
Alzheimer’s Disease and Type 2 Diabetes Mellitus: The Use of MCT Oil and a Ketogenic Diet

 and
and 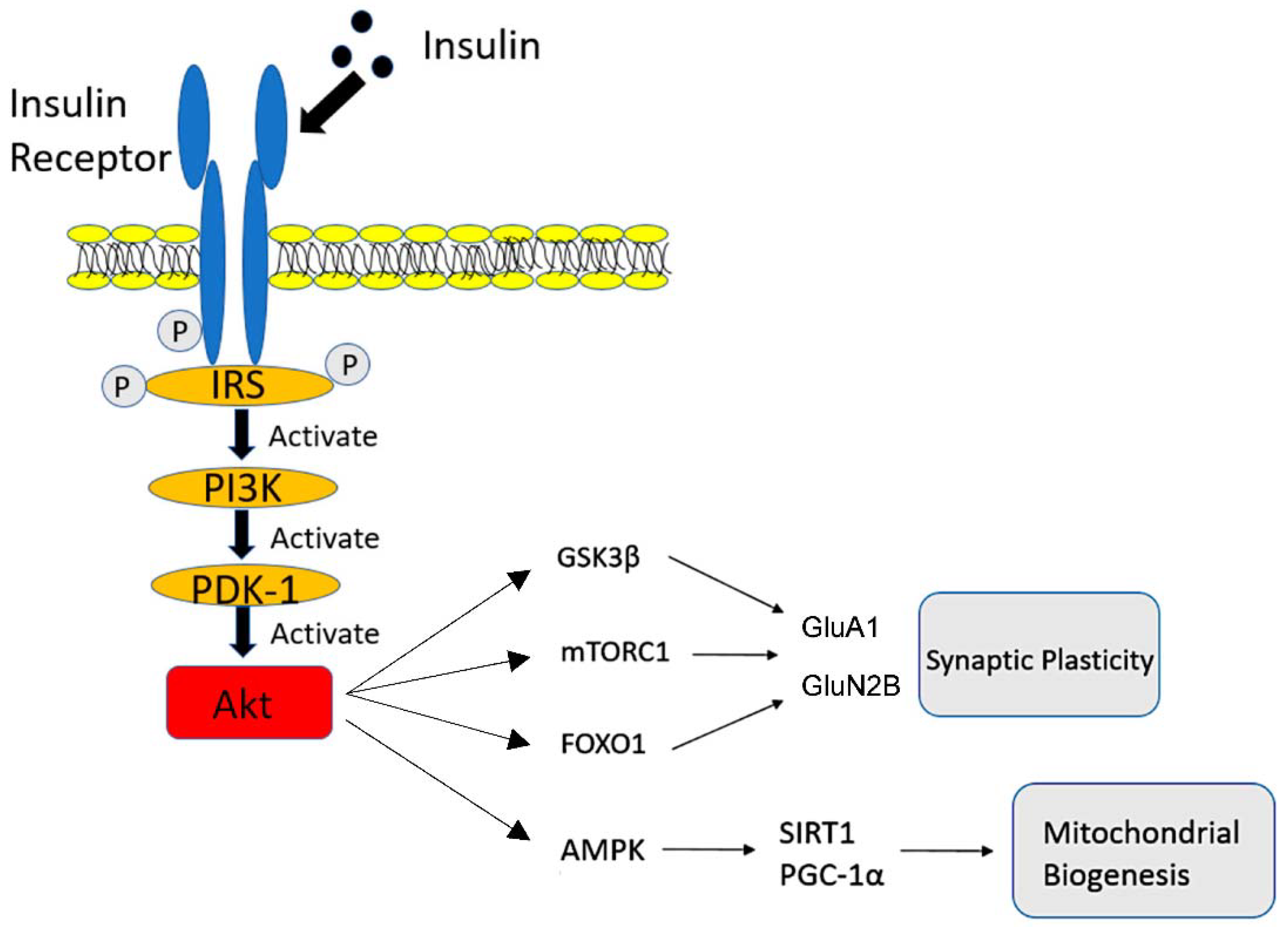{kind=link}
Abstract
:1. Introduction
2. Risk of AD
3. AD and DM: Mechanisms of Cognitive Decline Associated with Insulin Resistance
4. Declining Glucose Utilization and Preserving Ketone Metabolism in the Brain of AD
5. KD, CO, and MCT Oil
6. Influence of a KD and MCT Oil on AD
7. The Collateral Effects of MCT Oil for Cerebral Glucose Hypometabolism
8. The Hypothesis of Direct Effects of MCT Oil (MCFAs) on Cognitive Performance
8.1. Ligand for Peroxisome-Proliferator-Activated Receptor γ (PPARγ)
8.2. Lactate Shuttle
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s Disease. N. Eng. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Messier, C.; Teutenberg, K. The role of insulin, insulin growth factor, and insulin-degrading enzyme in brain aging and Alzheimer’s disease. Neural Plast. 2005, 12, 311–328. [Google Scholar] [CrossRef]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [Green Version]
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of dementia in diabetes mellitus: A systematic review. Lancet Neurol. 2006, 5, 64–74. [Google Scholar] [CrossRef]
- Cooper, C.; Sommerlad, A.; Lyketsos, C.; Livingston, G. Modifiable predictors of dementia in mild cognitive impairment: A systematic review and meta-analysis. Am. J. Psychiatry 2015, 172, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.V.F.; Loures, C.M.G.; Alves, L.C.V.; Souza, L.C.; Borges, K.B.G.; Carvalho, M.G. Alzheimer’s disease: Risk factors and potentially protective measures. J. Biomed. Sci. 2019, 26, 33. [Google Scholar] [CrossRef] [Green Version]
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Beydoun, M.A.; Beydoun, H.A.; Wang, Y. Obesity and central obesity as risk factors for incident dementia and its subtypes: A systematic review and meta-analysis. Obes. Rev. 2008, 9, 204–218. [Google Scholar] [CrossRef]
- Love, S.; Miners, J.S. Cerebrovascular disease in ageing and Alzheimer’s disease. Acta. Neuropathol. 2016, 131, 645–658. [Google Scholar] [CrossRef] [Green Version]
- Skoog, I.; Lernfelt, B.; Landahl, S.; Palmertz, B.; Andreasson, L.A.; Nilsson, L.; Persson, G.; Odén, A.; Svanborg, A. 15-Year Longitudinal Study of Blood Pressure and Dementia. Lancet 1996, 347, 1141–1145. [Google Scholar] [CrossRef]
- Mayeux, R.; Stern, Y. Epidemiology of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, 006239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kern, W.; Peters, A.; Fruehwald-Schultes, B.; Deininger, E.; Born, J.; Fehm, H.L. Improving Influence of Insulin on Cognitive Functions in Humans. Neuroendocrinology 2001, 74, 270–280. [Google Scholar] [CrossRef]
- Craft, S.; Newcomer, J.; Kanne, S.; Dagogo-Jack, S.; Cryer, P.; Sheline, Y.; Luby, J.; Dagogo-Jack, A.; Alderson, A. Memory improvement following induced hyperinsulinemia in Alzheimer’s disease. Neurobiol. Aging 1996, 17, 123–130. [Google Scholar] [CrossRef]
