
Design, Synthesis, and Anticancer Activity of a Selenium-Containing Galectin-3 and Galectin-9N Inhibitor

 ,
,  ,
,  ,
, 

 , ,
, , 
Abstract
:
1. Introduction
2. Results and Discussion
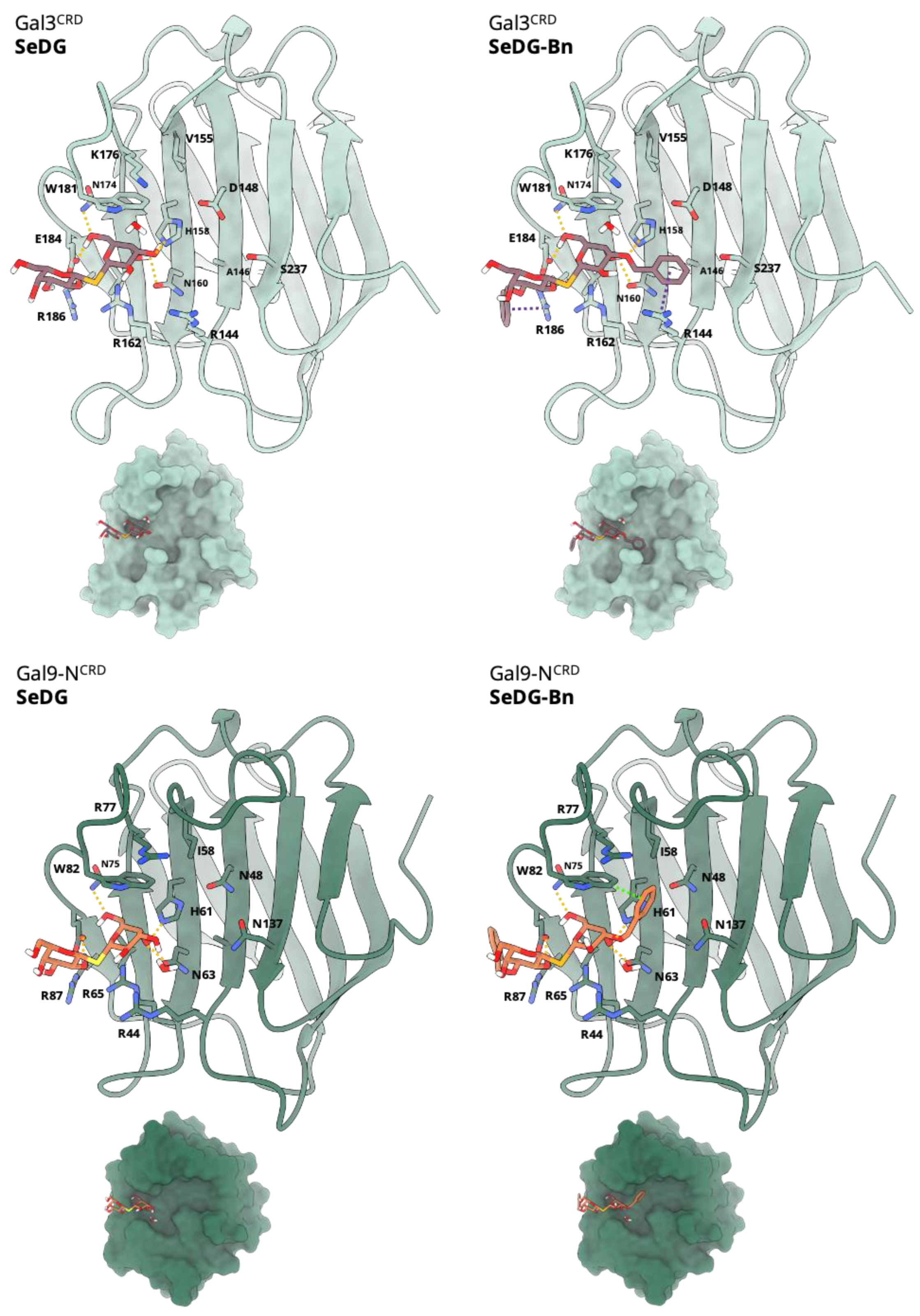
2.1. Modelling and Molecular Docking
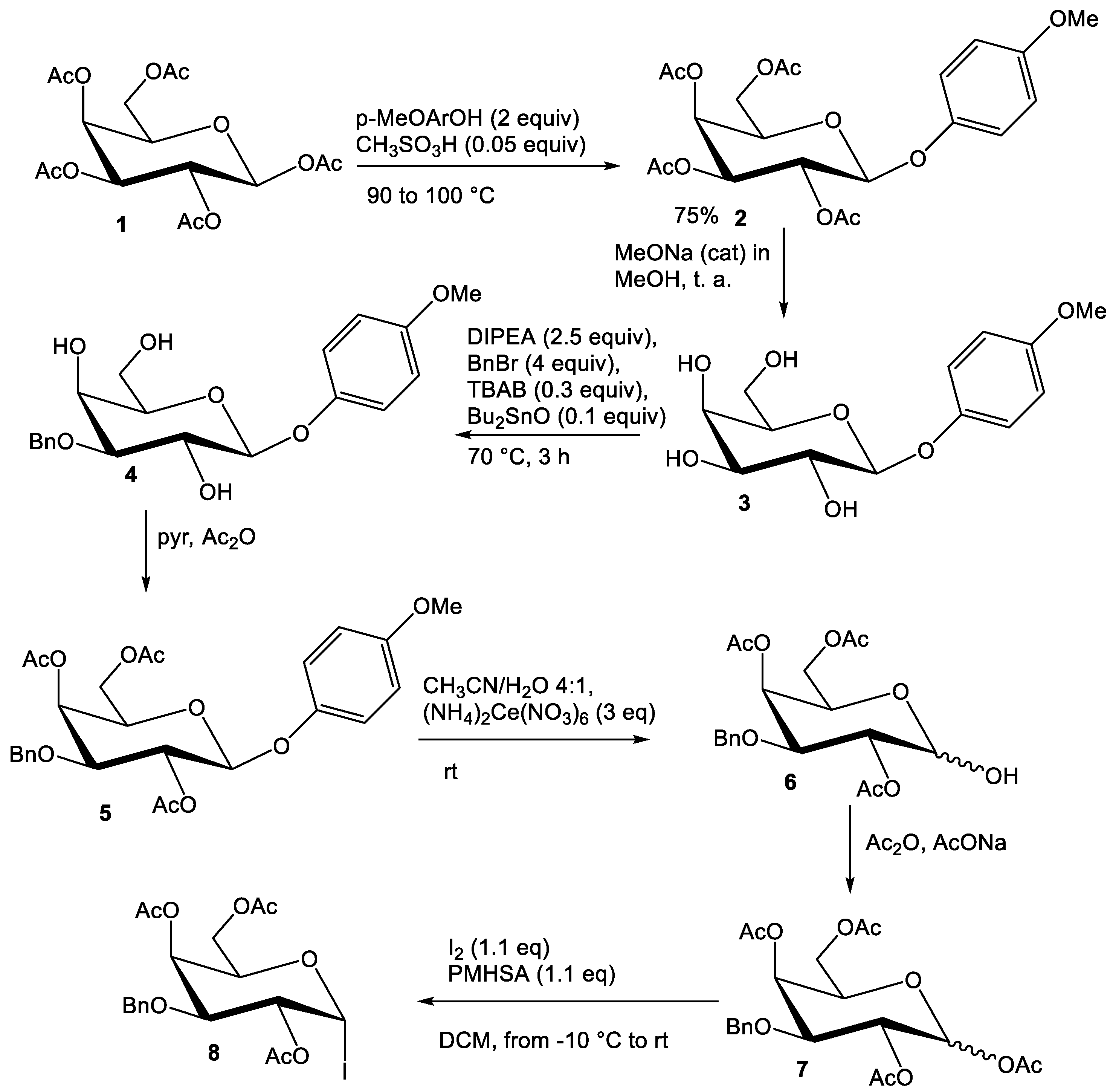
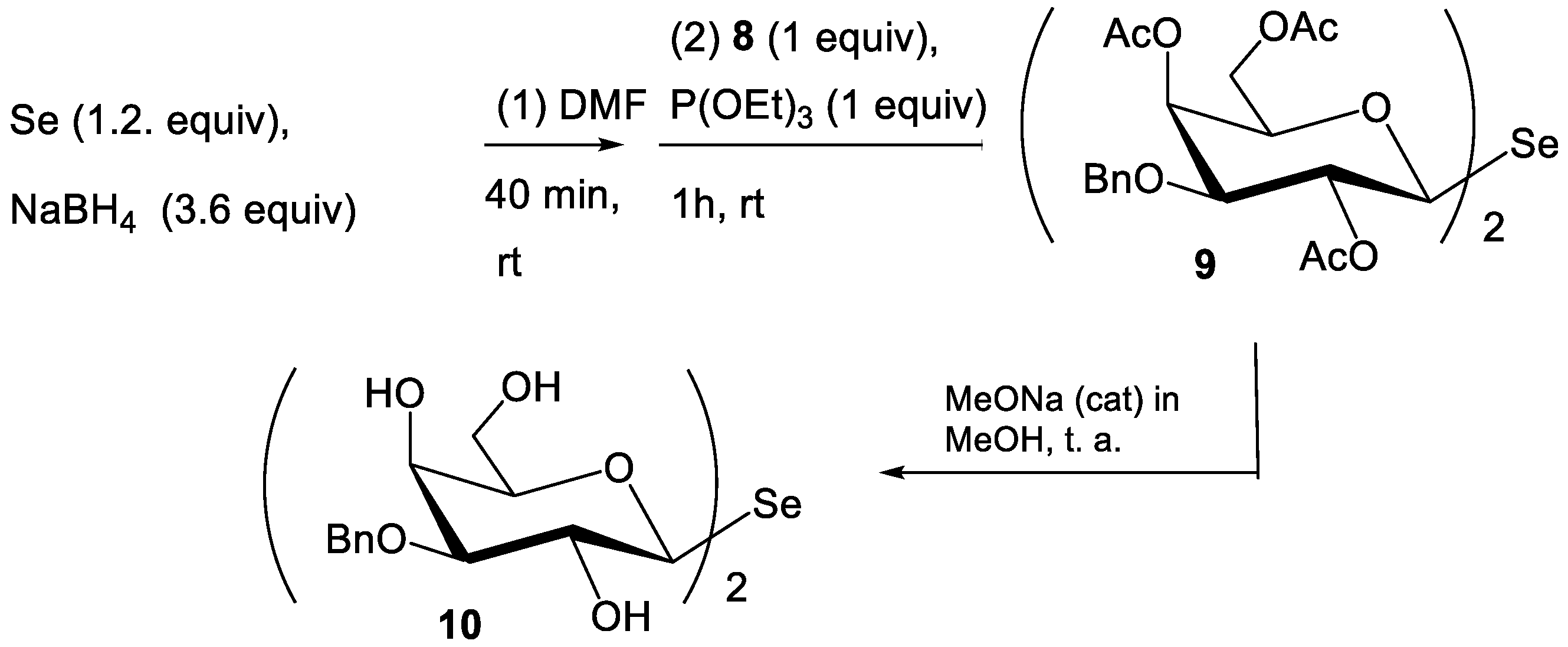
2.2. Synthesis of Compounds
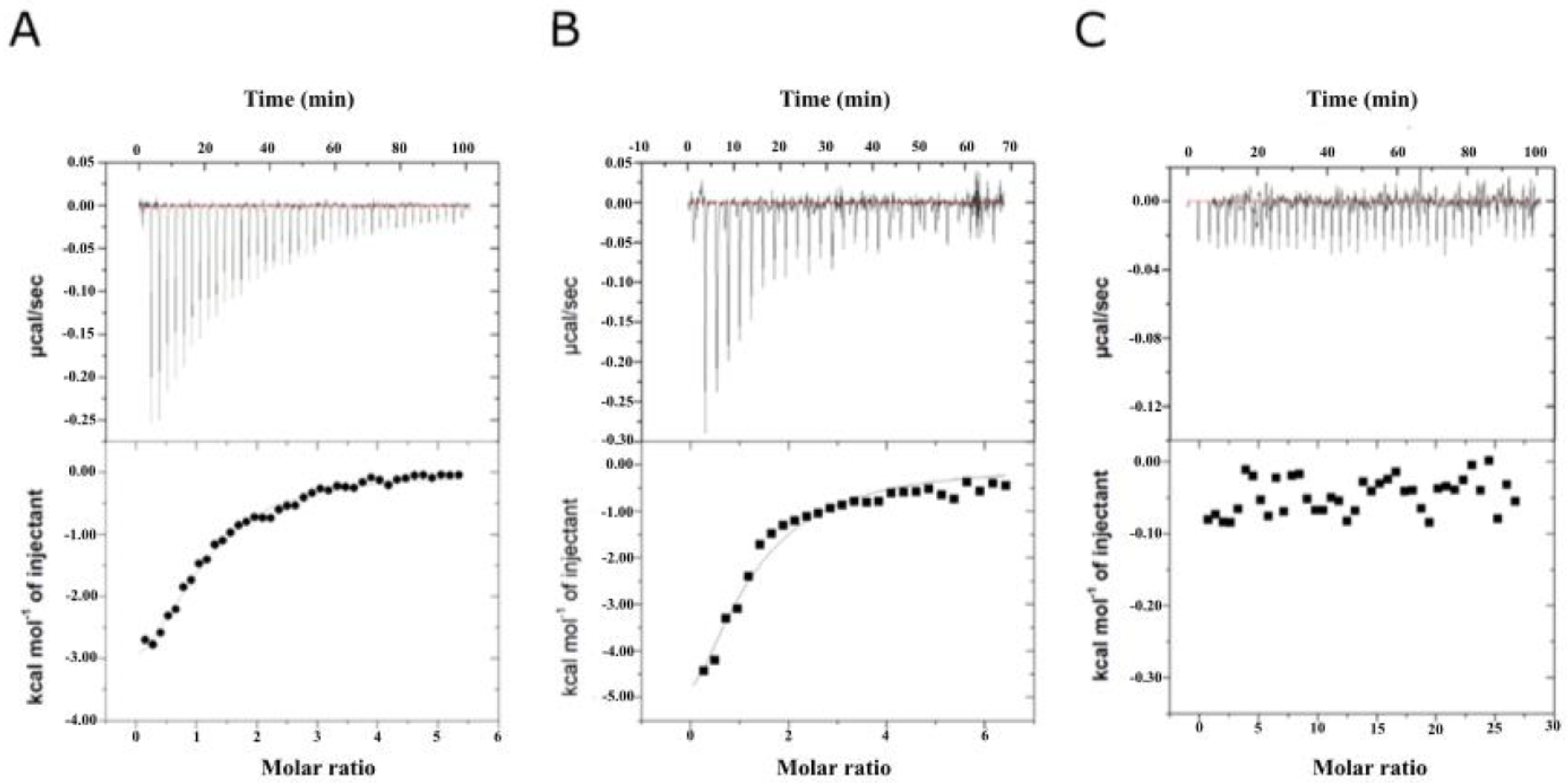
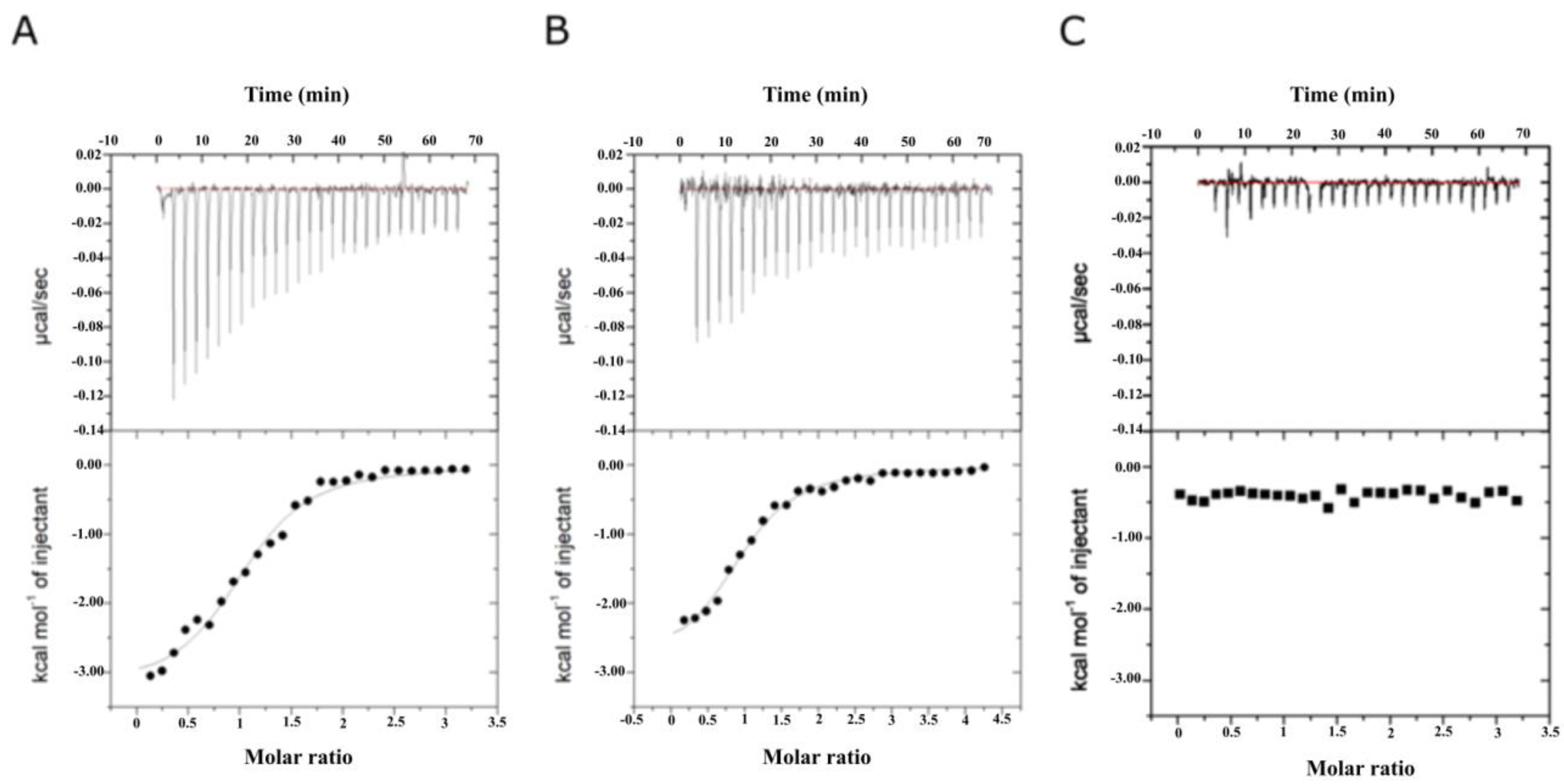
2.3. Binding Affinity Analysis of SeDG and SeDG-Bn to Gal-3CRD and Gal-9NCRD
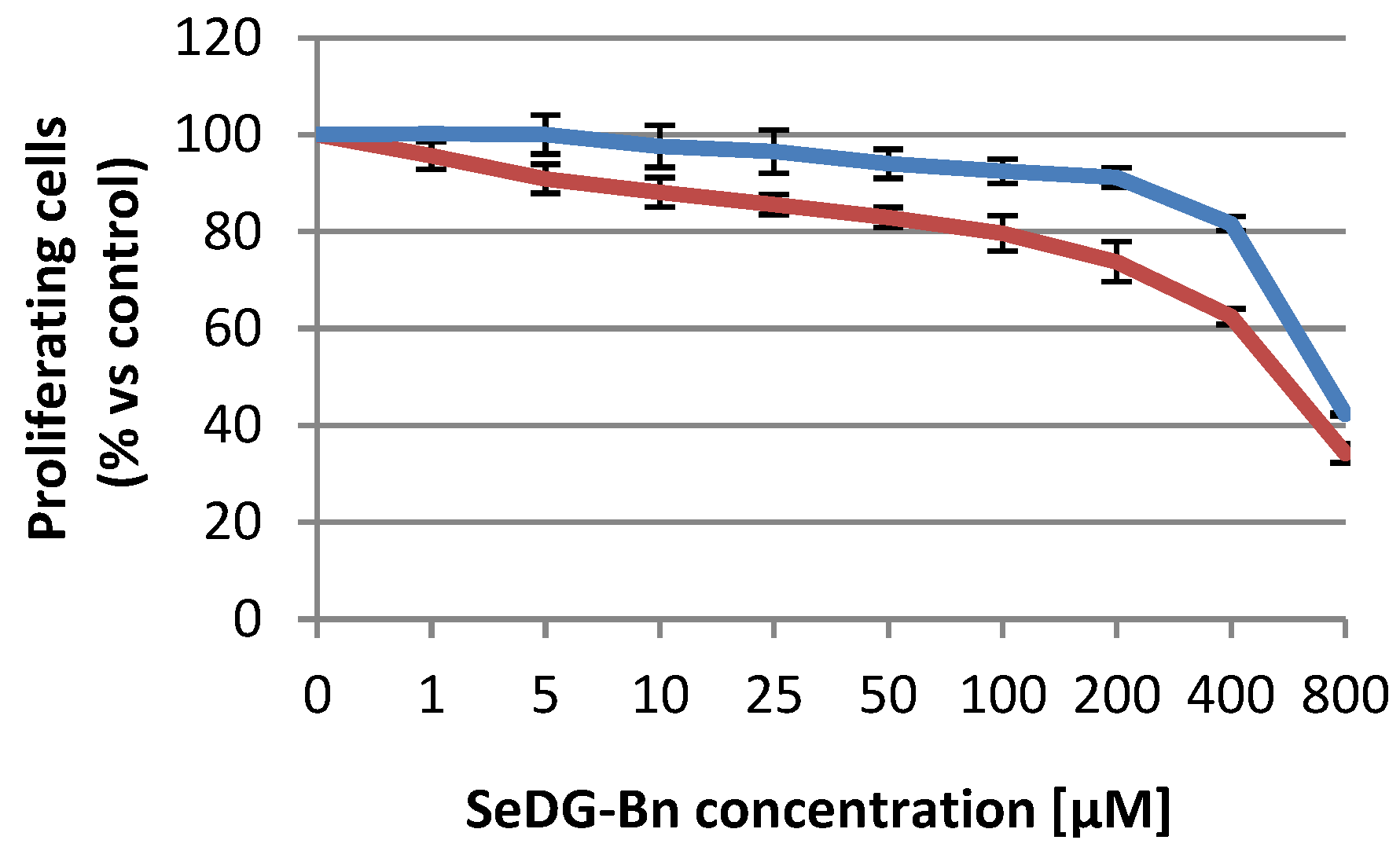
2.4. Citotoxicity Assays
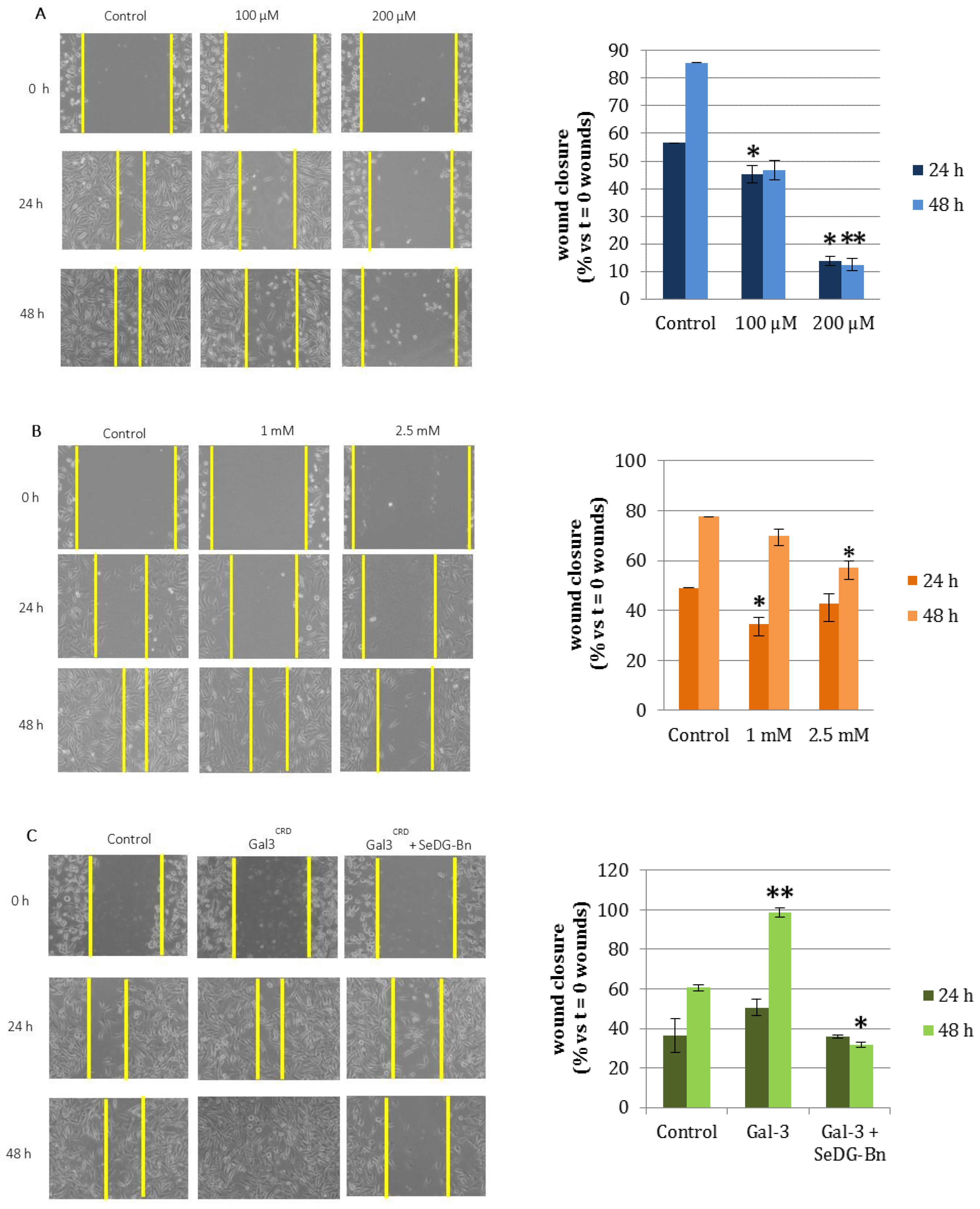
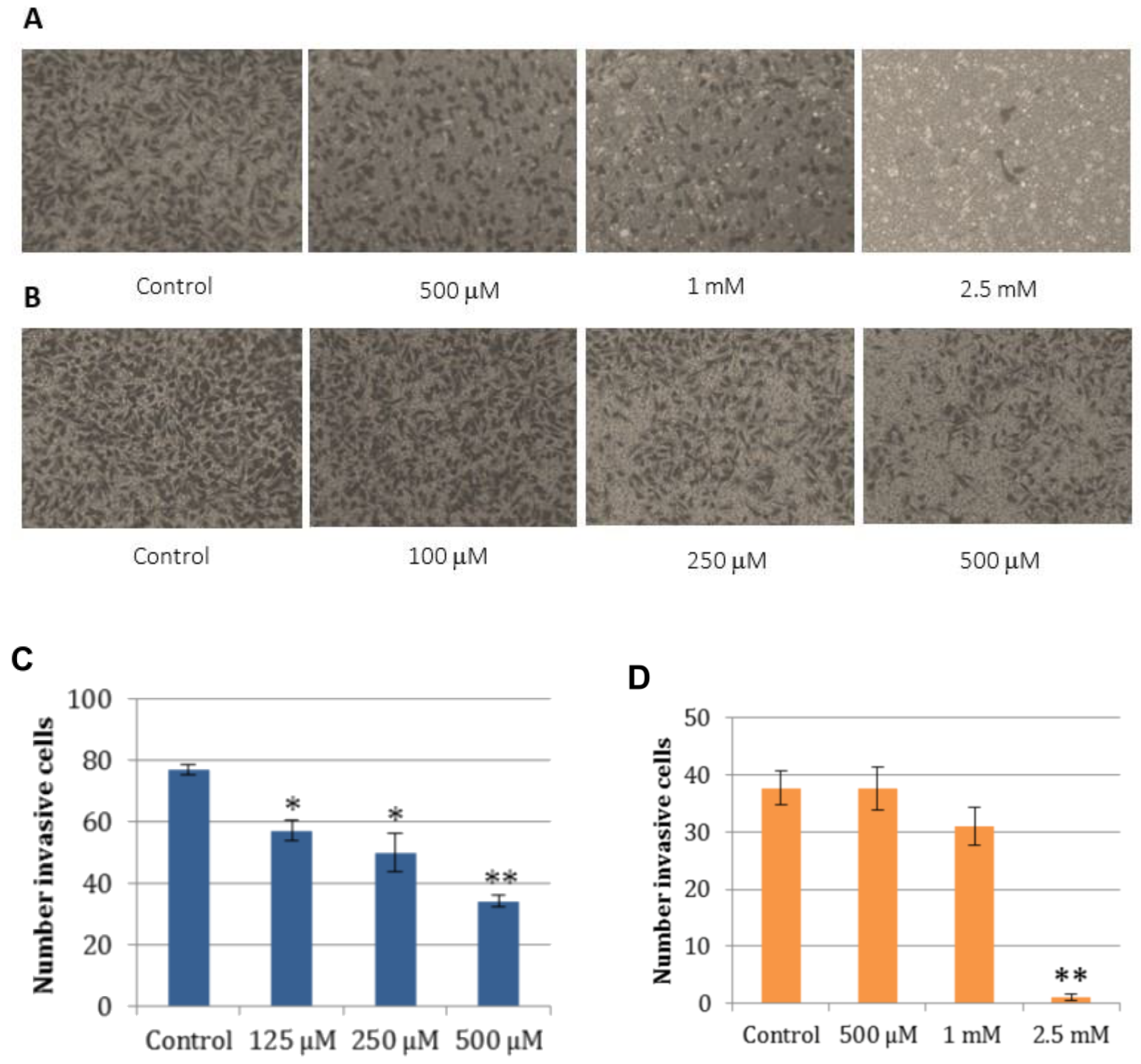
2.5. Cell Wound Healing and Invasion Assay
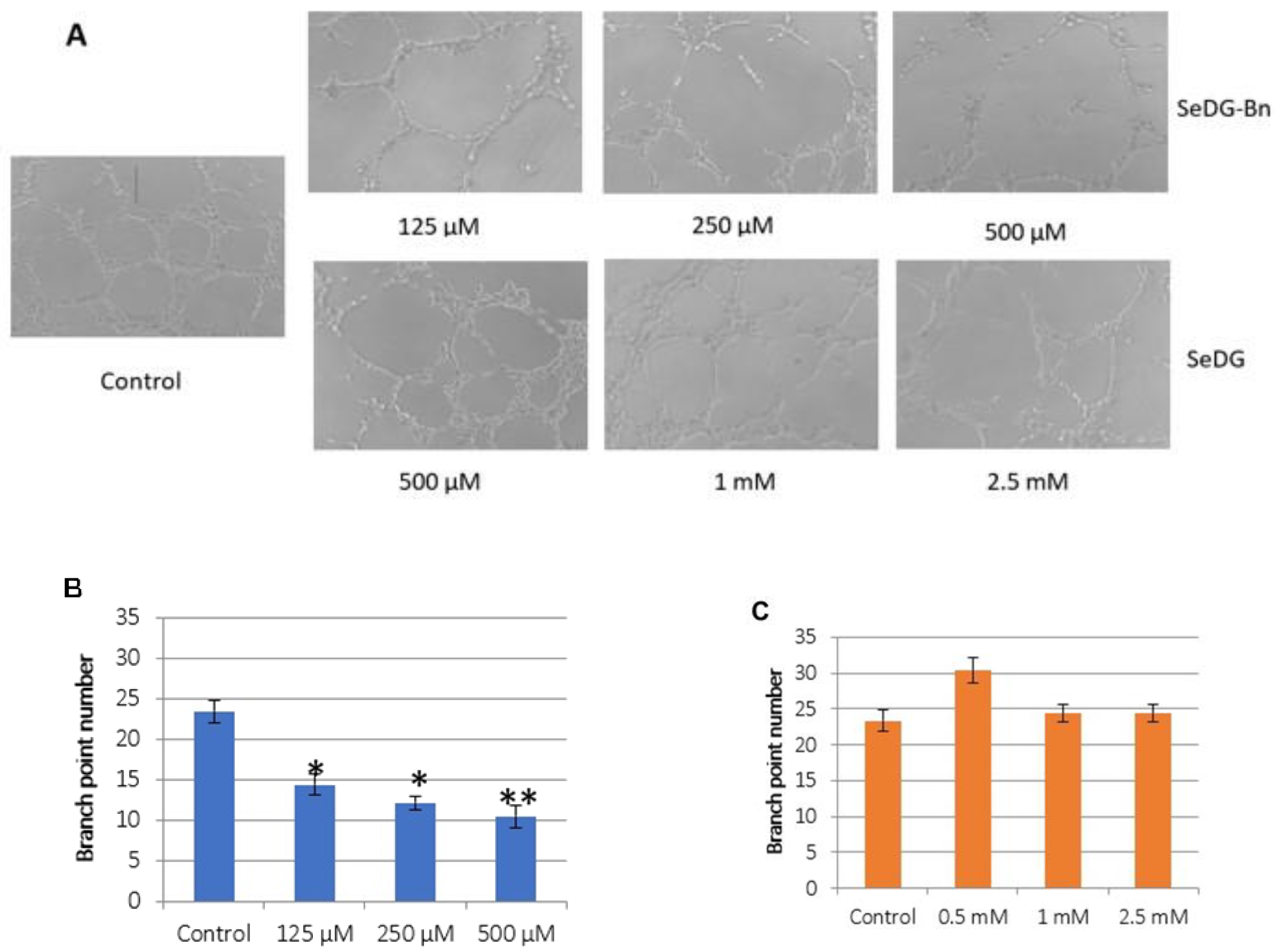
2.6. Angiogenesis Assay
3. Materials and Methods
3.1. Molecular Modeling and Docking
3.2. Compound Synthesis
3.2.1. Synthesis of 4
3.2.2. Synthesis of 5
3.2.3. Synthesis of 6
3.2.4. Synthesis of 7
3.2.5. Synthesis of 8
3.2.6. Synthesis of 9
3.2.7. Synthesis of 10 (SeDG-Bn)
3.3. Gal-3CRD and Gal-9NCRD Expression and Purification
3.4. Isothermal Titration Calorimetry
3.5. Cell Lines and Culture Conditions
3.6. Western Blotting
3.7. Cell Proliferation Assay
3.8. The Wound Healing Assay
3.9. Invasion Assay
3.10. Angiogenesis Assay
3.11. Statistical Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SeDG | Digalactosyl selenide |
| SeDG-Bn | 3-3′-O-dibenzyl Digalactosyl selenide with |
| IPTG | Isopropyl β-D-1-thiogalactopyranoside |
References
- Laaf, D.; Bojarová, P.; Elling, L.; Křen, V. Galectin-Carbohydrate Interactions in Biomedicine and Biotechnology. Trends Biotechnol. 2019, 37, 402–415. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.N. Galectinomics: Finding themes in complexity. Biochim. Biophys. Acta 2002, 1572, 209–231. [Google Scholar] [CrossRef]
- Houzelstein, D.; Gonçalves, I.R.; Fadden, A.J.; Sidhu, S.S.; Cooper, D.N.W.; Drickamer, K.; Leffler, H.; Poirier, F. Phylogenetic analysis of the vertebrate galectin family. Mol. Biol. Evol. 2004, 21, 1177–1187. [Google Scholar] [CrossRef] [Green Version]
- Ludger, J.; Jacob, R.; Leffler, H. Galectins at a glance. J. Cell Sci. 2018, 131, jcs208884. [Google Scholar]
