
Alzheimer’s Disease as Type 3 Diabetes: Common Pathophysiological Mechanisms between Alzheimer’s Disease and Type 2 Diabetes


 , ,
, , 


Abstract
:1. Introduction
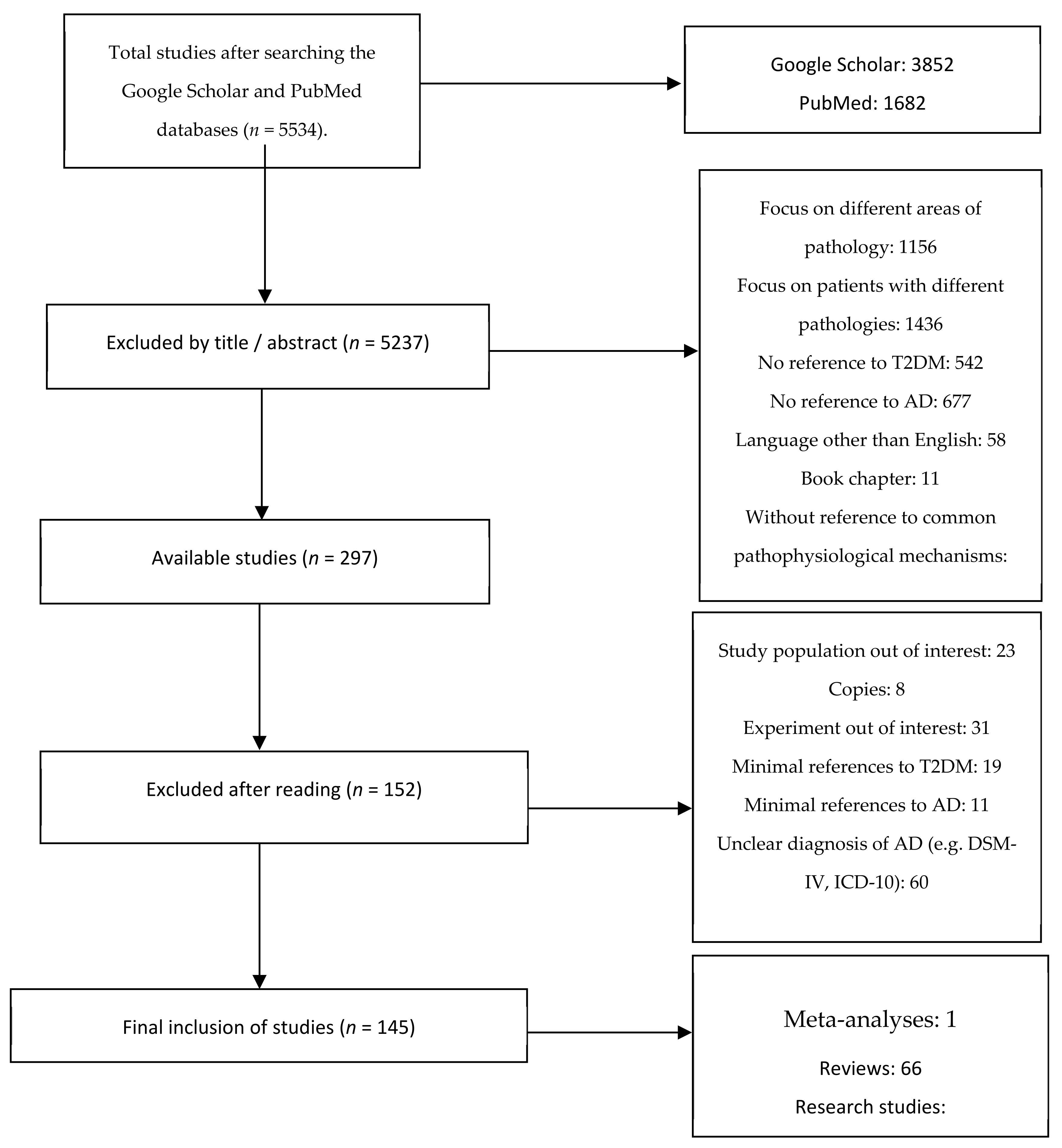
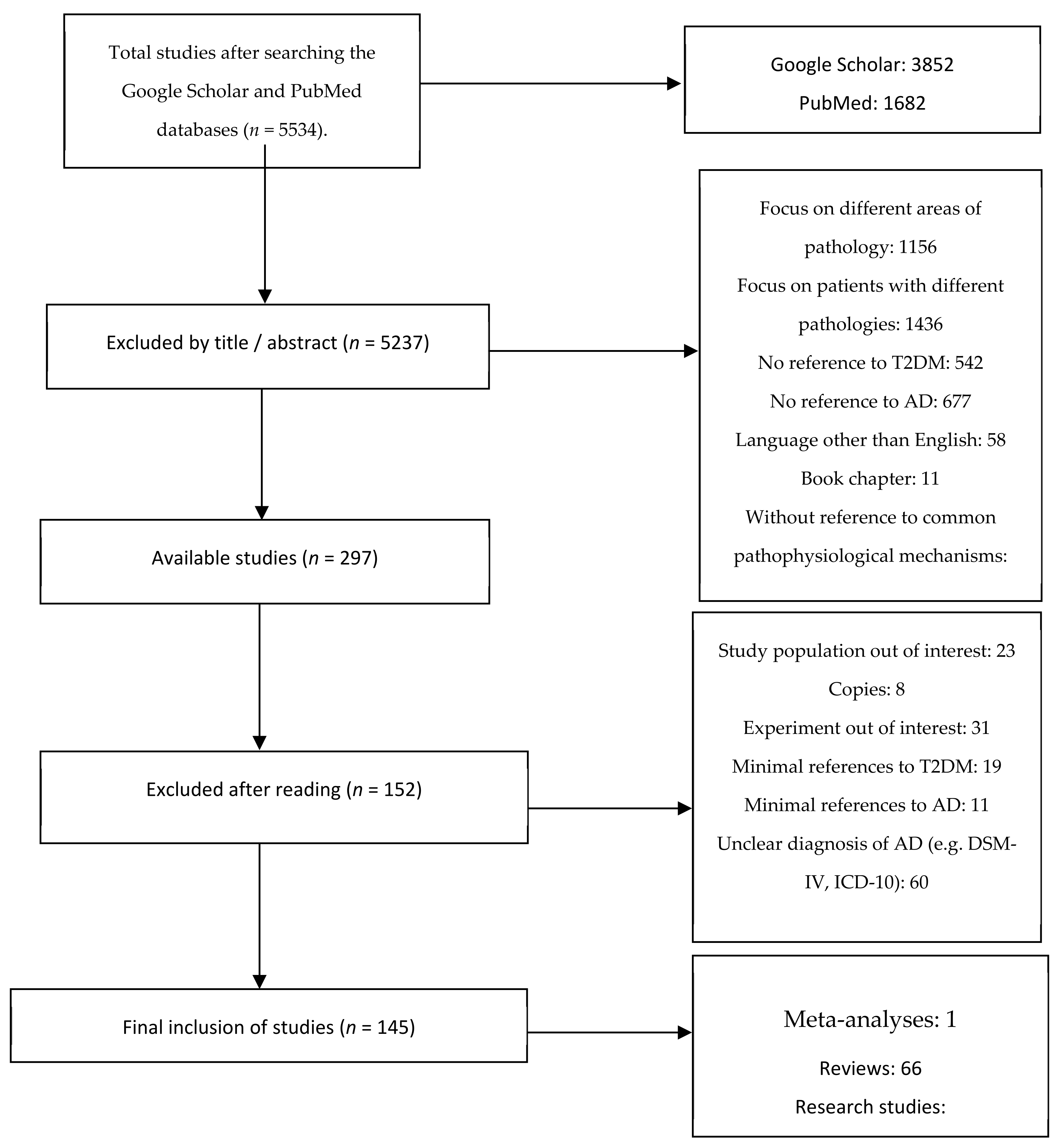
2. Materials and Methods
2.1. Search of the Literature
2.2. Inclusion and Exclusion Criteria
3. Results
3.1. Common Pathophysiological Mechanisms between AD and T2DM
3.2. Role and Production of Insulin in the Brain
3.3. Insulin Resistance and β-Amyloid
3.4. Insulin Resistance and Tau Protein
3.5. Neuroinflammation
3.6. Oxidative Stress and Mitochondrial Dysfunction
3.7. Advanced Glycosylation End Products (AGEs)
3.8. Metabolic Syndrome
3.9. Amylin
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Liu, P.-P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 1–22. [Google Scholar] [CrossRef]
- Alzheimer’s disease facts and figures. Alzheimers Dement. 2021, 17, 327–406.
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 2014, 88, 640–651. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.; Ortiz, A.; Castellino, J.; Kinney, J. Diabetes: Risk Factor and Translational Therapeutic Implications for Alzheimer’s Disease. Eur. J. Neurosci. 2022. [Google Scholar] [CrossRef]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [Green Version]
- Shinohara, M.; Kanekiyo, T.; Yang, L.; Linthicum, D.; Shinohara, M.; Fu, Y.; Price, L.; Frisch-Daiello, J.L.; Han, X.; Fryer, J.D.; et al. APOE2 eases cognitive decline during Aging: Clinical and preclinical evaluations. Ann. Neurol. 2016, 79, 758–774. [Google Scholar] [CrossRef]
- Getz, G.S.; Reardon, C.A. Apoprotein E and Reverse Cholesterol Transport. Int. J. Mol. Sci. 2018, 19, 3479. [Google Scholar] [CrossRef] [Green Version]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [Green Version]
- Stumvoll, M.; Goldstein, B.J.; van Haeften, T.W. Type 2 diabetes: Principles of pathogenesis and therapy. Lancet 2005, 365, 1333–1346. [Google Scholar] [CrossRef]
- Biessels, G.J.; Despa, F. Cognitive decline and dementia in diabetes mellitus: Mechanisms and clinical implications. Nat. Rev. Endocrinol. 2018, 14, 591–604. [Google Scholar] [CrossRef]
- Exalto, L.G.; Biessels, G.J.; Karter, A.J.; Huang, E.S.; Katon, W.J.; Minkoff, J.R.; Whitmer, R.A. Risk score for prediction of 10 year dementia risk in individuals with type 2 diabetes: A cohort study. Lancet Diabetes Endocrinol. 2013, 1, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Arvanitakis, Z.; Wilson, R.S.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Diabetes Mellitus and Risk of Alzheimer Disease and Decline in Cognitive Function. Arch. Neurol. 2004, 61, 661–666. [Google Scholar] [CrossRef]
- Wang, K.-C.; Woung, L.-C.; Tsai, M.-T.; Liu, C.-C.; Su, Y.-H.; Li, C.-Y. Risk of Alzheimer’s Disease in Relation to Diabetes: A Population-Based Cohort Study. Neuroepidemiology 2012, 38, 237–244. [Google Scholar] [CrossRef]
- Ott, A.; Stolk, R.P.; van Harskamp, F.; Pols, H.A.P.; Hofman, A.; Breteler, M.M.B. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999, 53, 1937. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar]
- De la Monte, S.M.; Tong, M.; Wands, J.R. The 20-Year Voyage Aboard the Journal of Alzheimer’s Disease: Docking at ‘Type 3 Diabetes’, Environmental/Exposure Factors, Pathogenic Mechanisms, and Potential Treatments. J. Alzheimers Dis. 2018, 62, 1381–1390. [Google Scholar]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1078–1089. [Google Scholar]
- Jarosz-Griffiths, H.H.; Noble, E.; Rushworth, J.V.; Hooper, N.M. Amyloid-β Receptors: The Good, the Bad, and the Prion Protein. J. Biol. Chem. 2016, 291, 3174–3183. [Google Scholar]
- Miklossy, J.; Qing, H.; Radenovic, A.; Kis, A.; Vileno, B.; Làszló, F.; Miller, L.; Martins, R.N.; Waeber, G.; Mooser, V.; et al. Beta amyloid and hyperphosphorylated tau deposits in the pancreas in type 2 diabetes. Neurobiol Aging. 2010, 31, 1503–1515. [Google Scholar]
- Rivera, E.J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J.R.; de la Monte, S.M. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. J. Alzheimer’s Dis. 2005, 8, 247–268. [Google Scholar] [CrossRef]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased Risk of Type 2 Diabetes in Alzheimer Disease. Diabetes 2004, 53, 474–481. [Google Scholar] [CrossRef] [Green Version]
