
The R369 Myosin Residue within Loop 4 Is Critical for Actin Binding and Muscle Function in Drosophila



Abstract
1. Introduction
2. Results
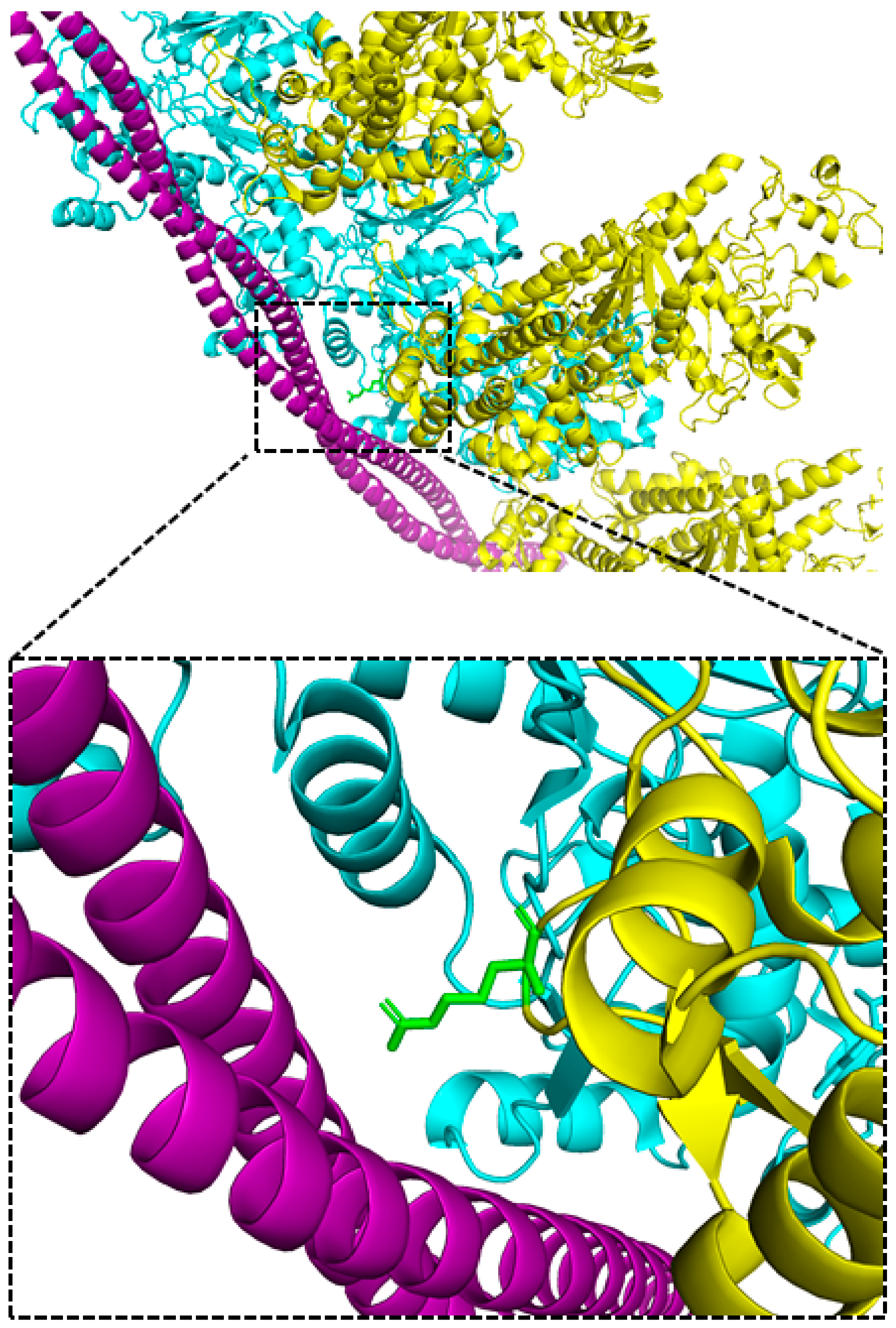
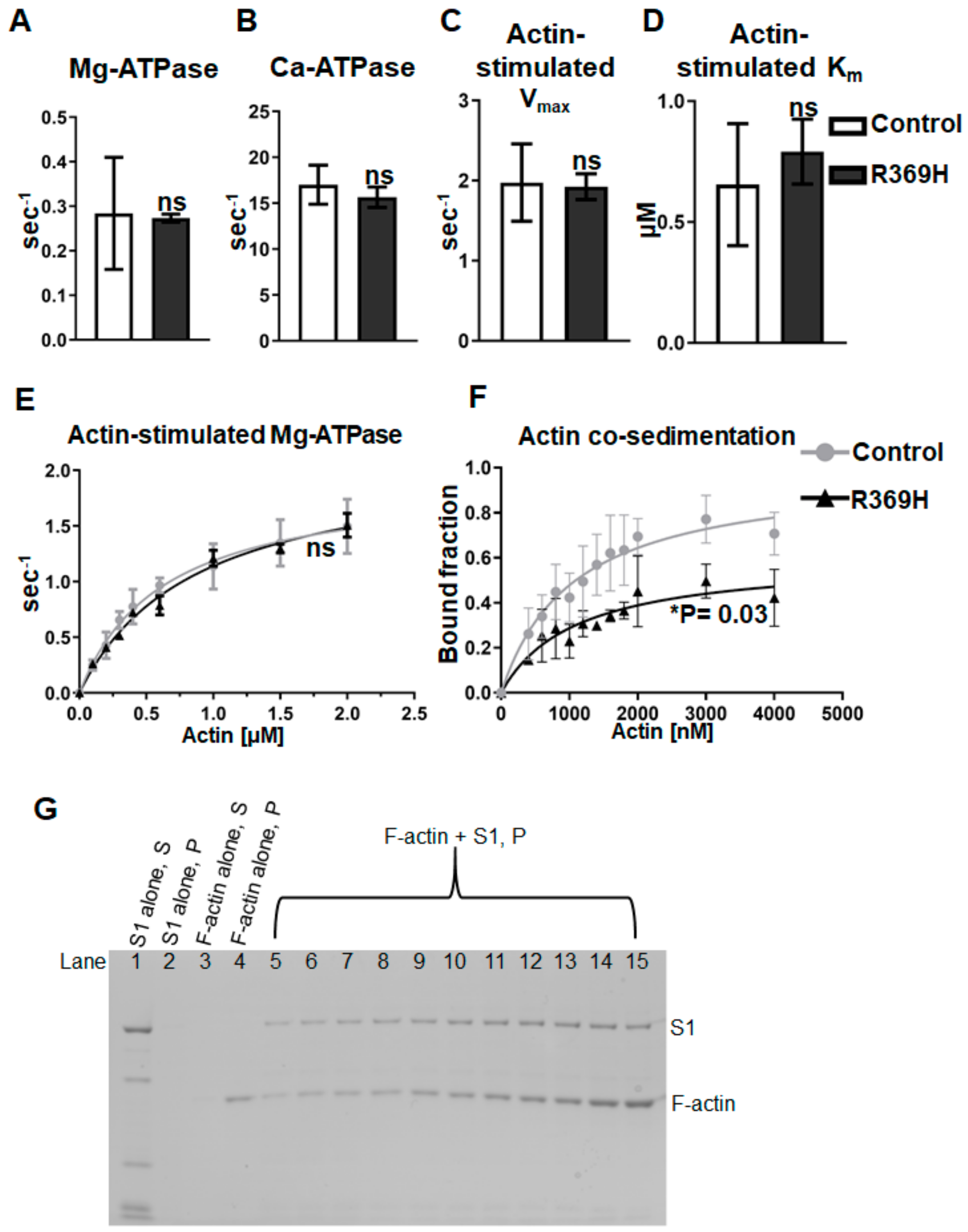
2.1. The R369 Myosin Residue Modulates Actin Binding
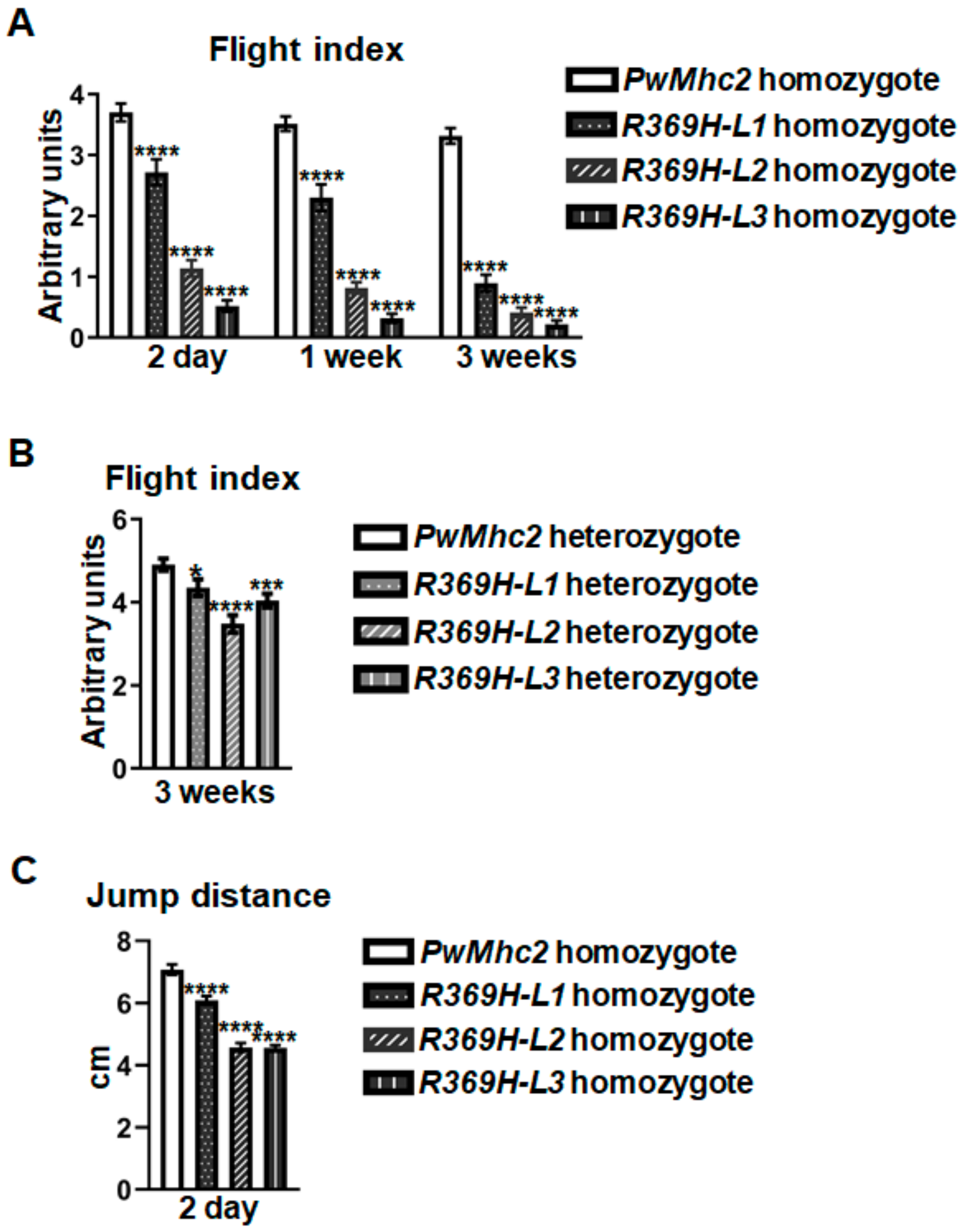
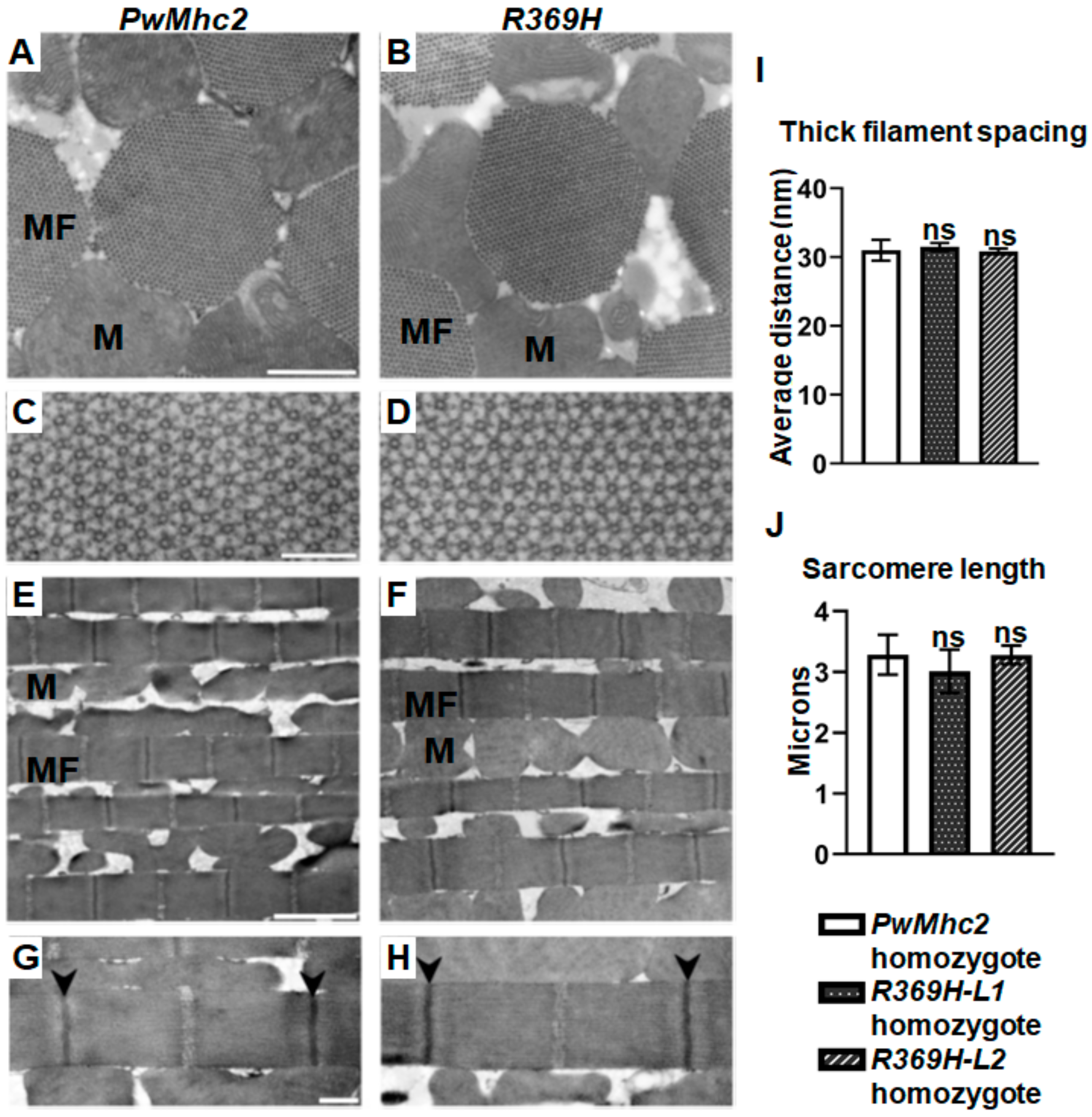
2.2. The R369H Myosin Mutation Reduces Locomotion without Affecting Skeletal Muscle Ultrastructure in Drosophila
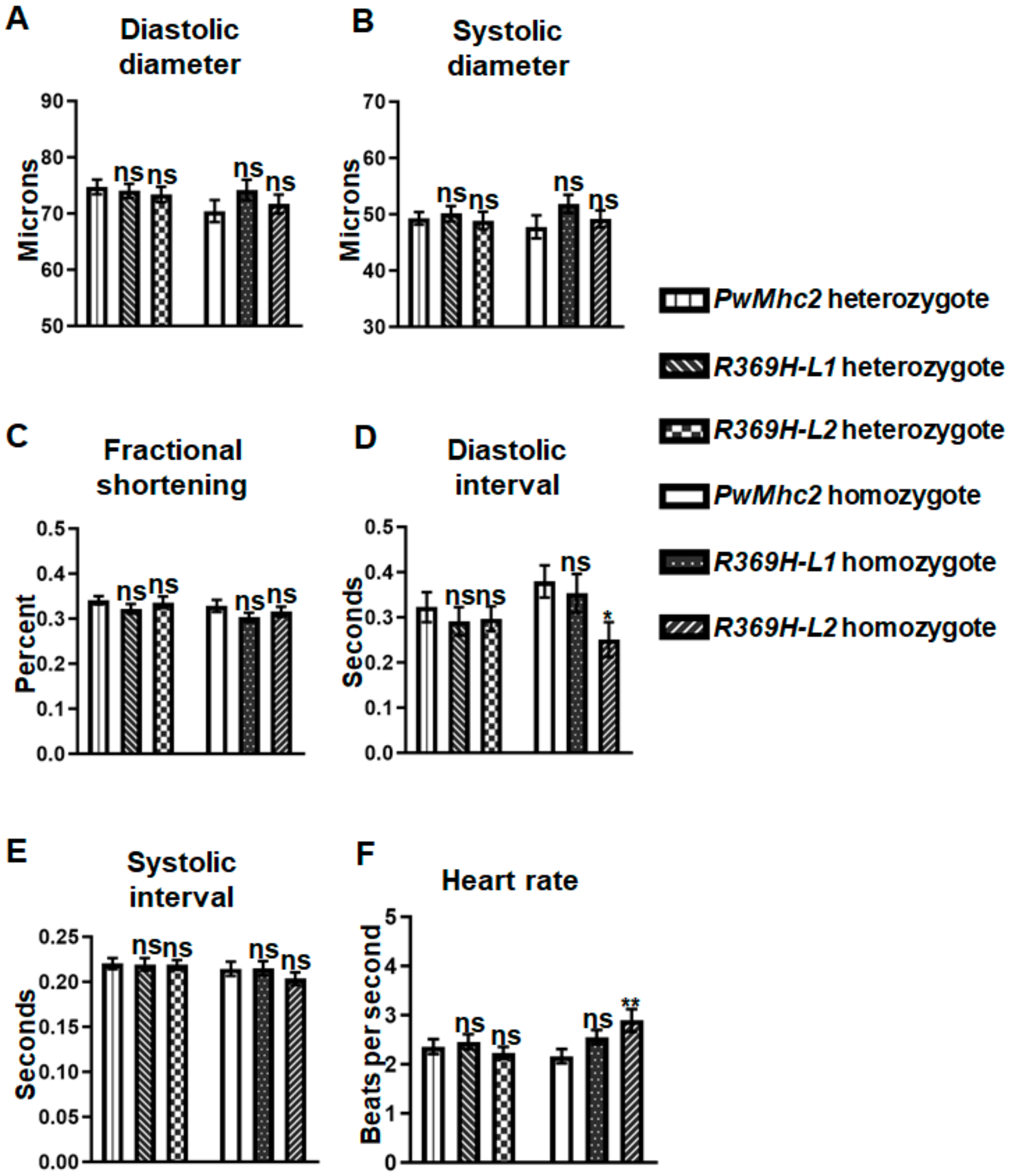
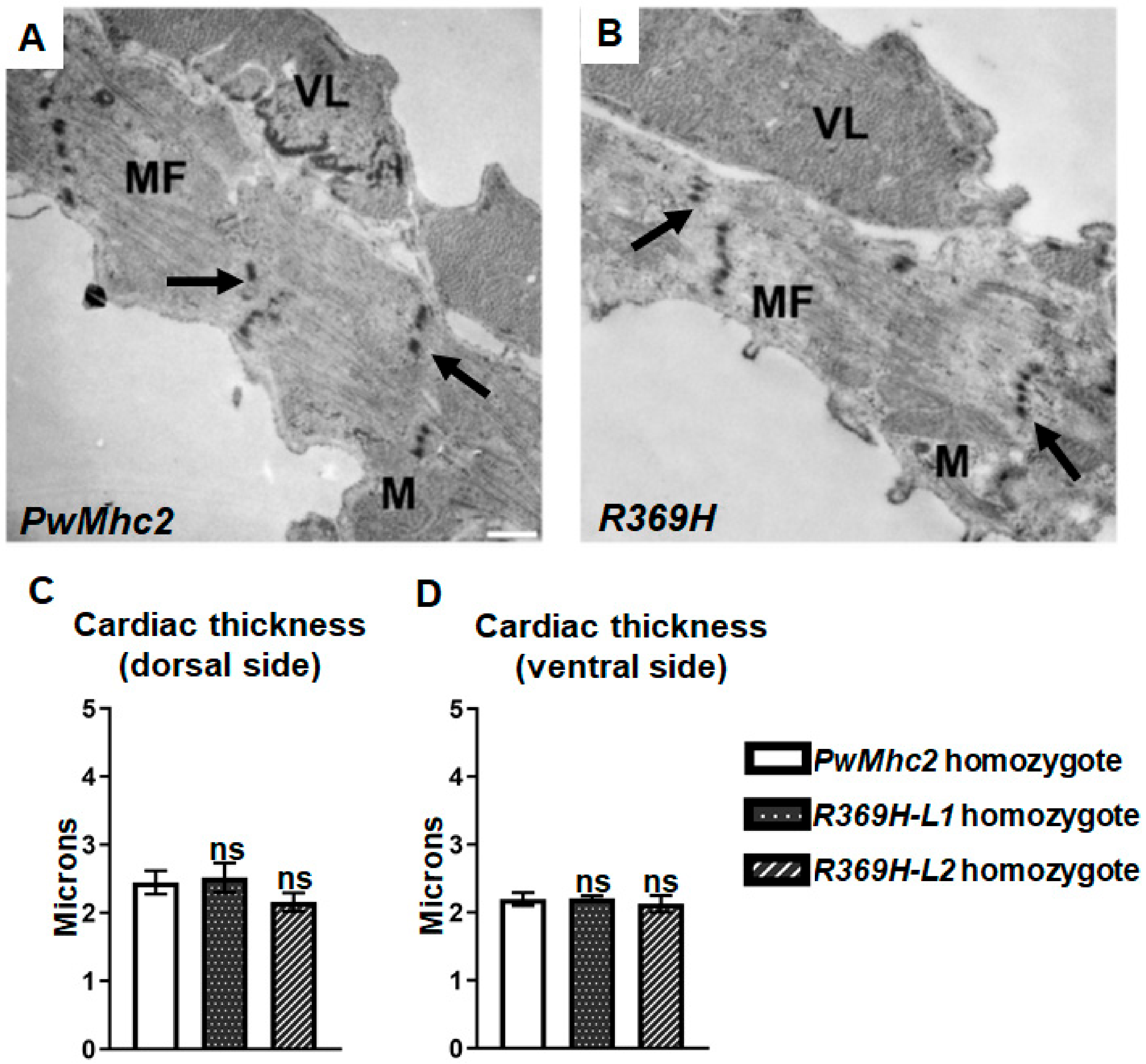
2.3. The R369H Myosin Mutation Does Not Affect Contractility, Dynamics or Ultrastructure of the Heart in Drosophila
3. Discussion
4. Methods
4.1. Generation of Mutant (Non His-Tagged) Fly Lines
4.2. Steady-State ATPase Activity
4.3. Generation of a Mutant His-Tagged Myosin Line and Protein Purification
4.4. Co-Sedimentation Assays
4.5. Transmission Electron Microscopy of IFMs and Hearts
4.6. Flight and Jump Tests
4.7. Cardiac Physiology
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moore, J.R.; Leinwand, L.; Warshaw, D.M. Understanding cardiomyopathy phenotypes based on the functional impact of mutations in the myosin motor. Circ. Res. 2012, 111, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Oldfors, A. Hereditary myosin myopathies. Neuromuscul. Disord. 2007, 17, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Doran, M.H.; Pavadai, E.; Rynkiewicz, M.J.; Walklate, J.; Bullitt, E.; Moore, J.R.; Regnier, M.; Geeves, M.A.; Lehman, W. Cryo-EM and molecular docking shows myosin loop 4 contacts actin and tropomyosin on thin filaments. Biophys. J. 2020, 119, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Dellefave, L.M.; Pytel, P.; Mewborn, S.; Mora, B.; Guris, D.L.; Fedson, S.; Waggoner, D.; Moskowitz, I.; McNally, E.M. Sarcomere mutations in cardiomyopathy with left ventricular hypertrabeculation. Circ. Cardiovasc. Genet. 2009, 2, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Risi, C.; Schäfer, L.U.; Belknap, B.; Pepper, I.; White, H.D.; Schröder, G.F.; Galkin, V.E. High-resolution cryo-EM structure of the cardiac actomyosin complex. Structure 2020, 29, 50–60.e4. [Google Scholar] [CrossRef]
- Kollmar, M.; Dürrwang, U.; Kliche, W.; Manstein, D.J.; Kull, F.J. Crystal structure of the motor domain of a class-I myosin. EMBO J. 2002, 21, 2517–2525. [Google Scholar] [CrossRef]
- Lorenz, M.; Holmes, K.C. The actin-myosin interface. Proc. Natl. Acad. Sci. USA 2010, 107, 12529–12534. [Google Scholar] [CrossRef] [PubMed]
- Gyimesi, M.; Tsaturyan, A.K.; Kellermayer, M.S.; Málnási-Csizmadia, A. kinetic characterization of the function of myosin loop 4 in the actin−myosin interaction. Biochemistry 2007, 47, 283–291. [Google Scholar] [CrossRef]
- Collier, V.L.; Kronert, W.A.; O’Donnell, P.T.; Edwards, K.A.; Bernstein, S.I. Alternative myosin hinge regions are utilized in a tissue-specific fashion that correlates with muscle contraction speed. Genes Dev. 1990, 4, 885–895. [Google Scholar] [CrossRef]
- Caldwell, J.T.; Melkani, G.C.; Huxford, T.; Bernstein, S.I. Transgenic expression and purification of myosin isoforms using the Drosophila melanogaster indirect flight muscle system. Methods 2012, 56, 25–32. [Google Scholar] [CrossRef][Green Version]