- Ludwig, A.K.; Michalak, M.; Xiao, Q.; Gilles, U.; Medrano, F.J.; Ma, H.; FitzGerald, F.G.; Hasley, W.D.; Melendez-Davila, A.; Liu, M.; et al. Design—Functionality relationships for adhesion/growth-regulatory galectins. Proc. Natl. Acad. Sci. USA 2019, 116, 2837–2842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stegmayr, J.; Zetterberg, F.; Carlsson, M.C.; Huang, X.; Sharma, G.; Kahl-Knutson, B.; Schambye, H.; Nilsson, U.J.; Oredsson, S.; Leffler, H. Extracellular and intracellular small-molecule galectin-3 inhibitors. Sci. Rep. 2019, 9, 2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciacchitano, S.; Lavra, L.; Morgante, A.; Ulivieri, A.; Magi, F.; De Francesco, G.P.; Bellotti, C.; Salehi, L.B.; Ricci, A. Galectin-3: One Molecule for an Alphabet of Diseases, from A to Z. Int. J. Mol. Sci. 2018, 19, 379. [Google Scholar] [CrossRef] [Green Version]
- Hughes, R.C. Galectins as modulators of cell adhesion. Biochimie 2001, 83, 667–676. [Google Scholar] [CrossRef]
- Etulain, J.; Negrotto, S.; Tribulatti, M.V.; Croci, D.O.; Carabelli, J.; Campetella, O.; Rabinovich, G.A.; Schattner, M. Control of Angiogenesis by Galectins Involves the Release of Platelet-Derived Proangiogenic Factors. PLos ONE 2014, 9, e96402. [Google Scholar] [CrossRef]
- De Oliveira, F.L.; Gatto, M.; Bassi, N.; Luisetto, R.; Ghirardello, A.; Punzi, L.; Doria, A. Galectin-3 in autoimmunity and autoimmune diseases. Exp. Biol. Med. 2015, 240, 1019–1028. [Google Scholar] [CrossRef] [Green Version]
- D’Alessandro, M.; De Vita, E.; Bergantini, L.; Mazzei, M.A.; di Valvasone, S.; Bonizzoli, M.; Peris, A.; Sestini, P.; Bargagli, E.; Bennett, D. Galactin-1, 3 and 9: Potential biomarkers in idiopathic pulmonary fibrosis and other interstitial lung diseases. Respir. Physiol. Neurobiol. 2020, 282, 103546. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.T.; Rabinovich, G.A. Galectins as modulators of tumour progression. Nat. Rev. Cancer 2005, 5, 29–41. [Google Scholar] [CrossRef]
- Pirone, L.; Del Gatto, A.; Di Gaetano, S.; Saviano, M.; Capasso, D.; Zaccaro, L.; Pedone, E. A Multi-Targeting Approach to Fight SARS-CoV-2 Attachment. Front. Mol. Biosci. 2020, 7, 186. [Google Scholar] [CrossRef]
- Di Gaetano, S.; Capasso, D.; Pirone, L.; Delre, P.; Mangiatordi, G.F.; Saviano, M.; Pedone, E. More is always better than one: The N-terminal domain of the spike protein as another emerging target for hampering the SARS-CoV-2 attachment to host cells. Int. J. Mol. Sci. 2021, 22, 6462. [Google Scholar] [CrossRef]
- Baum, L.G.; Garner, O.B.; Schaefer, K.; Lee, B. Microbe-host interactions are positively and negatively regulated by galectin-glycan interactions. Front. Immunol. 2014, 18, 284. [Google Scholar]
- Liu, F.T.; Patterson, R.J.; Wang, J.L. Intracellular functions of galectins. Biochim. Biophys. Acta 2002, 1572, 263–273. [Google Scholar] [CrossRef]
