hemophilia B. Hum. Mol. Genet. 1993, 2, 1309–1310. [Google Scholar] [CrossRef]
- Sauna, Z.E.; Kimchi-Sarfaty, C. Understanding the contribution of synonymous mutations to human disease. Nat. Rev. Genet. 2011, 12, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Knobe, K.E.; Sjorin, E.; Ljung, R.C. Why does the mutation G17736A/Val107Val (silent) in the F9 gene cause mild haemophilia B in five Swedish families? Haemophilia 2008, 14, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Simhadri, V.L.; Hamasaki-Katagiri, N.; Lin, B.C.; Hunt, R.; Jha, S.; Tseng, S.C.; Wu, A.; Bentley, A.A.; Zichel, R.; Lu, Q.; et al. Single synonymous mutation in factor IX alters protein properties and underlies haemophilia B. J. Med. Genet. 2017, 54, 338–345. [Google Scholar] [CrossRef]
- Pinotti, M.; Caruso, P.; Canella, A.; Campioni, M.; Tagariello, G.; Castaman, G.; Giacomelli, S.; Belvini, D.; Bernardi, F. Ribosome readthrough accounts for secreted full-length factor IX in hemophilia B patients with nonsense mutations. Hum. Mutat. 2012, 33, 1373–1376. [Google Scholar] [CrossRef]
- Branchini, A.; Ferrarese, M.; Campioni, M.; Castaman, G.; Mari, R.; Bernardi, F.; Pinotti, M. Specific factor IX mRNA and protein features favor drug-induced readthrough over recurrent nonsense mutations. Blood 2017, 129, 2303–2307. [Google Scholar] [CrossRef]
- Huang, L.; Li, L.; Lin, S.; Chen, J.; Li, K.; Fan, D.; Jin, W.; Li, Y.; Yang, X.; Xiong, Y.; et al. Molecular analysis of 76 Chinese hemophilia B pedigrees and the identification of 10 novel mutations. Mol. Genet. Genom. Med. 2020, 8, e1482. [Google Scholar] [CrossRef]
- Cooley, B.; Broze, G.J., Jr.; Mann, D.M.; Lin, F.C.; Pedersen, L.G.; Stafford, D.W. Dysfunctional endogenous FIX impairs prophylaxis in a mouse hemophilia B model. Blood 2019, 133, 2445–2451. [Google Scholar] [CrossRef] [PubMed]
- Quadros, L.; Ghosh, K.; Shetty, S. Novel mutations in factor IX gene from western India with reference to their phenotypic and haplotypic attributes. J. Pediatr. Hematol. Oncol. 2009, 31, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; Li, X.; Drost, J.; Thorland, E.C.; Liu, Q.; Lind, T.; Roberts, S.; Wang, H.Y.; Sommer, S.S. The human factor IX gene as germline mutagen test: Samples from Mainland China have the putatively endogenous pattern of mutation. Hum. Mutat. 2000, 16, 31–36. [Google Scholar] [CrossRef]
- Wu, P.C.; Hamaguchi, N.; Yu, Y.S.; Shen, M.C.; Lin, S.W. Hemophilia B with mutations at glycine-48 of factor IX exhibited delayed activation by the factor VIIa-tissue factor complex. Thromb. Haemost. 2000, 84, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Branchini, A.; Morfini, M.; Lunghi, B.; Belvini, D.; Radossi, P.; Bury, L.; Serino, M.L.; Giordano, P.; Cultrera, D.; Molinari, A.C.; et al. F9 missense mutations impairing factor IX activation are associated with pleiotropic plasma phenotypes. J. Thromb. Haemost. 2021, 20, 69–81. [Google Scholar] [CrossRef]
- Lenting, P.J.; ter Maat, H.; Clijsters, P.P.; Donath, M.J.; van Mourik, J.A.; Mertens, K. Cleavage at arginine 145 in human blood coagulation factor IX converts the zymogen into a factor VIII binding enzyme. J. Biol. Chem. 1995, 270, 14884–14890. [Google Scholar] [CrossRef]