- Kleinridders, A.; Cai, W.; Cappellucci, L.; Ghazarian, A.; Collins, W.R.; Vienberg, S.G.; Pothos, E.N.; Kahn, C.R. Insulin resistance in brain alters dopamine turnover and causes behavioral disorders. Proc. Natl. Acad. Sci. USA 2015, 112, 3463–3468. [Google Scholar]
- Maciejczyk, M.; Żebrowska, E.; Zalewska, A.; Chabowski, A. Redox Balance, Antioxidant Defense, and Oxidative Damage in the Hypothalamus and Cerebral Cortex of Rats with High Fat Diet-Induced Insulin Resistance. Oxidative Med. Cell. Longev. 2018, 2018, 6940515. [Google Scholar] [CrossRef] [Green Version]
- Whitmer, R.A.; Karter, A.J.; Yaffe, K.; Quesenberry, C.P.; Selby, J.V. Hypoglycemic Episodes and Risk of Dementia in Older Patients with Type 2 Diabetes Mellitus. JAMA 2009, 301, 1565–1572. [Google Scholar]
- Moran, C.; Phan, T.G.; Chen, J.; Blizzard, L.; Beare, R.; Venn, A.; Münch, G.; Wood, A.G.; Forbes, J.; Greenaway, T.M.; et al. Brain Atrophy in Type 2 Diabetes: Regional distribution and influence on cognition. Diabetes Care 2013, 36, 4036–4042. [Google Scholar] [CrossRef] [Green Version]
- An, Y.; Varma, V.R.; Varma, S.; Casanova, R.; Dammer, E.; Pletnikova, O.; Chia, C.W.; Egan, J.M.; Ferrucci, L.; Troncoso, J.; et al. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimers Dement. 2018, 14, 318–329. [Google Scholar]
- Sato, N.; Morishita, R. Brain alterations and clinical symptoms of dementia in diabetes: aβ/tau-dependent and independent mechanisms. Front. Endocrinol. 2014, 5, 143. [Google Scholar]
- Margolis, R.U.; Altszuler, N. Insulin in the Cerebrospinal Fluid. Nature 1967, 215, 1375–1376. [Google Scholar] [CrossRef]
- Bromander, S.; Anckarsäter, R.; Ahrén, B.; Kristiansson, M.; Blennow, K.; Holmäng, A.; Zetterberg, H.; Anckarsäter, H.; Wass, C.E. Cerebrospinal fluid insulin during non-neurological surgery. J. Neural Transm. 2010, 117, 1167–1170. [Google Scholar]
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin Action in Brain Regulates Systemic Metabolism and Brain Function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A.; Owen, J.B.; Erickson, M.A. Insulin in the brain: There and back again. Pharmacol. Ther. 2012, 136, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Blazquez, E.; Velã¡zquez, E.; Hurtado-Carneiro, V.; Ruiz-Albusac, J.M. Insulin in the Brain: Its Pathophysiological Implications for States Related with Central Insulin Resistance, Type 2 Diabetes and Alzheimer’s Disease. Front. Endocrinol. 2014, 5, 161. [Google Scholar] [CrossRef] [Green Version]
- Gray, S.M.; Meijer, R.I.; Barrett, E.J. Insulin regulates brain function, but how does it get there? Diabetes 2014, 63, 3992–3997. [Google Scholar]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimers disease–is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar]
- Deltour, L.; Leduque, P.; Blume, N.; Madsen, O.; Dubois, P.; Jami, J.; Bucchini, D. Differential expression of the two nonallelic proinsulin genes in the developing mouse embryo. Proc. Natl. Acad. Sci. USA 1993, 90, 527–531. [Google Scholar] [CrossRef] [Green Version]
- Devaskar, S.U.; Giddings, S.J.; Rajakumar, P.A.; Carnaghi, L.R.; Menon, R.K.; Zahm, D.S. Insulin gene expression and insulin synthesis in mammalian neuronal cells. J. Biol. Chem. 1994, 269, 8445–8454. [Google Scholar] [CrossRef]
- Schechter, R.; Whitmire, J.; Holtzclaw, L.; George, M.; Harlow, R.; Devaskar, S.U. Developmental regulation of insulin in the mammalian central nervous system. Brain Res. 1992, 582, 27–37. [Google Scholar] [CrossRef]
- Adamo, M.; Raizada, M.K.; LeRoith, D. Insulin and insulin-like growth factor receptors in the nervous system. Mol. Neurobiol. 1989, 3, 71–100. [Google Scholar] [CrossRef]
- Coker, G.T.; Studelska, D.; Harmon, S.; Burke, W.; O’Malley, K.L. Analysis of tyrosine hydroxylase and insulin transcripts in human neuroendocrine tissues. Mol. Brain Res. 1990, 8, 93–98. [Google Scholar] [CrossRef]
- Devaskar, S.U.; Singh, B.S.; Carnaghi, L.R.; Rajakumar, P.A.; Giddings, S.J. Insulin II gene expression in rat central nervous system. Regul. Pept. 1993, 48, 55–63. [Google Scholar] [CrossRef]
- Woods, S.C.; Seeley, R.; Baskin, D.; Schwartz, M. Insulin and the Blood-Brain Barrier. Curr. Pharm. Des. 2003, 9, 795–800. [Google Scholar] [CrossRef]