- Shimura, T.; Takenaka, Y.; Tsutsumi, S.; Hogan, V.; Kikuchi, A.; Raz, A. Galectin-3, a novel binding partner of beta-catenin. Cancer Res. 2004, 64, 6363–6367. [Google Scholar] [CrossRef] [Green Version]
- Nangia-Makker, P.; Nakahara, S.; Hogan, V.; Raz, A. Galectin-3 in apoptosis, a novel therapeutic target. J. Bioenerg. Biomembr. 2007, 39, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.T.; Hsu, D.K. The role of galectin-3 in promotion of the inflammatory response. Drug News Perspect. 2007, 20, 455–460. [Google Scholar] [CrossRef]
- Yang, R.Y.; Rabinovich, G.A.; Liu, F.T. Galectins: Structure, function and therapeutic potential. Expert Rev. Mol. Med. 2008, 13, e17. [Google Scholar]
- Dion, J.; Deshayes, F.; Storozhylova, N.; Advedissian, T.; Lambert, A.; Viguier, M.; Tellier, C.; Dussouy, C.; Poirier, F.; Grandjean, C. Lactosamine-Based Derivatives as Tools to Delineate the Biological Functions of Galectins: Application to Skin Tissue Repair. ChemBioChem. 2017, 18, 782–789. [Google Scholar] [CrossRef]
- Marchiori, M.F.; Souto, D.E.; Bortot, L.O.; Pereira, J.F.; Kubota, L.T.; Cummings, R.D.; Dias-Baruffi, M.; Carvalho, I.; Campo, V.L. Synthetic 1,2,3-triazole-linked glycoconjugates bind with high affinity to human galectin. Bioorg. Med. Chem. 2015, 23, 3414–3425. [Google Scholar] [CrossRef]
- Gordon-Alonso, M.; Hirsch, T.; Wildmann, C.; van der Bruggen, P. Galectin-3 captures interferon-gamma in the tumor matrix reducing chemokine gradient production and T-cell tumor infiltration. Nat. Commun. 2017, 8, 793. [Google Scholar] [CrossRef]
- Dange, M.C.; Agarwal, A.K.; Kalraiya, R.D. Extracellular galectin-3 induces MMP9 expression by activating p38 MAPK pathway via lysosome-associated membrane protein-1 (LAMP1). Mol. Cell. Biochem. 2015, 404, 79–86. [Google Scholar] [CrossRef]
- Laaf, D.; Bojarová, P.; Pelantová, H.; Křen, V.; Elling, L. Tailored Multivalent Neo-Glycoproteins: Synthesis, Evaluation, and Application of a Library of Galectin-3-Binding Glycan Ligands. Bioconjug. Chem. 2017, 28, 2832–2840. [Google Scholar] [CrossRef]
- Cumpstey, I.; Carlsson, S.; Leffler, H.; Nilsson, U.J. Synthesis of a phenyl thio-[small beta]-d-galactopyranoside library from 1,5-difluoro-2,4-dinitrobenzene: Discovery of efficient and selective monosaccharide inhibitors of galectin-Org. Biomol. Chem. 2005, 3, 1922–1932. [Google Scholar] [CrossRef]