- Trujillo, A.S.; Hsu, K.H.; Puthawala, J.; Viswanathan, M.C.; Loya, A.; Irving, T.C.; Cammarato, A.; Swank, D.M.; Bernstein, S.I. Myosin dilated cardiomyopathy mutation S532P disrupts actomyosin interactions, leading to altered muscle kinetics, reduced locomotion, and cardiac dilation in Drosophila. Mol. Biol. Cell 2021, 32, 1690–1706. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.S.; Kronert, W.A.; Guo, Y.; Hsu, K.H.; Sarsoza, F.; Bernstein, S.I. Reductions in ATPase activity, actin sliding velocity, and myofibril stability yield muscle dysfunction in Drosophila models of myosin-based Freeman–Sheldon syndrome. Mol. Biol. Cell 2019, 30, 30–41. [Google Scholar] [CrossRef]
- Viswanathan, M.C.; Tham, R.C.; Kronert, W.A.; Sarsoza, F.; Trujillo, A.S.; Cammarato, A.; Bernstein, S.I. Myosin storage myopathy mutations yield defective myosin filament assembly in vitro and disrupted myofibrillar structure and function in vivo. Hum. Mol. Genet. 2017, 26, 4799–4813. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, P.T.; Bernstein, S. Molecular and ultrastructural defects in a Drosophila myosin heavy chain mutant: Differential effects on muscle function produced by similar thick filament abnormalities. J. Cell Biol. 1988, 107, 2601–2612. [Google Scholar] [CrossRef] [PubMed]
- Achal, M.; Trujillo, A.S.; Melkani, G.C.; Farman, G.P.; Ocorr, K.; Viswanathan, M.C.; Kaushik, G.; Newhard, C.S.; Glasheen, B.M.; Melkani, A.; et al. A restrictive cardiomyopathy mutation in an invariant proline at the myosin head/rod junction enhances head flexibility and function, yielding muscle defects in Drosophila. J. Mol. Biol. 2016, 428, 2446–2461. [Google Scholar] [CrossRef]