- Bharadwaj, P.; Wijesekara, N.; Liyanapathirana, M.; Newsholme, P.; Ittner, L.; Fraser, P.; Verdile, G. The Link between Type 2 Diabetes and Neurodegeneration: Roles for Amyloid-β, Amylin, and Tau Proteins. J. Alzheimers Dis. 2017, 59, 421–432. [Google Scholar]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef]
- Miller, Y.; Ma, B.; Nussinov, R. Synergistic Interactions between Repeats in Tau Protein and Aβ Amyloids May Be Responsible for Accelerated Aggregation via Polymorphic States. Biochemistry 2011, 50, 5172–5181. [Google Scholar] [CrossRef]
- Nussbaum, J.M.; Schilling, S.; Cynis, H.; Silva, A.; Swanson, E.; Wangsanut, T.; Tayler, K.; Wiltgen, B.; Hatami, A.; Rönicke, R.; et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature 2012, 485, 651–655. [Google Scholar]
- Nussbaum, J.M.; Seward, M.E.; Bloom, G.S. Alzheimer disease: A tale of two prions. Prion 2013, 7, 14–19. [Google Scholar]
- Pauwels, K.; Williams, T.L.; Morris, K.L.; Jonckheere, W.; Vandersteen, A.; Kelly, G.; Schymkowitz, J.; Rousseau, F.; Pastore, A.; Serpell, L.; et al. Structural Basis for Increased Toxicity of Pathological Aβ42:Aβ40 Ratios in Alzheimer Disease. J. Biol. Chem. 2013, 287, 5650–5660. [Google Scholar] [CrossRef] [Green Version]
- Hamley, I.W. The Amyloid Beta Peptide: A Chemist’s Perspective. Role in Alzheimer’s and Fibrillization. Chem. Rev. 2012, 112, 5147–5192. [Google Scholar]
- Bosco, D.; Fava, A.; Plastino, M.; Montalcini, T.; Pujia, A. Possible implications of insulin resistance and glucose metabolism in Alzheimer’s disease pathogenesis. J. Cell. Mol. Med. 2011, 15, 1807–1821. [Google Scholar] [CrossRef] [Green Version]
- Emmerling, M.R.; Spiegel, M.K.; Watson, D. Inhibiting the formation of classical C3-convertase on the Alzheimer’s β-amyloid peptide. Immunopharmacology 1997, 38, 101–109. [Google Scholar] [CrossRef]
- Blasko, I.; Stampfer-Kountchev, M.; Robatscher, P.; Veerhuis, R.; Eikelenboom, P.; Grubeck-Loebenstein, B. How chronic inflammation can affect the brain and support the development of Alzheimer’s disease in old age: The role of microglia and astrocytes. Aging Cell 2004, 3, 169–176. [Google Scholar] [CrossRef]
- Tumminia, A.; Vinciguerra, F.; Parisi, M.; Frittitta, L. Type 2 Diabetes Mellitus and Alzheimer’s Disease: Role of Insulin Signalling and Therapeutic Implications. Int. J. Mol. Sci. 2018, 19, 3306. [Google Scholar] [CrossRef] [Green Version]
- Kulas, J.A.; Franklin, W.F.; Smith, N.A.; Manocha, G.; Puig, K.L.; Nagamoto-Combs, K.; Hendrix, R.; Taglialatela, G.; Barger, S.W.; Combs, C.K. Ablation of amyloid precursor protein increases insulin-degrading enzyme levels and activity in brain and peripheral tissues. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E106–E120. [Google Scholar] [CrossRef]
- Watson, G.S.; Peskind, E.R.; Asthana, S.; Purganan, K.; Wait, C.; Chapman, D.; Schwartz, M.W.; Plymate, S.; Craft, S. Insulin increases CSF A 42 levels in normal older adults. Neurology 2003, 60, 1899–1903. [Google Scholar] [CrossRef]
- Kurochkin, I.V.; Guarnera, E.; Berezovsky, I.N. Insulin-Degrading Enzyme in the Fight against Alzheimer’s Disease. Trends Pharmacol. Sci. 2018, 39, 49–58. [Google Scholar] [CrossRef]
- Qiu, W.Q.; Walsh, D.M.; Ye, Z.; Vekrellis, K.; Zhang, J.; Podlisny, M.B.; Rosner, M.R.; Safavi, A.; Hersh, L.B.; Selkoe, D.J. Insulin-degrading Enzyme Regulates Extracellular Levels of Amyloid β-Protein by Degradation. J. Biol. Chem. 1998, 273, 32730–32738. [Google Scholar] [CrossRef] [Green Version]
- Starks, E.J.; O’Grady, J.P.; Hoscheidt, S.M.; Racine, A.M.; Carlsson, C.M.; Zetterberg, H.; Blennow, K.; Okonkwo, O.C.; Puglielli, L.; Asthana, S.; et al. Insulin Resistance is Associated with Higher Cerebrospinal Fluid Tau Levels in Asymptomatic APOE ɛ4 Carriers. J. Alzheimer’s Dis. 2015, 46, 525–533. [Google Scholar] [CrossRef] [Green Version]
- Shiiki, T.; Ohtsuki, S.; Kurihara, A.; Naganuma, H.; Nishimura, K.; Tachikawa, M.; Hosoya, K.; Terasaki, T. Brain insulin impairs amyloid-β (1-40) clearance from the brain. J. Neurosci. 2004, 24, 9632–9637. [Google Scholar]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guenette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar]
- Edland, S.D. Insulin-Degrading Enzyme, Apolipoprotein E, and Alzheimers Disease. J. Mol. Neurosci. 2004, 23, 213–218. [Google Scholar]