- Vašíček, T.; Spiwok, V.; Červený, J.; Petrásková, L.; Bumba, L.; Vrbata, D.; Pelantová, H.; Křen, V.; Bojarová, P. Regioselective 3-O-Substitution of Unprotected Thiodigalactosides: Direct Route to Galectin Inhibitors. Chemistry 2020, 26, 9620–9631. [Google Scholar] [CrossRef]
- Cumpstey, I.; Sundin, A.; Leffler, H.; Nilsson, U.J. C2-symmetrical thiodigalactoside bis-benzamido derivatives as high-affinity inhibitors of galectin-3: Efficient lectin inhibition through double arginine-arene interactions. Angew. Chem. 2005, 44, 5110–5112. [Google Scholar] [CrossRef]
- Delaine, T.; Collins, P.; MacKinnon, A.; Sharma, G.; Stegmayr, J.; Rajput, V.K.; Mandal, S.; Cumstey, I.; Larumbe, A.; Salameh, B.A.; et al. Galectin-3-Binding Glycomimetics that Strongly Reduce Bleomycin-Induced Lung Fibrosis and Modulate Intracellular Glycan Recognition. ChemBioChem 2016, 17, 1759–1770. [Google Scholar] [CrossRef]
- Salameh, A.; Cumpstey, I.; Sundin, A.; Leffler, H.; Nilsson, U.J. 1H-1,2,3-triazol-1-yl thiodigalactoside derivatives as high affinity galectin-3 inhibitors. Bioorg. Med. Chem. 2010, 18, 5367–5378. [Google Scholar] [CrossRef]
- Rajput, V.K.; MacKinnon, A.; Mandal, S.; Collins, P.; Blanchard, H.; Leffler, H.; Sethi, T.; Schambye, H.; Mukhopadhyay, B.; Nilsson, U.J. A Selective Galactose-Coumarin-Derived Galectin-3 Inhibitor Demonstrates Involvement of Galectin-3-glycan Interactions in a Pulmonary Fibrosis Model. J. Med. Chem. 2016, 59, 8141. [Google Scholar] [CrossRef] [PubMed]
- Di Gaetano, S.; Bedini, E.; Landolfi, A.; Pedone, E.; Pirone, L.; Saviano, M.; Traboni, S.; Capasso, D.; Iadonisi, A. Synthesis of diglycosylated (di)sulfides and comparative evaluation of their antiproliferative effect against tumor cell lines: A focus on the nature of sugar-recognizing mediators involved. Carbohydr. Res. 2019, 482, 107740. [Google Scholar] [CrossRef] [PubMed]
- Mangiavacchi, F.; Coelho Dias, I.F.; Di Lorenzo, I.; Grzes, P.; Palomba, M.; Rosati, O.; Bagnoli, L.; Marini, F.; Santi, C.; Lenardao, E.J.; et al. Sweet Selenium: Synthesis and Properties of Selenium-Containing Sugars and Derivatives. Pharmaceuticals 2020, 13, 211. [Google Scholar] [CrossRef] [PubMed]
- Sanmartín, C.; Plano, D.; Sharma, A.K.; Palop, J.A. Selenium compounds, apoptosis and other types of cell death: An overview for cancer therapy. Int. J. Mol. Sci. 2012, 13, 9649–9672. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.P.; Gandin, V. Selenium compounds as therapeutic agents in cancer. Biophys. Acta 2014, 1850, 1642–1660. [Google Scholar] [CrossRef] [PubMed]