- Bhide, S.; Trujillo, A.S.; O’Connor, M.T.; Young, G.H.; Cryderman, D.E.; Chandran, S.; Nikravesh, M.; Wallrath, L.L.; Melkani, G.C. Increasing autophagy and blocking Nrf2 suppress laminopathy-induced age-dependent cardiac dysfunction and shortened lifespan. Aging Cell 2018, 17, e12747. [Google Scholar] [CrossRef]
- Kaushik, G.; Spenlehauer, A.; Sessions, A.O.; Trujillo, A.S.; Fuhrmann, A.; Fu, Z.; Venkatraman, V.; Pohl, D.; Tuler, J.; Wang, M.; et al. Vinculin network–mediated cytoskeletal remodeling regulates contractile function in the aging heart. Sci. Transl. Med. 2015, 7, 292ra99. [Google Scholar] [CrossRef]
- Kronert, W.A.; Bell, K.M.; Viswanathan, M.C.; Melkani, G.C.; Trujillo, A.S.; Huang, A.; Melkani, A.; Cammarato, A.; Swank, D.M.; Bernstein, S.I. Prolonged cross-bridge binding triggers muscle dysfunction in a Drosophila model of myosin-based hypertrophic cardiomyopathy. eLife 2018, 7, e38064. [Google Scholar] [CrossRef]
- Melkani, G.C.; Trujillo, A.S.; Ramos, R.; Bodmer, R.; Bernstein, S.I.; Ocorr, K. Huntington’s Disease induced cardiac amyloidosis is reversed by modulating protein folding and oxidative stress pathways in the Drosophila heart. PLoS Genet. 2013, 9, e1004024. [Google Scholar] [CrossRef]
- Ajtai, K.; Halstead, M.F.; Nyitrai, M.; Penheiter, A.R.; Zheng, Y.; Burghardt, T.P. The myosin C-Loop is an allosteric actin contact sensor in actomyosin. Biochemistry 2009, 48, 5263–5275. [Google Scholar] [CrossRef]