- Cook, D.G.; Leverenz, J.B.; McMillan, P.J.; Kulstad, J.J.; Ericksen, S.; Roth, R.A.; Schellenberg, G.D.; Jin, L.-W.; Kovacina, K.S.; Craft, S. Reduced Hippocampal Insulin-Degrading Enzyme in Late-Onset Alzheimer’s Disease Is Associated with the Apolipoprotein E-ε4 Allele. Am. J. Pathol. 2003, 162, 313–319. [Google Scholar] [CrossRef]
- Kim, M.; Hersh, L.B.; Leissring, M.A.; Ingelsson, M.; Matsui, T.; Farris, W.; Lu, A.; Hyman, B.T.; Selkoe, D.J.; Bertram, L.; et al. Decreased Catalytic Activity of the Insulin-degrading Enzyme in Chromosome 10-Linked Alzheimer Disease Families. J. Biol. Chem. 2007, 282, 7825–7832. [Google Scholar]
- El Khoury, N.B.; Gratuze, M.; Papon, M.-A.; Bretteville, A.; Planel, E. Insulin dysfunction and Tau pathology. Front. Cell. Neurosci. 2014, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Lacovich, V.; Espindola, S.L.; Alloatti, M.; Devoto, V.P.; Cromberg, L.E.; Čarná, M.E.; Forte, G.; Gallo, J.-M.; Bruno, L.; Stokin, G.B.; et al. Tau Isoforms Imbalance Impairs the Axonal Transport of the Amyloid Precursor Protein in Human Neurons. J. Neurosci. 2016, 37, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The Many Faces of Tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2015, 17, 22–35. [Google Scholar] [CrossRef]
- Mohandas, E.; Rajmohan, V.; Raghunath, B. Neurobiology of Alzheimer’s disease. Indian J. Psychiatry 2009, 51, 55–61. [Google Scholar]
- Gonçalves, R.A.; Wijesekara, N.; Fraser, P.E.; De Felice, F.G. The Link Between Tau and Insulin Signaling: Implications for Alzheimer’s Disease and Other Tauopathies. Front. Cell. Neurosci. 2019, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Daval, M.; Bedrood, S.; Gurlo, T.; Huang, C.-J.; Costes, S.; Butler, P.C.; Langen, R. The effect of curcumin on human islet amyloid polypeptide misfolding and toxicity. Amyloid 2010, 17, 118–128. [Google Scholar] [CrossRef]
- Schubert, M.; Gautam, D.; Surjo, D.; Ueki, K.; Baudler, S.; Schubert, D.; Kondo, T.; Alber, J.; Galldiks, N.; Küstermann, E.; et al. Role for neuronal insulin resistance in neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 2004, 101, 3100–3105. [Google Scholar] [CrossRef] [Green Version]
- De La Monte, S.M.; Tong, M.; Bowling, N.; Moskal, P. si-RNA inhibition of brain insulin or insulin-like growth factor receptors causes developmental cerebellar abnormalities: Relevance to fetal alcohol spectrum disorder. Mol. Brain 2011, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Bhat, R.; Xue, Y.; Berg, S.; Hellberg, S.; Ormö, M.; Nilsson, Y.; Radesäter, A.-C.; Jerning, E.; Markgren, P.-O.; Borgegård, T.; et al. Structural Insights and Biological Effects of Glycogen Synthase Kinase 3-specific Inhibitor AR-A014418. J. Biol. Chem. 2003, 278, 45937–45945. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Lee, V.M.-Y. Insulin and Insulin-like Growth Factor-1 Regulate Tau Phosphorylation in Cultured Human Neurons. J. Biol. Chem. 1997, 272, 19547–19553. [Google Scholar] [CrossRef] [Green Version]
- Clodfelder-Miller, B.J.; Zmijewska, A.A.; Johnson, G.V.; Jope, R.S. Tau Is Hyperphosphorylated at Multiple Sites in Mouse Brain In Vivo After Streptozotocin-Induced Insulin Deficiency. Diabetes 2006, 55, 3320–3325. [Google Scholar] [CrossRef] [Green Version]
- Ragaglia, V.V.; Schuck, T.; Trojanowski, J.Q.; Lee, V.M.-Y. PP2A mRNA Expression Is Quantitatively Decreased in Alzheimer’s Disease Hippocampus. Exp. Neurol. 2001, 168, 402–412. [Google Scholar] [CrossRef]
- Eikelenboom, P.; Van Exel, E.; Hoozemans, J.J.; Veerhuis, R.; Rozemuller, A.J.M.; Van Gool, W.A. Neuroinflammation–An Early Event in Both the History and Pathogenesis of Alzheimer’s Disease. Neurodegener. Dis. 2010, 7, 38–41. [Google Scholar] [CrossRef]
- Eikelenboom, P.; Van Gool, W.A. Neuroinflammatory perspectives on the two faces of Alzheimer’s disease. J. Neural Transm. 2004, 111, 281–294. [Google Scholar] [CrossRef]
- Tuppo, E.E.; Arias, H.R. The role of inflammation in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2005, 37, 289–305. [Google Scholar] [CrossRef]
- Markesbery, W.R.; Carney, J.M. Oxidative Alterations in Alzheimer’s Disease. Brain Pathol. 2006, 9, 133–146. [Google Scholar] [CrossRef]
- Laurent, C.; Buée, L.; Blum, D. Tau and neuroinflammation: What impact for Alzheimer’s Disease and Tauopathies? Biomed. J. 2018, 41, 21–33. [Google Scholar]