- Mugesh, G.; du Mont, W.W.; Sies, H. Chemistry of biologically important synthetic organoselenium compounds. Chem. Rev. 2001, 101, 2125–2179. [Google Scholar] [CrossRef]
- Nogueira, C.W.; Zeni, G.; Rocha, J.B. Organoselenium and organotellurium compounds: Toxicology and pharmacology. Chem. Rev. 2004, 104, 6255–6285. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Mugesh, G.; Panda, A.; Singh, H.B.; Punekar, N.S.; Butcher, R.J. Glutathione peroxidase-like antioxidant activity of diaryl diselenides: A Mechanistic Study. J. Am. Chem. Soc. 2001, 123, 839–850. [Google Scholar] [CrossRef]
- Mugesh, G.; Panda, A.; Singh, H.B.; Punekar, N.S.; Butcher, R.J. Diferrocenyl diselenides: Excellent thiol peroxidase-like antioxidants. Chem. Commun. 1998, 20, 2227–2228. [Google Scholar] [CrossRef]
- Shechter, S.; Zomer, E.; Traber, P.G.; Nir, R.; Johnson, J.M.; Ryan, G. Selenogalactoside Compounds for the Prevention and Treatment of Diseases Associated with Galectin and the Use Thereof. U.S. Patent WO 2017/152048A, 8 September 2017. [Google Scholar]
- André, S.; Kövér, K.E.; Gabius, H.J.; Szilágyi, L. Thio- and selenoglycosides as ligands for biomedically relevant lectins: Valency-activity correlations for benzene-based dithiogalactoside clusters and first assessment for (di)selenodigalactosides. Bioorg. Med. Chem. Lett. 2015, 25, 931–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iadonisi, A.; Traboni, S.; Capasso, D.; Bedini, E.; Cuomo, S.; Di Gaetano, S.; Vessella, G. Switchable synthesis of glycosyl selenides or diselenides with direct use of selenium as selenating agent. Org. Chem. Front. 2021, 8, 823–1829. [Google Scholar] [CrossRef]
- Lee, Y.S.; Rho, E.S.; Min, Y.K.; Kim, B.T.; Kim, K.H. Practical β-stereoselective O-glycosylation of phenols with penta-O-acetyl-β-D-Gglucopyranose. J. Carbohydr. Chem. 2001, 20, 503–506. [Google Scholar] [CrossRef]
- Giordano, M.; Iadonisi, A. Polymethylhydrosiloxane (PMHS): A convenient option for synthetic applications of the iodine/silane combined reagent. Straightforward entries to 2-hydroxyglycals and useful building-blocks of glucuronic acid and glucosamine. Eur. J. Org. Chem. 2013, 2013, 125–131. [Google Scholar] [CrossRef]
- Adinolfi, M.; Iadonisi, A.; Ravidà, A.; Schiattarella, M. Efficient and direct synthesis of saccharidic 1,2-ethylidenes, orthoesters, and glycals from peracetylated sugars via the in situ generation of glycosyl iodides with I2/Et3SiH. Tetraedron Lett. 2003, 44, 7863–7866. [Google Scholar] [CrossRef]