- Holmes, K.C.; Angert, I.; Kull, F.J.; Jahn, W.; Schröder, R.R. Electron cryo-microscopy shows how strong binding of myosin to actin releases nucleotide. Nature 2003, 425, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Holmes, K.C.; Schröder, R.R.; Sweeney, H.L.; Houdusse, A. The structure of the rigor complex and its implications for the power stroke. Philos. Trans. R. Soc. B Biol. Sci. 2004, 359, 1819–1828. [Google Scholar] [CrossRef] [PubMed]
- Houdusse, A.; Sweeney, H.L. How myosin generates force on actin filaments. Trends Biochem. Sci. 2016, 41, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Namba, K. Structure of actomyosin rigour complex at 5.2 Å resolution and insights into the ATPase cycle mechanism. Nat. Commun. 2017, 8, 13969. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, O. Direct sarcomere modulators are promising new treatments for cardiomyopathies. Int. J. Mol. Sci. 2019, 21, 226. [Google Scholar] [CrossRef] [PubMed]
- Ajtai, K.; Garamszegi, S.P.; Watanabe, S.; Ikebe, M.; Burghardt, T.P. The myosin cardiac loop participates functionally in the actomyosin interaction. J. Biol. Chem. 2004, 279, 23415–23421. [Google Scholar] [CrossRef]
- Pospich, S.; Sweeney, H.L.; Houdusse, A.; Raunser, S. High-resolution structures of the actomyosin-V complex in three nucleotide states provide insights into the force generation mechanism. eLife 2021, 10, e73724. [Google Scholar] [CrossRef]
- Rayment, I.; Rypniewski, W.R.; Schmidt-Base, K.; Smith, R.; Tomchick, D.R.; Benning, M.M.; Winkelmann, D.A.; Wesenberg, G.; Holden, H.M. Three-dimensional structure of myosin subfragment-1: A molecular motor. Science 1993, 261, 50–58. [Google Scholar] [CrossRef]
- Coureux, P.-D.; Sweeney, H.L.; Houdusse, A. Three myosin V structures delineate essential features of chemo-mechanical transduction. EMBO J. 2004, 23, 4527–4537. [Google Scholar] [CrossRef]