- Diuguid, D.L.; Rabiet, M.J.; Furie, B.C.; Furie, B. Molecular defects of factor IX Chicago-2 (Arg 145----His) and prothrombin Madrid (Arg 271----cys): Arginine mutations that preclude zymogen activation. Blood 1989, 74, 193–200. [Google Scholar] [CrossRef]
- Liddell, M.B.; Peake, I.R.; Taylor, S.A.; Lillicrap, D.P.; Giddings, J.C.; Bloom, A.L. Factor IX Cardiff: A variant factor IX protein that shows abnormal activation is caused by an arginine to cysteine substitution at position 145. Br. J. Haematol. 1989, 72, 556–560. [Google Scholar] [CrossRef]
- Belvini, D.; Salviato, R.; Radossi, P.; Pierobon, F.; Mori, P.; Castaldo, G.; Tagariello, G. Molecular genotyping of the Italian cohort of patients with hemophilia B. Haematologica 2005, 90, 635–642. [Google Scholar]
- Huang, M.N.; Kasper, C.K.; Roberts, H.R.; Stafford, D.W.; High, K.A. Molecular defect in factor IXHilo, a hemophilia Bm variant: Arg----Gln at the carboxyterminal cleavage site of the activation peptide. Blood 1989, 73, 718–721. [Google Scholar] [CrossRef] [PubMed]
- Usharani, P.; Warn-Cramer, B.J.; Kasper, C.K.; Bajaj, S.P. Characterization of three abnormal factor IX variants (Bm Lake Elsinore, Long Beach, and Los Angeles) of hemophilia-B. Evidence for defects affecting the latent catalytic site. J. Clin. Investig. 1985, 75, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Monroe, D.M.; McCord, D.M.; Huang, M.N.; High, K.A.; Lundblad, R.L.; Kasper, C.K.; Roberts, H.R. Functional consequences of an arginine180 to glutamine mutation in factor IX Hilo. Blood 1989, 73, 1540–1544. [Google Scholar] [CrossRef] [PubMed]
- Radic, C.P.; Rossetti, L.C.; Abelleyro, M.M.; Candela, M.; Perez Bianco, R.; de Tezanos Pinto, M.; Larripa, I.B.; Goodeve, A.; De Brasi, C. Assessment of the F9 genotype-specific FIX inhibitor risks and characterisation of 10 novel severe F9 defects in the first molecular series of Argentinian patients with haemophilia B. Thromb. Haemost. 2013, 109, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Dai, J.; Liu, H.; Ding, Q.; Lu, Y.; Wang, H.; Wang, X.; Fu, Q. Spectrum of F9 mutations in Chinese haemophilia B patients: Identification of 20 novel mutations. Pathology 2012, 44, 342–347. [Google Scholar] [CrossRef]
- Weinmann, A.F.; Murphy, M.E.; Thompson, A.R. Consequences of factor IX mutations in 26 families with haemophilia B. Br. J. Haematol. 1998, 100, 58–61. [Google Scholar] [CrossRef]
- Bajaj, S.P. Region of factor IXa protease domain that interacts with factor VIIIa: Analysis of select hemophilia B mutants. Thromb. Haemost. 1999, 82, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Mathur, A.; Zhong, D.; Sabharwal, A.K.; Smith, K.J.; Bajaj, S.P. Interaction of factor IXa with factor VIIIa. Effects of protease domain Ca2+ binding site, proteolysis in the autolysis loop, phospholipid, and factor X. J. Biol. Chem. 1997, 272, 23418–23426. [Google Scholar] [CrossRef]
- Rodriguez-Merchan, E.C.; De Pablo-Moreno, J.A.; Liras, A. Gene Therapy in Hemophilia: Recent Advances. Int. J. Mol. Sci. 2021, 22, 7647. [Google Scholar] [CrossRef]