- Abbott, M.A.; Wells, D.G.; Fallon, J.R. The insulin receptor tyrosine kinase substrate p58/53 and the insulin receptor are components of CNS Synapses. J. Neurosci. 1999, 19, 7300–7308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Chen, H.; Xu, H.; Moore, E.; Meiri, N.; Quon, M.J.; Alkon, D.L. Brain insulin receptors and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation, and signaling molecules in the hippocampus of water maze trained rats. J. Biol. Chem. 1999, 274, 34893–34902. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A.; Owen, J.B.; Erickson, M.A. Insulin in the brain: There and back again. Pharmacol. Ther. 2012, 136, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, W.A.; Jaspan, J.B.; Kastin, A.J. Selective Physiological Transport of Insulin Across the Blood-Brain Barrier: Novel Demonstration by Species-Specific Radioimmunoassays. Peptides 1997, 18, 1257–1262. [Google Scholar] [CrossRef]
- Hers, I.; Vincent, E.E.; Tavaré, J.M. Akt signalling in health and disease. Cell Signal. 2011, 23, 1515–1527. [Google Scholar] [CrossRef] [PubMed]
- Tumminia, A.; Vinciguerra, F.; Parisi, M.; Frittitta, L. Type 2 diabetes mellitus and alzheimer’s disease: Role of insulin signalling and therapeutic implications. Int. J. Mol. Sci. 2018, 19, 3306. [Google Scholar] [CrossRef] [Green Version]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, S.L.; Chen, C.M.; Cline, H.T. Insulin Receptor Signaling Regulates Synapse Number, Dendritic Plasticity and Circuit Function in Vivo. Neuron 2008, 58, 708–719. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.L. Sirt1 and the mitochondria. Mol. Cells 2016, 39, 87–95. [Google Scholar]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1α. J. Biol. Chem. 2005, 280, 16456–16460. [Google Scholar] [CrossRef] [Green Version]
- Zilliox, L.A.; Chadrasekaran, K.; Kwan, J.Y.; Russell, J.W. Diabetes and Cognitive Impairment. Curr. Diabetes Rep. 2016, 16, 87. [Google Scholar] [CrossRef] [Green Version]
- Sartorius, T.; Peter, A.; Heni, M.; Maetzler, W.; Fritsche, A.; Haring, H.U.; Hennige, A.M. The Brain Response to Peripheral Insulin Declines with Age: A Contribution of the Blood-Brain Barrier? PLoS ONE 2015, 10, e0126804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, M.W.; Figlewicz, D.F.; Kahn, S.E.; Baskin, D.G.; Greenwood, M.R.C.; Porte, D., Jr. Insulin Binding to Brain Capillaries Is Reduced in Genetically Obese, Hyperinsulinemic Zucker Rats. Peptides 1990, 11, 467–472. [Google Scholar] [CrossRef]
- Frazier, H.N.; Ghoweri, A.O.; Anderson, K.L.; Lin, R.L.; Porter, N.M.; Thibault, O. Broadening the definition of brain insulin resistance in aging and Alzheimer’s disease. Exp. Neurol. 2019, 313, 79–87. [Google Scholar] [CrossRef]