- Behrmann, E.; Müller, M.; Penczek, P.A.; Mannherz, H.G.; Manstein, D.J.; Raunser, S. Structure of the rigor actin-tropomyosin-myosin complex. Cell 2012, 150, 327–338. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- George, E.L.; Ober, M.B.; Emerson, C.P., Jr. Functional domains of the Drosophila melanogaster muscle myosin heavy-chain gene are encoded by alternatively spliced exons. Mol. Cell. Biol. 1989, 9, 2957–2974. [Google Scholar] [CrossRef] [PubMed]
- Cammarato, A.; Dambacher, C.M.; Knowles, A.F.; Kronert, W.A.; Bodmer, R.; Ocorr, K.; Bernstein, S.I. Myosin transducer mutations differentially affect motor function, myofibril structure, and the performance of skeletal and cardiac muscles. Mol. Biol. Cell 2008, 19, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, M.C.; Schmidt, W.; Rynkiewicz, M.J.; Agarwal, K.; Gao, J.; Katz, J.; Lehman, W.; Cammarato, A. distortion of the actin A-Triad results in contractile disinhibition and cardiomyopathy. Cell Rep. 2017, 20, 2612–2625. [Google Scholar] [CrossRef]
- Viswanathan, M.C.; Kaushik, G.; Engler, A.J.; Lehman, W.; Cammarato, A. A Drosophila melanogaster model of diastolic dysfunction and cardiomyopathy based on impaired troponin-T function. Circ. Res. 2014, 114, e6-17. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, M.C.; Schmidt, W.; Franz, P.; Rynkiewicz, M.J.; Newhard, C.S.; Madan, A.; Lehman, W.; Swank, D.M.; Preller, M.; Cammarato, A. A role for actin flexibility in thin filament-mediated contractile regulation and myopathy. Nat. Commun. 2020, 11, 2417. [Google Scholar] [CrossRef]