- Giovannini, M.G.; Scali, C.; Prosperi, C.; Bellucci, A.; Vannucchi, M.G.; Rosi, S.; Pepeu, G.; Casamenti, F. β-Amyloid-Induced Inflammation and Cholinergic Hypofunction in the Rat Brain in Vivo: Involvement of the p38MAPK Pathway. Neurobiol. Dis. 2002, 11, 257–274. [Google Scholar] [CrossRef] [Green Version]
- Dandrea, M.R.; Reiser, P.A.; Gumula, N.A.; Hertzog, B.M.; Andrade-Gordon, P. Application of triple immunohistochemistry to characterize amyloid plaque-associated inflammation in brains with Alzheimers disease. Biotech. Histochem. 2001, 76, 97–106. [Google Scholar]
- Mehlhorn, G.; Hollborn, M.; Schliebs, R. Induction of cytokines in glial cells surrounding cortical β-amyloid plaques in transgenic Tg2576 mice with Alzheimer pathology. Int. J. Dev. Neurosci. 2000, 18, 423–431. [Google Scholar] [CrossRef]
- De La Monte, S.M.; Longato, L.; Tong, M.; Wands, J.R. Insulin resistance and neurodegeneration: Roles of obesity, type 2 diabetes mellitus and non-alcoholic steatohepatitis. Curr. Opin. Investig. Drugs 2009, 10, 1049–1060. [Google Scholar]
- De la Monte, S.M. Insulin resistance and Alzheimer’s disease. BMB Rep. 2009, 42, 475–481. [Google Scholar]
- Rosenberg, P.B. Clinical aspects of inflammation in Alzheimer’s disease. Int. Rev. Psychiatry 2005, 17, 503–514. [Google Scholar] [CrossRef]
- Kiecolt-Glaser, J.K.; Preacher, K.J.; MacCallum, R.C.; Atkinson, C.; Malarkey, W.B.; Glaser, R. Chronic stress and age-related increases in the proinflammatory cytokine IL-6. Proc. Natl. Acad. Sci. USA 2003, 100, 9090–9095. [Google Scholar] [CrossRef] [Green Version]
- Reiman, E.M.; Chen, K.; Alexander, G.E.; Caselli, R.J.; Bandy, D.; Osborne, D.; Saunders, A.M.; Hardy, J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl. Acad. Sci. USA 2004, 101, 284–289. [Google Scholar]
- Erol, A. An Integrated and Unifying Hypothesis for the Metabolic Basis of Sporadic Alzheimer’s Disease. J. Alzheimer’s Dis. 2008, 13, 241–253. [Google Scholar] [CrossRef]
- Lee, S.; Tong, M.; Hang, S.; Deochand, C.; de la Monte, S. CSF and Brain Indices of Insulin Resistance, Oxidative Stress and Neuro-Inflammation in Early versus Late Alzheimer’s Disease. J. Alzheimers Dis. Parkinsonism 2013, 3, 128. [Google Scholar]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef]
- Erejuwa, O. Oxidative Stress in Diabetes Mellitus: Is There a Role for Hypoglycemic Drugs and/or Antioxidants? Oxidative Stress Dis. 2012, 217, 246. [Google Scholar] [CrossRef] [Green Version]
- Henriksen, E.J.; Diamond-Stanic, M.K.; Marchionne, E.M. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic. Biol. Med. 2011, 51, 993–999. [Google Scholar] [CrossRef] [Green Version]
- Żukowski, P.; Maciejczyk, M.; Matczuk, J.; Kurek, K.; Waszkiel, D.; Żendzian-Piotrowska, M.; Zalewska, A. Effect of N-Acetylcysteine on Antioxidant Defense, Oxidative Modification, and Salivary Gland Function in a Rat Model of Insulin Resistance. Oxidative Med. Cell. Longev. 2018, 2018, 6581970. [Google Scholar] [CrossRef]
- Rains, J.L.; Jain, S.K. Oxidative stress, insulin signaling, and diabetes. Free Radic. Biol. Med. 2011, 50, 567–575. [Google Scholar]
- Sripetchwandee, J.; Chattipakorn, N.; Chattipakorn, S.C. Links Between Obesity-Induced Brain Insulin Resistance, Brain Mitochondrial Dysfunction, and Dementia. Front. Endocrinol. 2018, 31, 496. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Wang, X.; Michaelis, E.K. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010, 2, 12. [Google Scholar]
- Cha, M.-Y.; Han, S.-H.; Son, S.; Hong, H.-S.; Choi, Y.-J.; Byun, J.; Mook-Jung, I. Mitochondria-Specific Accumulation of Amyloid β Induces Mitochondrial Dysfunction Leading to Apoptotic Cell Death. PLoS ONE 2012, 7, e34929. [Google Scholar] [CrossRef]
- Gu, F.; Zhu, M.; Shi, J.; Hu, Y.; Zhao, Z. Enhanced oxidative stress is an early event during development of Alzheimer-like pathologies in presenilin conditional knock-out mice. Neurosci. Lett. 2008, 440, 44–48. [Google Scholar] [CrossRef]
- Nunomura, A.; Castellani, R.J.; Zhu, X.; Moreira, P.I.; Perry, G.; Smith, M.A. Involvement of Oxidative Stress in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2006, 65, 631–641. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.-J.; Xu, J.; Lahousse, S.A.; Caggiano, N.L.; de la Monte, S.M. Transient hypoxia causes Alzheimer-type molecular and biochemical abnormalities in cortical neurons: Potential strategies for neuroprotection. J. Alzheimer’s Dis. 2003, 5, 209–228. [Google Scholar] [CrossRef]