- Trinchero, A.; Sholzberg, M.; Matino, D. The Evolution of Hemophilia Care: Clinical and Laboratory Advances, Opportunities, and Challenges. Hamostaseologie 2020, 40, 311–321. [Google Scholar] [CrossRef]
- Naryshkin, N.A.; Weetall, M.; Dakka, A.; Narasimhan, J.; Zhao, X.; Feng, Z.; Ling, K.K.; Karp, G.M.; Qi, H.; Woll, M.G.; et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 2014, 345, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Wen, W.H.; Qin, W.J. Bispecific Antibodies as a Development Platform for New Concepts and Treatment Strategies. Int. J. Mol. Sci. 2017, 18, 48. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Regions | MTs | UMs | % of UMs | PN | % of PN | |
|---|---|---|---|---|---|---|
| Noncoding | Promoter * | Point | 23 | 2.10 | 86 | 2.32 |
| Deletion | 2 | 0.18 | 2 | 0.05 | ||
| Polymorphism | 5 | 0.46 | 5 | 0.13 | ||
| Intron | Point | 86 | 7.86 | 226 | 6.09 | |
| Deletion | 14 | 1.28 | 23 | 0.62 | ||
| Insertion | 2 | 0.18 | 8 | 0.22 | ||
| Indel | 1 | 0.09 | 1 | 0.03 | ||
| Polymorphism | 33 | 3.02 | 33 | 0.89 | ||
| 3′ UTR | Point | 2 | 0.18 | 22 | 0.59 | |
| Duplication | 1 | 0.09 | 1 | 0.03 | ||
| Polymorphism | 2 | 0.18 | 3 | 0.08 | ||
| Coding | Point | 689 | 62.98 | 2937 | 79.10 | |
| Deletion | 145 | 13.25 | 204 | 5.49 | ||
| Insertion | 33 | 3.02 | 38 | 1.02 | ||
| Indel | 15 | 1.37 | 17 | 0.46 | ||
| Duplication | 4 | 0.37 | 4 | 0.11 | ||
| Polymorphism | 11 | 1.01 | 18 | 0.48 | ||
| Multiple Regions | Deletion | 19 | 1.75 | 78 | 2.10 | |
| Insertion | 1 | 0.09 | 1 | 0.03 | ||
| Indel | 1 | 0.09 | 1 | 0.03 | ||
| Complex | 5 | 0.46 | 5 | 0.13 | ||
| Grand total | 1094 | 100 | 3713 | 100 | ||
| Regions | Mutation Effects | Unique Mutations | Patient Number | % of Total Patients |
|---|---|---|---|---|
| Promoter | Leyden/NA | 23 | 86 | 2.32 |
| Exons | Missense | 586 | 2422 | 65.23 |
| Nonsense | 87 | 469 | 12.63 | |
| Silent | 16 | 46 | 1.24 | |
| Introns | Splice | 86 | 226 | 6.09 |
| 3′ UTR | NA | 2 | 22 | 0.59 |
| Grand total | 800 | 3271 | 88.1 |
| Exon 1 | Exon 2 | Exon 3 | Exon 4 | Exon 5 | Exon 6 | Exon 7 | Exon 8 | |
|---|---|---|---|---|---|---|---|---|
| Missense | 43 | 351 | 22 | 205 | 164 | 365 | 177 | 1095 |
| Nonsense | 5 | 85 | 6 | 16 | 37 | 33 | 5 | 282 |
| Silent | 7 | 0 | 0 | 0 | 20 | 7 | 8 | 4 |
| PN | 55 | 436 | 28 | 221 | 221 | 405 | 190 | 1381 |
| PN/LCS | 0.63 | 2.66 | 1.12 | 1.94 | 1.71 | 2 | 1.65 | 2.52 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, G.; Gao, M.; Cao, Q.; Li, W. The Molecular Basis of FIX Deficiency in Hemophilia B. Int. J. Mol. Sci. 2022, 23, 2762. https://doi.org/10.3390/ijms23052762
Shen G, Gao M, Cao Q, Li W. The Molecular Basis of FIX Deficiency in Hemophilia B. International Journal of Molecular Sciences. 2022; 23(5):2762. https://doi.org/10.3390/ijms23052762
Chicago/Turabian StyleShen, Guomin, Meng Gao, Qing Cao, and Weikai Li. 2022. "The Molecular Basis of FIX Deficiency in Hemophilia B" International Journal of Molecular Sciences 23, no. 5: 2762. https://doi.org/10.3390/ijms23052762
APA StyleShen, G., Gao, M., Cao, Q., & Li, W. (2022). The Molecular Basis of FIX Deficiency in Hemophilia B. International Journal of Molecular Sciences, 23(5), 2762. https://doi.org/10.3390/ijms23052762