- Pastore, A.; Valerio, S.; Adinolfi, M.; Iadonisi, A. An Easy and Versatile Approach for the Regioselective De-O-benzylation of Protected Sugars Based on the I2/Et3SiH Combined System. Chem. Eur. J. 2011, 17, 5881–5889. [Google Scholar] [CrossRef]
- Ito, K.; Scott, S.A.; Cutler, S.; Dong, L.F.; Neuzil, J.; Blanchard, H.; Ralph, S.J. Thiodigalactoside inhibits murine cancers by concurrently blocking effects of galectin-1 on immune dysregulation, angiogenesis and protection against oxidative stress. Angiogenesis 2011, 14, 293–307. [Google Scholar] [CrossRef] [Green Version]
- Duckworth, C.A.; Scott, E.; Guimond, S.E.; Sindrewicz, P.; Hughes, A.J.; French, N.S.; Lian, L.Y.; Yates, E.A.; Pritchard, D.M.; Rhodes, J.M.; et al. Chemically modified, non-anticoagulant heparin derivatives are potent galectin-3 binding inhibitors and inhibit circulating galectin-3-promoted metastasis. Oncotarget 2015, 6, 23671–23687. [Google Scholar] [CrossRef] [Green Version]
- Astorgues-Xerri, L.; Riveiro, M.E.; Tijeras-Raballand, A.; Serova, M.; Neuzillet, C.; Albert, S.; Raymond, E.; Faivre, S. Unraveling galectin-1 as a novel therapeutic target for cancer. Cancer Treat. Rev. 2014, 40, 307–319. [Google Scholar] [CrossRef]
- Delaine, T.; Cumpstey, I.; Ingrassia, L.; Le Mercier, M.; Okechukwu, P.; Leffler, H.; Kiss, R.; Ulf, J.; Nilsson, U.J. Galectin-Inhibitory Thiodigalactoside Ester Derivatives Have Antimigratory Effects in Cultured Lung and Prostate Cancer Cells. J. Med. Chem. 2008, 51, 8109–8114. [Google Scholar] [CrossRef]
- Polli, C.D.; Toledo, K.A.; Franco, L.H.; Mariano, V.S.; de Oliveira, L.L.; Bernardes, E.S.; Roque-Barreira, M.C. Gabriela Pereira-da-Silva, G. Monocyte migration driven by galectin-3 occurs through distinct mechanisms involving selective interactions with the extracellular matrix. ISRN Inflamm. 2013, 2013, 259256. [Google Scholar]
- Thijssen, V.L.; Griffioen, A.W. Galectin-1 and -9 in angiogenesis: A sweet couple. Glycobiology 2014, 24, 915–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos, S.N.; Sheldon, H.; Pereira, J.X.; Paluch, C.; Bridges, E.M.; El-Cheikh, M.C.; Harris, A.L.; Bernardes, E.S. Galectin-3 acts as an angiogenic switch to induce tumor angiogenesis via Jagged-1/Notch activation. Oncotarget 2017, 8, 49484–49501. [Google Scholar] [CrossRef] [Green Version]
- Nangia-Makker, P.; Honjo, Y.; Sarvis, R.; Akahani, S.; Hogan, V.; Pienta, K.J.; Raz, A. Galectin-3 induces endothelial cell morphogenesis and angiogenesis. Am. J. Pathol. 2000, 156, 899–909. [Google Scholar]
- Mahabeleshwar, G.H.; Byz, T.V. Angiogenesis in Melanoma. Semin. Oncol. 2007, 34, 555–565. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef]