- Grünblatt, E.; Salkovic-Petrisic, M.; Osmanovic, J.; Riederer, P.; Hoyer, S. Brain insulin system dysfunction in streptozotocin intracerebroventricularly treated rats generates hyperphosphorylated tau protein. J. Neurochem. 2007, 101, 757–770. [Google Scholar] [CrossRef]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced Glycation End Products. Circulation 2006, 114, 597–605. [Google Scholar]
- Yamagishi, S.I. Diabetes and Advanced Glycation End Products. In Diabetes and Aging-Related Complications; Springer: Singapore, 2018; pp. 201–212. [Google Scholar]
- Yamagishi, S.-I. Role of advanced glycation end products (AGEs) and receptor for AGEs (RAGE) in vascular damage in diabetes. Exp. Gerontol. 2011, 46, 217–224. [Google Scholar] [CrossRef]
- Sato, T.; Shimogaito, N.; Wu, X.; Kikuchi, S.; Yamagishi, S.-I.; Takeuchi, M. Toxic Advanced Glycation End Products (TAGE) Theory in Alzheimer’s Disease. Am. J. Alzheimer’s Dis. Other Dementiasr 2006, 21, 197–208. [Google Scholar] [CrossRef]
- Woltjer, R.L.; Maezawa, I.; Ou, J.J.; Montine, K.S.; Montine, T.J. Advanced glycation endproduct precursor alters intracellular amyloid- β/AβPP carboxy-terminal fragment aggregation and cytotoxicity. J. Alzheimer’s Dis. 2004, 5, 467–476. [Google Scholar] [CrossRef]
- Kuhla, B.; Haase, C.; Flach, K.; Lüth, H.-J.; Arendt, T.; Münch, G. Effect of Pseudophosphorylation and Cross-linking by Lipid Peroxidation and Advanced Glycation End Product Precursors on Tau Aggregation and Filament Formation. J. Biol. Chem. 2007, 282, 6984–6991. [Google Scholar] [CrossRef] [Green Version]
- Gudala, K.; Bansal, D.; Schifano, F.; Bhansali, A. Diabetes mellitus and risk of dementia: A meta-analysis of prospective observational studies. J. Diabetes Investig. 2013, 4, 640–650. [Google Scholar] [CrossRef] [Green Version]
- Ott, C.; Jacobs, K.; Haucke, E.; Santos, A.N.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef] [Green Version]
- Cutler, R.G.; Kelly, J.; Storie, K.; Pedersen, W.A.; Tammara, A.; Hatanpaa, K.; Troncoso, J.C.; Mattson, M.P. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2070–2075. [Google Scholar]
- Debette, S.; Beiser, A.; Hoffmann, U.; DeCarli, C.; O’Donnell, C.J.; Massaro, J.M.; Au, R.; Himali, J.J.; Wolf, P.A.; Fox, C.S.; et al. Visceral fat is associated with lower brain volume in healthy middle-aged adults. Ann. Neurol. 2010, 68, 136–144. [Google Scholar] [CrossRef]
- Frisardi, V.; Solfrizzi, V.; Capurso, C.; Imbimbo, B.P.; Vendemiale, G.; Seripa, D.; Pilotto, A.; Panza, F. Is Insulin Resistant Brain State a Central Feature of the Metabolic-Cognitive Syndrome? J. Alzheimers Dis. 2010, 21, 57–63. [Google Scholar]
- Yates, K.F.; Sweat, V.; Yau, P.L.; Turchiano, M.M.; Convit, A. Impact of metabolic syndrome on cognition and brain: A selected review of the literature. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2060–2067. [Google Scholar]
- Tan, Z.S.; Beiser, A.S.; Fox, C.S.; Au, R.; Himali, J.J.; Debette, S.; Decarli, C.; Vasan, R.S.; Wolf, P.A.; Seshadri, S. Association of metabolic dysregulation with volumetric brain magnetic resonance imaging and cognitive markers of subclinical brain aging in middle-aged adults: The Framingham Offspring Study. Diabetes Care 2011, 34, 1766–1770. [Google Scholar]
- Burns, J.M.; Honea, R.A.; Vidoni, E.D.; Hutfles, L.J.; Brooks, W.M.; Swerdlow, R.H. Insulin is differentially related to cognitive decline and atrophy in Alzheimer’s disease and aging. Biochim. Biophysica Acta 2012, 1822, 333–339. [Google Scholar]
- Baker, L.D.; Cross, D.J.; Minoshima, S.; Belongia, D.; Watson, G.S.; Craft, S. Insulin Resistance and Alzheimer-like Reductions in Regional Cerebral Glucose Metabolism for Cognitively Normal Adults with Prediabetes or Early Type 2 Diabetes. Arch. Neurol. 2011, 68, 51–57. [Google Scholar] [CrossRef]
- Roberts, R.O.; Knopman, D.S.; Cha, R.H.; Mielke, M.M.; Pankratz, V.S.; Boeve, B.F.; Kantarci, K.; Geda, Y.E.; Jack, C.R., Jr.; Petersen, R.C.; et al. Diabetes and elevated hemoglobin A1c levels are associated with brain hypometabolism but not amyloid accumulation. J. Nucl. Med. 2014, 55, 759–764. [Google Scholar]