- Talbot, K.; Wang, H.; Kazi, H.; Han, L.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 4, 1316–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarchoan, M.; Toledo, J.B.; Lee, E.B.; Arvanitakis, Z.; Kazi, H.; Ying, L.; Natalia, H.; Virginia, L.; Sangwon, M.Y.L.; Trojanowski, J.Q.; et al. Abnormal serine phosphorylation of insulin receptor substrate 1 is associated with tau pathology in Alzheimer’s disease and tauopathies. Acta Neuropathol. 2014, 128, 679–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frölich, L.; Blum-Degen, D.; Bernstein, H.G.; Engelsberger, S.; Humrich, J.; Laufer, S.; Muschner, D.; Thalheimer, A.; Türk, A.; Hoyer, S.; et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J. Neural Transm. 1998, 105, 423–438. [Google Scholar] [CrossRef] [PubMed]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Du Yan, S. Mitochondrial Aβ: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosconi, L.; Pupi, A.; Leon, M.J.D. Brain Glucose Hypometabolism and Oxidative Stress in Preclinical Alzheimer’s Disease. Ann. N.Y. Acad. Sci. 2008, 1147, 180–195. [Google Scholar] [CrossRef] [PubMed]
- Croteau, E.; Castellano, C.A.; Fortier, M.; Bocti, C.; Fulop, T.; Paquet, N.; Cunnane, S.C. A cross-sectional comparison of brain glucose and ketone metabolism in cognitively healthy older adults, mild cognitive impairment and early Alzheimer’s disease. Exp. Gerontol. 2018, 107, 18–26. [Google Scholar] [CrossRef]
- Castellano, C.A.; Nugent, S.; Paquet, N.; Tremblay, S.; Bocti, C.; Lacombe, G.; Imbeault, H.; Turcotte, É.; Fulop, T.; Cunnane, S.C. Lower brain 18F-fluorodeoxyglucose uptake but normal 11C-acetoacetate metabolism in mild Alzheimer’s disease dementia. J. Alzheimers Dis. 2015, 43, 1343–1353. [Google Scholar] [CrossRef]
- Nugent, S.; Tremblay, S.; Chen, K.W.; Ayutyanont, N.; Roontiva, A.; Castellano, C.A.; Fortier, M.; Roy, M.; Courchesne-Loyer, A.; Bocti, C.; et al. Brain glucose and acetoacetate metabolism: A comparison of young and older adults. Neurobiol. Aging 2014, 35, 1386–1395. [Google Scholar] [CrossRef]
- Roy, M.; Nugent, S.; Tremblay-Mercier, J.; Tremblay, S.; Courchesne-Loyer, A.; Beaudoin, J.F.; Tremblay, L.; Descoteaux, M.; Lecomte, R.; Cunnane, S.C. The ketogenic diet increases brain glucose and ketone uptake in aged rats: A dual tracer PET and volumetric MRI study. Brain Res. 2012, 1488, 14–23. [Google Scholar] [CrossRef]
- Pifferi, F.; Tremblay, S.; Croteau, E.; Fortier, M.; Tremblay-Mercier, J.; Lecomte, R.; Cunnane, S.C. Mild experimental ketosis increases brain uptake of 11C-acetoacetate and 18F-fluorodeoxyglucose: A dual-tracer PET imaging study in rats. Nutr. Neurosci. 2011, 14, 51–58. [Google Scholar] [CrossRef]
- Courchesne-Loyer, A.; Croteau, E.; Castellano, C.A.; St-Pierre, V.; Hennebelle, M.; Cunnane, S.C. Inverse relationship between brain glucose and ketone metabolism in adults during short-term moderate dietary ketosis: A dual tracer quantitative positron emission tomography study. J. Cereb. Blood Flow Metab. 2017, 37, 2485–2493. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.K.; Swerdlow, R.H.; Burns, J.M.; Sullivan, D.K. An experimental ketogenic diet for Alzheimer disease was nutritionally dense and rich in vegetables and avocado. Curr. Dev. Nutr. 2019, 3, nzz003. [Google Scholar] [CrossRef]