- Bischof, J.; Maeda, R.K.; Hediger, M.; Karch, F.; Basler, K. An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc. Natl. Acad. Sci. USA 2007, 104, 3312–3317. [Google Scholar] [CrossRef]
- Swank, D.M.; Bartoo, M.L.; Knowles, A.F.; Iliffe, C.; Bernstein, S.I.; Molloy, J.E.; Sparrow, J.C. Alternative exon-encoded regions of Drosophila myosin heavy chain modulate ATPase rates and actin sliding velocity. J. Biol. Chem. 2001, 276, 15117–15124. [Google Scholar] [CrossRef]
- Suggs, J.A.; Cammarato, A.; Kronert, W.A.; Nikkhoy, M.; Dambacher, C.M.; Megighian, A.; Bernstein, S.I. Alternative S2 hinge regions of the myosin rod differentially affect muscle function, myofibril dimensions and myosin tail length. J. Mol. Biol. 2007, 367, 1312–1329. [Google Scholar] [CrossRef]
- Bell, K.M.; Kronert, W.A.; Huang, A.; Bernstein, S.I.; Swank, D.M. The R249Q hypertrophic cardiomyopathy myosin mutation decreases contractility in Drosophila by impeding force production. J. Physiol. 2019, 597, 2403–2420. [Google Scholar] [CrossRef]
- Drummond, D.; Hennessey, E.S.; Sparrow, J.C. Characterisation of missense mutations in the Act88F gene of Drosophila melanogaster. Mol. Gen. Genet. 1991, 226, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Fink, M.; Callol-Massot, C.; Chu, A.; Ruiz-Lozano, P.; Belmonte, J.C.I.; Giles, W.; Bodmer, R.; Ocorr, K. A new method for detection and quantification of heartbeat parameters in Drosophila, zebrafish, and embryonic mouse hearts. BioTechniques 2009, 46, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Rossi, A.C.; Smerdu, V.; Leinwand, L.A.; Reggiani, C. Developmental myosins: Expression patterns and functional significance. Skelet. Muscle 2015, 5, 22. [Google Scholar]