- Amato, T.; Virgilio, A.; Pirone, L.; Vellecco, V.; Bucci, M.; Pedone, E.; Esposito, V.; Galeone, A. Investigating the properties of TBA variants with twin thrombin binding domains. Sci. Rep. 2019, 9, 9184. [Google Scholar] [CrossRef] [Green Version]
- Vicidomini, C.; Cioffi, F.; Broersen, K.; Roviello, V.; Riccardi, C.; Montesarchio, D.; Capasso, D.; Di Gaetano, S.; Musumeci, D.; Roviello, G.N. Benzodifurans for biomedical applications: BZ4, a selective anti-proliferative and anti-amyloid lead compound. Future Med. Chem. 2019, 11, 285–302. [Google Scholar] [CrossRef]
- Zhang, K.; Dong, C.; Chen, M.; Yang, T.; Wang, X.; Gao, Y.; Wang, L.; Wen, Y.; Chen, G.; Wang, X.; et al. Extracellular vesicle-mediated delivery of miR-101 inhibits lung metastasis in osteosarcoma. Theranostics 2020, 10, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.S.; Sarnella, A.; Capasso, D.; Comegna, D.; Del Gatto, A.; Gramanzini, M.; Albanese, S.; Saviano, M.; Zaccaro, L.; Zannetti, A. Therapeutic Potential of a Novel αvβ₃ Antagonist to Hamper the Aggressiveness of Mesenchymal Triple Negative Breast Cancer Sub-Type. Cancers 2019, 11, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comegna, D.; Zannetti, A.; Del Gatto, A.; de Paola, I.; Russo, L.; Di Gaetano, S.; Liguoro, A.; Capasso, D.; Saviano, M.; Zaccaro, L. Chemical Modification for Proteolytic Stabilization of the Selective αvβ3 Integrin RGDechi Peptide: In Vitro and in Vivo Activities on Malignant Melanoma Cells. J. Med.Chem. 2017, 60, 9874–9884. [Google Scholar] [CrossRef] [PubMed]
- Capasso, D.; Di Gaetano, S.; Celentano, V.; Diana, D.; Festa, L.; Di Stasi, R.; De Rosa, L.; Fattorusso, R.; D’Andrea, L.D. Unveiling a VEGF-mimetic peptide sequence in IQGAP1 protein. Mol. BioSyst. 2017, 13, 1619. [Google Scholar] [CrossRef]
- Seguin, L.; Odouard, S.; Corlazzoli, F.; Al Haddad, S.; Mondrot, L.; Calvo Tardón, M.; Yebra, M.; Koval, A.; Marinari, E.; Bes, V.; et al. Macropinocytosis requires Gal-3 in a subset of patient-derived glioblastoma stem cells. Commun. Biol. 2021, 4, 718. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Wang, X.; Wang, F.; Feng, E.; You, G. Galectin-9: A predictive biomarker negatively regulating immune response in glioma patients. World Neurosurg. 2019, 132, e455–e462. [Google Scholar] [CrossRef]