- Nevin, K.G.; Rajamohan, T. Beneficial effects of virgin coconut oil on lipid parameters and in vitro LDL oxidation. Clin. Biochem. 2004, 37, 830–835. [Google Scholar] [CrossRef]
- Khaw, K.T.; Sharp, S.J.; Finikarides, L.; Afzal, I.; Lentjes, M.; Luben, R.; Forouhi, N.G. Randomised trial of coconut oil, olive oil or butter on blood lipids and other cardiovascular risk factors in healthy men and women. BMJ Open 2018, 8, e020167. [Google Scholar] [CrossRef] [Green Version]
- Teng, M.; Zhao, Y.J.; Khoo, A.L.; Yeo, T.C.; Yong, Q.W.; Lim, B.P. Impact of coconut oil consumption on cardiovascular health: A systematic review and meta-Analysis. Nutr. Rev. 2020, 78, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Eyres, L.; Eyres, M.F.; Chisholm, A.; Brown, R.C. Coconut oil consumption and cardiovascular risk factors in humans. Nutr. Rev. 2016, 74, 267–280. [Google Scholar] [CrossRef] [Green Version]
- DebMandal, M.; Mandal, S. Coconut (Cocos nucifera L.: Arecaceae): In health promotion and disease prevention. Asian Pac. J. Trop. Med. 2011, 4, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Ulbricht, T.L.V.; Southgate, D.A.T. Coronary heart disease and dietary factors. Lancet 1991, 338, 985–992. [Google Scholar] [CrossRef]
- Jayawardena, R.; Swarnamali, H.; Lanerolle, P.; Ranasinghe, P. Effect of coconut oil on cardio-metabolic risk: A systematic review and meta-analysis of interventional studies. Diabetes Metab. Syndr. 2020, 14, 2007–2020. [Google Scholar] [CrossRef]
- Fernando, W.M.A.D.B.; Martins, I.J.; Goozee, K.G.; Brennan, C.S.; Jayasena, V.; Martins, R.N. The role of dietary coconut for the prevention and treatment of Alzheimer’s disease: Potential mechanisms of action. Br. J. Nutr. 2015, 114, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Traul, K.A.; Driedger, A.; Ingle, D.L.; Nakhasi, D. Review of the toxicologic properties of medium-chain triglycerides. Food Chem. Toxicol. 2000, 38, 79–98. [Google Scholar] [CrossRef]
- Bach, A.C.; Babayan, V.K. Medium-chain triglycerides: An update. Am. J. Clin. Nutr. 1982, 36, 950–962. [Google Scholar] [CrossRef] [PubMed]
- Greenberger, N.J.; Skillman, T.G. Medium-chain triglycerides. N. Engl. J. Med. 1969, 280, 1045–1058. [Google Scholar] [CrossRef] [Green Version]
- Wheless, J.W. History of the ketogenic diet. Epilepsia 2008, 49, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Augustin, K.; Khabbush, A.; Williams, S.; Eaton, S.; Orford, M.; Cross, J.H.; Heales, S.J.R.; Walker, M.C.; Williams, R.S.B. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 2018, 17, 84–93. [Google Scholar] [CrossRef]
- Sampaio, L.P.D.B. Ketogenic diet for epilepsy treatment. Arq. Neuropsiquiatr. 2016, 74, 842–848. [Google Scholar] [CrossRef] [Green Version]