- The Genome Aggregation Database. Available online: https://gnomad.broadinstitute.org/ (accessed on 28 January 2022).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Effect |
|---|---|
| Myosin ATPase activity | Unaffected |
| Actin binding | Reduced |
| Indirect flight muscle function | Reduced |
| Jump muscle function | Reduced |
| Cardiac function | Unaffected |
| Cardiac and skeletal muscle structure | Unaffected |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trujillo, A.S.; Hsu, K.H.; Viswanathan, M.C.; Cammarato, A.; Bernstein, S.I. The R369 Myosin Residue within Loop 4 Is Critical for Actin Binding and Muscle Function in Drosophila. Int. J. Mol. Sci. 2022, 23, 2533. https://doi.org/10.3390/ijms23052533
Trujillo AS, Hsu KH, Viswanathan MC, Cammarato A, Bernstein SI. The R369 Myosin Residue within Loop 4 Is Critical for Actin Binding and Muscle Function in Drosophila. International Journal of Molecular Sciences. 2022; 23(5):2533. https://doi.org/10.3390/ijms23052533
Chicago/Turabian StyleTrujillo, Adriana S., Karen H. Hsu, Meera C. Viswanathan, Anthony Cammarato, and Sanford I. Bernstein. 2022. "The R369 Myosin Residue within Loop 4 Is Critical for Actin Binding and Muscle Function in Drosophila" International Journal of Molecular Sciences 23, no. 5: 2533. https://doi.org/10.3390/ijms23052533
APA StyleTrujillo, A. S., Hsu, K. H., Viswanathan, M. C., Cammarato, A., & Bernstein, S. I. (2022). The R369 Myosin Residue within Loop 4 Is Critical for Actin Binding and Muscle Function in Drosophila. International Journal of Molecular Sciences, 23(5), 2533. https://doi.org/10.3390/ijms23052533