- Li, W.; Risacher, S.L.; Huang, E.; Saykin, A.J. Alzheimer’s Disease Neuroimaging Initiative Type 2 diabetes mellitus is associated with brain atrophy and hypometabolism in the ADNI cohort. Neurology 2016, 87, 595–600. [Google Scholar]
- Cunnane, S.; Nugent, S.; Roy, M.; Courchesne-Loyer, A.; Croteau, E.; Tremblay, S.; Castellano, A.; Pifferi, F.; Bocti, C.; Paquet, N.; et al. Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 2011, 27, 3–20. [Google Scholar]
- Herholz, K.; Salmon, E.; Perani, D.; Baron, J.C.; Holthoff, V.; Frölich, L.; Schönknecht, P.; Ito, K.; Mielke, R.; Kalbe, E.; et al. Discrimination between Alzheimer Dementia and Controls by Automated Analysis of Multicenter FDG PET. Neuroimage 2002, 17, 302–316. [Google Scholar]
- Newberg, A.; Alavi, A.; Reivich, M. Determination of regional cerebral function with FDG-PET imaging in neuropsychiatric disorders. Semin. Nucl. Med. 2002, 32, 13–34. [Google Scholar] [CrossRef]
- Kalaria, R.N.; Harik, S.I. Abnormalities of the glucose transporter at the blood-brain barrier and in the brain in Alzheimer’s disease. Prog. Clin. Biol. Res. 1989, 317, 415–421. [Google Scholar]
- Simpson, I.A.; Chundu, K.R.; Davies-Hill, T.; Honer, W.G.; Davies, P. Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann. Neurol. 1994, 35, 546–551. [Google Scholar] [CrossRef]
- Langbaum, J.B.S.; Chen, K.; Caselli, R.J.; Lee, W.; Reschke, C.; Bandy, D.; Alexander, G.E.; Burns, C.M.; Kaszniak, A.W.; Reeder, S.A.; et al. Hypometabolism in Alzheimer-Affected Brain Regions in Cognitively Healthy Latino Individuals Carrying the Apolipoprotein E ε4 Allele. Arch. Neurol. 2010, 67, 462–468. [Google Scholar] [CrossRef] [Green Version]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar]
- Lutz, T.A. The interaction of amylin with other hormones in the control of eating. Diabetes Obes. Metab. 2012, 15, 99–111. [Google Scholar] [CrossRef]
- Hay, D.L.; Chen, S.; Lutz, T.A.; Parkes, D.G.; Roth, J.D. Amylin: Pharmacology, Physiology, and Clinical Potential. Pharmacol. Rev. 2015, 67, 564–600. [Google Scholar]
- Lutz, T.A.; Meyer, U. Amylin at the interface between metabolic and neurodegenerative disorders. Front. Neurosci. 2015, 9, 216. [Google Scholar] [CrossRef]
- Westermark, P.; Andersson, A.; Westermark, G.T. Islet Amyloid Polypeptide, Islet Amyloid, and Diabetes Mellitus. Physiol. Rev. 2011, 91, 795–826. [Google Scholar]
- Fu, W.; Ruangkittisakul, A.; MacTavish, D.; Shi, J.Y.; Ballanyi, K.; Jhamandas, J.H. Amyloid β (Aβ) Peptide Directly Activates Amylin-3 Receptor Subtype by Triggering Multiple Intracellular Signaling Pathways. J. Biol. Chem. 2012, 287, 18820–18830. [Google Scholar] [CrossRef] [Green Version]
- Bs, K.J.; Barisone, G.A.; Diaz, E.; Jin, L.-W.; DeCarli, C.; Despa, F. Amylin deposition in the brain: A second amyloid in Alzheimer disease? Ann. Neurol. 2013, 74, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Oskarsson, M.E.; Paulsson, J.F.; Schultz, S.W.; Ingelsson, M.; Westermark, P.; Westermark, G.T. In Vivo Seeding and Cross-Seeding of Localized Amyloidosis. Am. J. Pathol. 2015, 185, 834–846. [Google Scholar] [CrossRef]
- Rorbach-Dolata, A.; Piwowar, A. Neurometabolic Evidence Supporting the Hypothesis of Increased Incidence of Type 3 Diabetes Mellitus in the 21st Century. BioMed Res. Int. 2019, 2019, 1435276. [Google Scholar] [CrossRef] [Green Version]
- Andreetto, E.; Yan, L.-M.; Tatarek-Nossol, M.; Velkova, A.; Frank, R.; Kapurniotu, A. Identification of Hot Regions of the Aβ-IAPP Interaction Interface as High-Affinity Binding Sites in both Cross- and Self-Association. Angew. Chem. Int. Ed. 2010, 49, 3081–3085. [Google Scholar] [CrossRef]
- Berhanu, W.M.; Yaşar, F.; Hansmann, U.H.E. In Silico Cross Seeding of Aβ and Amylin Fibril-like Oligomers. ACS Chem. Neurosci. 2013, 4, 1488–1500. [Google Scholar] [CrossRef] [Green Version]
- Fawver, J.; Ghiwot, Y.; Koola, C.; Carrera, W.; Rodriguez-Rivera, J.; Hernandez, C.; Dineley, K.T.; Kong, Y.; Li, J.; Jhamandas, J.; et al. Islet Amyloid Polypeptide (IAPP): A Second Amyloid in Alzheimer’s Disease. Curr. Alzheimer Res. 2014, 11, 928–940. [Google Scholar]
- Lundmark, K.; Westermark, G.T.; Olsen, A.; Westermark, P. Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: Cross-seeding as a disease mechanism. Proc. Natl. Acad. Sci. USA 2005, 102, 6098–6102. [Google Scholar] [CrossRef] [Green Version]