- Kossoff, E.H. More fat and fewer seizures: Dietary therapies for epilepsy. Lancet Neurol. 2004, 3, 415–420. [Google Scholar] [CrossRef]
- Dashti, H.M.; Mathew, T.C.; Khadada, M.; Al-Mousawi, M.; Talib, H.; Asfar, S.K.; Behbahani, A.I.; Al-Zaid, N.S. Beneficial effects of ketogenic diet in obese diabetic subjects. Mol. Cell. Biochem. 2007, 302, 249–256. [Google Scholar] [CrossRef]
- Hussain, T.A.; Mathew, T.C.; Dashti, A.A.; Asfar, S.; Al-Zaid, N.; Dashti, H.M. Effect of low-calorie versus low-carbohydrate ketogenic diet in type 2 diabetes. Nutrition 2012, 28, 1016–1021. [Google Scholar] [CrossRef]
- Ota, M.; Matsuo, J.; Ishida, I.; Takano, H.; Yokoi, Y.; Hori, H.; Yoshida, S.; Ashida, K.; Nakamura, K.; Takahashi, T.; et al. Effects of a medium-chain triglyceride-based ketogenic formula on cognitive function in patients with mild-to-moderate Alzheimer’s disease. Neurosci. Lett. 2019, 690, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.C.L.; Deprez, L.M.; Mortimer, G.M.N.; Murtagh, D.K.J.; McCoy, S.; Mylchreest, R.; Gilbertson, L.J.; Clark, K.M.; Simpson, P.V.; McManus, E.J.; et al. Randomized crossover trial of a modified ketogenic diet in Alzheimer’s disease. Alzheimer’s Res. Ther. 2021, 13, 51. [Google Scholar] [CrossRef] [PubMed]
- Avgerinos, K.I.; Egan, J.M.; Mattson, M.P.; Kapogiannis, D. Medium Chain Triglycerides induce mild ketosis and may improve cognition in Alzheimer’s disease. A systematic review and meta-analysis of human studies. Ageing Res. Rev. 2020, 58, 101001. [Google Scholar] [CrossRef] [PubMed]
- Abe, S.; Ezaki, O.; Suzuki, M. Medium-chain triglycerides (8:0 and 10:0) increase mini-mental state examination (mmse) score in frail elderly adults in a randomized controlled trial. J. Nutr. 2020, 150, 2383–2390. [Google Scholar] [CrossRef]
- Rho, J.M. Hoe does the ketogenic diet induce anti-seizure effects? Neurosci. Lett. 2017, 637, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Kinzig, K.P.; Honors, M.A.; Hargrave, S.L. Insulin sensitivity and glucose tolerance are altered by maintenance on a ketogenic diet. Endocrinology 2010, 151, 3105–3114. [Google Scholar] [CrossRef] [Green Version]
- Fukao, T.; Mitchell, G.; Sass, J.O.; Hori, T.; Orii, K.; Aoyama, Y. Ketone body metabolism and its defects. J. Inherit. Metab. Dis. 2014, 37, 541–551. [Google Scholar] [CrossRef]
- Fujino, T.; Kondo, J.; Ishikawa, M.; Morikawa, K.; Yamamoto, T.T. Acetyl-CoA Synthetase 2, a Mitochondrial Matrix Enzyme Involved in the Oxidation of Acetate. J. Biol. Chem. 2001, 276, 11420–11426. [Google Scholar] [CrossRef] [Green Version]
- Pierre, K.; Pellerin, L. Monocarboxylate transporters in the central nervous system: Distribution, regulation and function. J. Neurochem. 2005, 94, 1–14. [Google Scholar] [CrossRef]
- Morris, A.A.M. Cerebral ketone body metabolism. J. Inherit. Metab. Dis. 2005, 28, 109–121. [Google Scholar] [CrossRef]