- Labrie, M.; De Araujo, L.O.F.; Communal, L.; Mes-Masson, A.-M.; St-Pierre, Y. Tissue and plasma levels of galectins in patients with high grade serous ovarian carcinoma as new predictive biomarkers. Sci. Rep. 2017, 7, 13244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, A.M.; Reiche, C.; Heiduk, M.; Tannert, A.; Meinecke, A.-C.; Baier, S.; von Renesse, J.; Kahlert, C.; Distler, M.; Welsch, T.; et al. Detection of pancreatic ductal adenocarcinoma with galectin-9 serum levels. Oncogene 2020, 39, 3102–3113. [Google Scholar] [CrossRef] [Green Version]
- Wdowiak, K.; Gallego-Colon, E.; Francuz, T.; Czajka-Francuz, P.; Ruiz-Agamez, N.; Kubeczko, M.; Grochoła, I.; Wybraniec, M.; Chudek, J.; Wojnar, J. Increased serum levels of Galectin-9 in patients with chronic lymphocytic leukemia. Oncol. Lett. 2018, 17, 1019–1029. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Kd (μM) | ΔG (kcal/mol) | ΔH (kcal/mol) | −TΔS (kcal/mol) | n |
|---|---|---|---|---|---|
| Gal-3CRD | |||||
| SeDG | 20.2 ± 2.2 | −6.31 | −4.32 ± 0.27 | −1.99 | 1.09 ± 0.05 |
| SeDG-Bn | 2.2 ± 0.4 | −7.69 | −3.25 ± 0.12 | −4.44 | 1.07 ± 0.03 |
| Gal-9NCRD | |||||
| SeDG | 22.4 ± 4.4 | −7.10 | −5.64 ± 0.36 | −1.46 | 1.01 ± 0.05 |
| SeDG-Bn | 2.9 ± 0.3 | −7.53 | −2.91 ± 0.12 | −4.62 | 0.99 ± 0.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Gaetano, S.; Pirone, L.; Galdadas, I.; Traboni, S.; Iadonisi, A.; Pedone, E.; Saviano, M.; Gervasio, F.L.; Capasso, D. Design, Synthesis, and Anticancer Activity of a Selenium-Containing Galectin-3 and Galectin-9N Inhibitor. Int. J. Mol. Sci. 2022, 23, 2581. https://doi.org/10.3390/ijms23052581
Di Gaetano S, Pirone L, Galdadas I, Traboni S, Iadonisi A, Pedone E, Saviano M, Gervasio FL, Capasso D. Design, Synthesis, and Anticancer Activity of a Selenium-Containing Galectin-3 and Galectin-9N Inhibitor. International Journal of Molecular Sciences. 2022; 23(5):2581. https://doi.org/10.3390/ijms23052581
Chicago/Turabian StyleDi Gaetano, Sonia, Luciano Pirone, Ioannis Galdadas, Serena Traboni, Alfonso Iadonisi, Emilia Pedone, Michele Saviano, Francesco Luigi Gervasio, and Domenica Capasso. 2022. "Design, Synthesis, and Anticancer Activity of a Selenium-Containing Galectin-3 and Galectin-9N Inhibitor" International Journal of Molecular Sciences 23, no. 5: 2581. https://doi.org/10.3390/ijms23052581
APA StyleDi Gaetano, S., Pirone, L., Galdadas, I., Traboni, S., Iadonisi, A., Pedone, E., Saviano, M., Gervasio, F. L., & Capasso, D. (2022). Design, Synthesis, and Anticancer Activity of a Selenium-Containing Galectin-3 and Galectin-9N Inhibitor. International Journal of Molecular Sciences, 23(5), 2581. https://doi.org/10.3390/ijms23052581