- Ono, K.; Takahashi, R.; Ikeda, T.; Mizuguchi, M.; Hamaguchi, T.; Yamada, M. Exogenous amyloidogenic proteins function as seeds in amyloid β-protein aggregation. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 646–653. [Google Scholar]
- O’Nuallain, B.; Williams, A.D.; Westermark, P.; Wetzel, R. Seeding Specificity in Amyloid Growth Induced by Heterologous Fibrils. J. Biol. Chem. 2004, 279, 17490–17499. [Google Scholar]
- Verma, N.; Ly, H.; Liu, M.; Chen, J.; Zhu, H.; Chow, M.; Hersh, L.B.; Despa, F. Intraneuronal Amylin Deposition, Peroxidative Membrane Injury and Increased IL-1β Synthesis in Brains of Alzheimer’s Disease Patients with Type-2 Diabetes and in Diabetic HIP Rats. J. Alzheimers Dis. 2016, 53, 259–272. [Google Scholar]
- Rawlings, A.M.; Sharrett, A.R.; Mosley, T.H.; Ballew, S.H.; Deal, J.A.; Selvin, E. Glucose Peaks and the Risk of Dementia and 20-Year Cognitive Decline. Diabetes Care 2017, 40, 879–886. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, A.M.; Torres-Alemán, I. The many faces of insulin-like peptide signalling in the brain. Nat. Rev. Neurosci. 2012, 13, 225–239. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Tong, M. Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 548–559. [Google Scholar]
- Hoyer, S.; Nitsch, R. Cerebral excess release of neurotransmitter amino acids subsequent to reduced cerebral glucose metabolism in early-onset dementia of Alzheimer type. J. Neural Transm. 1989, 75, 227–232. [Google Scholar] [CrossRef]

{kind=link}
| Study | Mechanism | Synopsis |
|---|---|---|
| Low insulin and a decrease in choline acetyltransferase | Low insulin levels and sensitivity can lead to AD through a decrease in acetylcholine synthesis. |
| Amyloid deposition in islet and brain cells | Higher levels of islet amyloid in AD patients than in control subjects. No greater brain amyloid in DM patients compared with control subjects. |
| Amyloid beta and hyperphosphorylated tau | The presence of tau protein and β-amyloid in the pancreas after analysis of pancreatic tissue from 21 autopsy cases of patients with T2DM. |
| Insulin resistance in the brain | Demonstrating a potential molecular link between central insulin resistance and behavioral disorders (in mice). |
| Oxidative stress | Significant increase in the oxidative damage markers (AGE, 4-HNE, MDA, 8-OHdG, and OSI) in the cerebral cortex of insulin-resistant mice. |
| Hypoglycemia | In older patients with T2DM, history of severe hypoglycemic episodes was associated with a greater risk of dementia. |
| T2DM and brain atrophy | T2DM is associated with reduced volume of the hippocampus. |
| Impaired glucose metabolism | Abnormalities in the metabolism of brain glucose may be intrinsic to the pathology of AD. |
| Mitochondrial function in the brain | T2DM impairs neurite outgrowth through JAK/STAT3 modulation of mitochondrial bioenergetics. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michailidis, M.; Moraitou, D.; Tata, D.A.; Kalinderi, K.; Papamitsou, T.; Papaliagkas, V. Alzheimer’s Disease as Type 3 Diabetes: Common Pathophysiological Mechanisms between Alzheimer’s Disease and Type 2 Diabetes. Int. J. Mol. Sci. 2022, 23, 2687. https://doi.org/10.3390/ijms23052687
Michailidis M, Moraitou D, Tata DA, Kalinderi K, Papamitsou T, Papaliagkas V. Alzheimer’s Disease as Type 3 Diabetes: Common Pathophysiological Mechanisms between Alzheimer’s Disease and Type 2 Diabetes. International Journal of Molecular Sciences. 2022; 23(5):2687. https://doi.org/10.3390/ijms23052687
Chicago/Turabian StyleMichailidis, Michalis, Despina Moraitou, Despina A. Tata, Kallirhoe Kalinderi, Theodora Papamitsou, and Vasileios Papaliagkas. 2022. "Alzheimer’s Disease as Type 3 Diabetes: Common Pathophysiological Mechanisms between Alzheimer’s Disease and Type 2 Diabetes" International Journal of Molecular Sciences 23, no. 5: 2687. https://doi.org/10.3390/ijms23052687
APA StyleMichailidis, M., Moraitou, D., Tata, D. A., Kalinderi, K., Papamitsou, T., & Papaliagkas, V. (2022). Alzheimer’s Disease as Type 3 Diabetes: Common Pathophysiological Mechanisms between Alzheimer’s Disease and Type 2 Diabetes. International Journal of Molecular Sciences, 23(5), 2687. https://doi.org/10.3390/ijms23052687