- Chatterjee, P.; Fernando, M.; Fernando, B.; Dias, C.B.; Shah, T.; Silva, R.; Williams, S.; Pedrini, S.; Hillebrandt, H.; Goozee, K.; et al. Potential of coconut oil and medium chain triglycerides in the prevention and treatment of Alzheimer’s disease. Mech. Ageing Dev. 2020, 186, 111209. [Google Scholar] [CrossRef]
- Veech, R.L.; Chance, B.; Kashiwaya, Y.; Lardy, H.A.; Cahill, G.F. Ketone bodies, potential therapeutic uses. IUBMB Life 2001, 51, 241–247. [Google Scholar]
- Lomb, D.J.; Laurent, G.; Haigis, M.C. Sirtuins regulate key aspects of lipid metabolism. Biochim. Biophys. Acta 2010, 1804, 1652–1657. [Google Scholar] [CrossRef] [PubMed]
- Bough, K.J.; Wetherington, J.; Hassel, B.; Pare, J.F.; Gawryluk, J.W.; Greene, J.G.; Shaw, R.; Smith, Y.; Geiger, J.D.; Dingledine, R.J. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 2006, 60, 223–235. [Google Scholar] [CrossRef]
- Hasan-Olive, M.M.; Lauritzen, K.H.; Ali, M.; Rasmussen, L.J.; Storm-Mathisen, J.; Bergersen, L.H. A Ketogenic Diet Improves Mitochondrial Biogenesis and Bioenergetics via the PGC1α-SIRT3-UCP2 Axis. Neurochem. Res. 2019, 44, 22–37. [Google Scholar] [CrossRef]
- Dabke, P.; Das, A.M. Mechanism of action of ketogenic diet treatment: Impact of decanoic acid and beta—hydroxybutyrate on sirtuins and energy metabolism in hippocampal murine neurons. Nutrients 2020, 12, 2379. [Google Scholar] [CrossRef]
- Malapaka, R.R.V.; Khoo, S.K.; Zhang, J.; Choi, J.H.; Zhou, X.E.; Xu, Y.; Gong, Y.; Li, J.; Yong, E.L.; Chalmers, M.J.; et al. Identification and mechanism of 10-carbon fatty acid as modulating ligand of peroxisome proliferator-activated receptors. J. Biol. Chem. 2012, 287, 183–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stumvoll, M.; Goldstein, B.J.; van Haeften, T.W. Type 2 diabetes: Principles of pathogenesis and therapy. Lancet 2005, 365, 1333–1346. [Google Scholar] [CrossRef]
- Miglio, G.; Rosa, A.C.; Rattazzi, L.; Collino, M.; Lombardi, G.; Fantozzi, R. PPARγ stimulation promotes mitochondrial biogenesis and prevents glucose deprivation-induced neuronal cell loss. Neurochem. Int. 2009, 55, 496–504. [Google Scholar] [CrossRef]
- Kanabus, M.; Fassone, E.; Hughes, S.D.; Bilooei, S.F.; Rutherford, T.; O’Donnell, M.; Heales, S.J.R.; Rahman, S. The pleiotropic effects of decanoic acid treatment on mitochondrial function in fibroblasts from patients with complex I deficient Leigh syndrome. J. Inherit. Metab. Dis. 2016, 39, 415–426. [Google Scholar] [CrossRef] [Green Version]
- Hughes, S.D.; Kanabus, M.; Anderson, G.; Hargreaves, I.P.; Rutherford, T.; O’Donnell, M.; Cross, J.H.; Rahman, S.; Eaton, S.; Heales, S.J.R. The ketogenic diet component decanoic acid increases mitochondrial citrate synthase and complex I activity in neuronal cells. J. Neurochem. 2014, 129, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.S.; Rogatzki, M.J.; Goodwin, M.L.; Kane, D.A.; Rightmire, Z.; Gladden, L.B. Lactate metabolism: Historical context, prior misinterpretations, and current understanding. Euro. J. Appl. Physiol. 2018, 118, 691–728. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A. Brain lactate metabolism: The discoveries and the controversies. J. Cereb. Blood Flow Metab. 2012, 32, 1107–1138. [Google Scholar] [CrossRef] [Green Version]
- Gladden, L.B. Lactate metabolism: A new paradigm for the third millennium. J. Physiol. 2004, 558, 5–30. [Google Scholar] [CrossRef]
- Pellerin, L.; Bouzier-Sore, A.K.; Aubert, A.; Serres, S.; Merle, M.; Costalat, R.; Magistretti, P.J. Activity-dependent regulation of energy metabolism by astrocytes: An update. Glia 2007, 55, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, M.; Blázquez, C. Is there an astrocyte- neuron ketone body shuttle? Trends Endocrinol. Metab. 2001, 12, 169–173. [Google Scholar] [CrossRef]
- Deitmer, J.W. Glial strategy for metabolic shuttling and neuronal function. BioEssays 2000, 22, 747–752. [Google Scholar] [CrossRef]
- Thevenet, J.; De Marchi, U.; Domingo, J.S.; Christinat, N.; Bultot, L.; Lefebvre, G.; Sakamoto, K.; Descombes, P.; Masoodi, M.; Wiederkehr, A. Medium-chain fatty acids inhibit mitochondrial metabolism in astrocytes promoting astrocyte-neuron lactate and ketone body shuttle systems. FASEB J. 2016, 30, 1913–1926. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.L. Brain activity-induced neuronal glucose uptake/glycolysis: Is the lactate shuttle not required? Brain Res. Bull. 2018, 137, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A. Lack of appropriate stoichiometry: Strong evidence against an energetically important astrocyte–neuron lactate shuttle in brain. J. Neurosci. Res. 2017, 95, 2103–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takeishi, J.; Tatewaki, Y.; Nakase, T.; Takano, Y.; Tomita, N.; Yamamoto, S.; Mutoh, T.; Taki, Y. Alzheimer’s Disease and Type 2 Diabetes Mellitus: The Use of MCT Oil and a Ketogenic Diet. Int. J. Mol. Sci. 2021, 22, 12310. https://doi.org/10.3390/ijms222212310
Takeishi J, Tatewaki Y, Nakase T, Takano Y, Tomita N, Yamamoto S, Mutoh T, Taki Y. Alzheimer’s Disease and Type 2 Diabetes Mellitus: The Use of MCT Oil and a Ketogenic Diet. International Journal of Molecular Sciences. 2021; 22(22):12310. https://doi.org/10.3390/ijms222212310
Chicago/Turabian StyleTakeishi, Junpei, Yasuko Tatewaki, Taizen Nakase, Yumi Takano, Naoki Tomita, Shuzo Yamamoto, Tatsushi Mutoh, and Yasuyuki Taki. 2021. "Alzheimer’s Disease and Type 2 Diabetes Mellitus: The Use of MCT Oil and a Ketogenic Diet" International Journal of Molecular Sciences 22, no. 22: 12310. https://doi.org/10.3390/ijms222212310
APA StyleTakeishi, J., Tatewaki, Y., Nakase, T., Takano, Y., Tomita, N., Yamamoto, S., Mutoh, T., & Taki, Y. (2021). Alzheimer’s Disease and Type 2 Diabetes Mellitus: The Use of MCT Oil and a Ketogenic Diet. International Journal of Molecular Sciences, 22(22), 12310. https://doi.org/10.3390/ijms222212310