
Epigenetic Studies in the Male APP/BIN1/COPS5 Triple-Transgenic Mouse Model of Alzheimer’s Disease





Abstract
1. Introduction
2. Results
2.1. APP/BIN1/COPS5 3xTg-AD Mice Strongly Express AD-Related Pathological Hallmarks
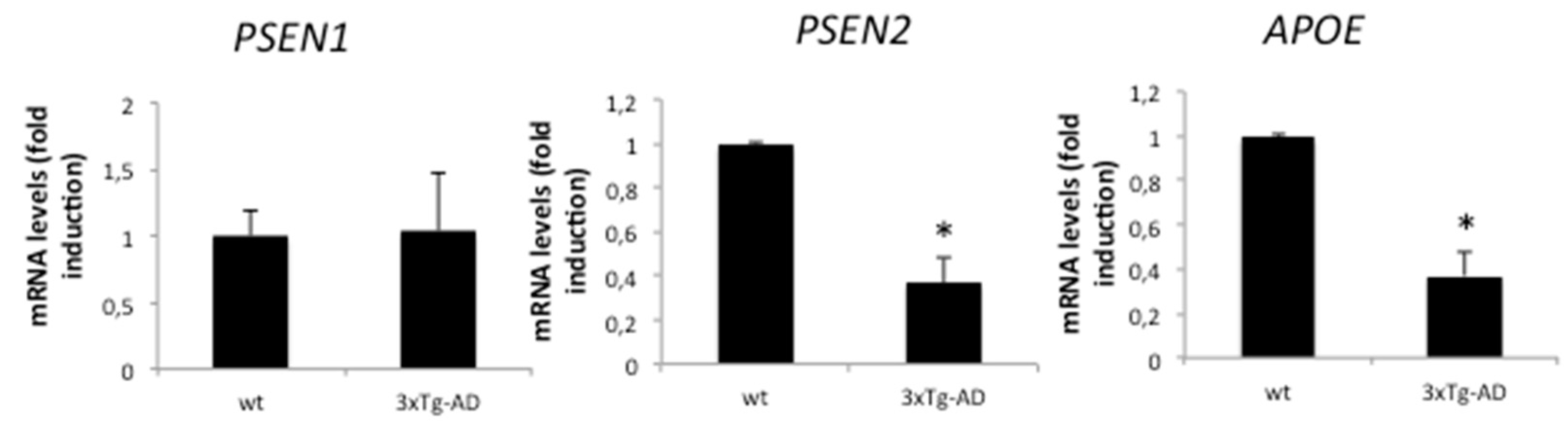
2.2. AD-Related Gene Expression Is Regulated in APP/BIN1/COPS5 3xTg-AD Mice
2.3. Sirtuin Activity and Expression Are Regulated in APP/BIN1/COPS5 3xTg-AD Mice
2.4. HDAC Activity and Expression Are Regulated in APP/BIN1/COPS5 3xTg-AD Mice
3. Discussion
4. Materials and Methods
4.1. Animal Model
4.2. Tissue Collection and Preparation
4.3. Immunofluorescence
4.4. Quantitative Real Time RT-PCR (qPCR)
4.4.1. RNA Extraction
4.4.2. Nuclear Protein Extraction
4.4.3. Quantification of Sirtuin and HDAC Activity
4.5. Statistical Analysis
5. Conclusions
6. Limitations of the Study
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Cacabelos, R.; Teijido, O.; Carril, J.C. Can cloud-based tools accelerate Alzheimer’s drug discovery? Expert Opin. Drug Discov. 2016, 11, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 391–460. [Google Scholar] [CrossRef] [PubMed]
- Brodaty, H.; Seeher, K.; Gibson, L. Dementia time to death: A systematic literature review on survival time and years of life lost in people with dementia. Int. J. Psychogeriatr. 2012, 24, 1034–1045. [Google Scholar] [CrossRef]
- Knopman, D.S.; Amieva, H.; Petesen, R.C.; Chetelat, G.; Holtzman, D.; Hyman, B.; Nixon, R.; Jones, D. Alzheimer Disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2021 Alzheimer’s Diseases Facts and Figures. Alzheimer’s Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Have there been improvements in Alzheimer’s disease drug discovery over the past 5 years? Expert Opin. Drug Discov. 2018, 13, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Fernandez-Novoa, L.; Lombardi, V.; Kubota, Y.; Takeda, M. Molecular genetics of Alzheimer’s disease and aging. Methods Find. Exp. Clin. Pharmacol. 2005, 27 (Suppl. A), 1–573. [Google Scholar]
- Cacabelos, R.; Torrelas, C.; Teijido, O.; Carril, J.C. Pharmacogenetic considerations in the treatment of Alzheimer’s disease. Pharmacogenomics 2016, 17, 1041–1074. [Google Scholar] [CrossRef]
- Cacabelos, R.; Meyyazhagan, A.; Carril, J.C.; Cacabelos, P.; Teijido, O. Pharmacogenetics of vascular risk factors in Alzheimer’s disease. J. Pers. Med. 2018, 8, 3. [Google Scholar] [CrossRef]
- de la Torre, J.C. Cerebral perfusion enhancing intervention: A new strategy for the prevention of Alzheimer’s disease. Brain Pathol. 2016, 26, 618–631. [Google Scholar] [CrossRef]
- Sterniczuk, R.; Antle, M.C.; Laferla, F.M.; Dyck, R.H. Characterization of the 3xTg-AD mouse model of Alzheimer’s Disease: Part Behavioral and cognitive changes. Brain Res. 2010, 1348, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.A.; Jung, A.R.; Lakshmana, M.K.; Bedrossian, A.; Lim, Y.; Bu, J.H.; Park, S.A.; Koo, E.H.; Mook-Jung, I.; Kang, D.E. Pivotal role of the RanBP9-cofilin pathway in Ab-induced apoptosis and neurodegeneratiom. Cell Death Differ. 2012, 19, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Palavicini, J.P.; Wang, H.; Minond, D.; Bianchi, E.; Xu, S.; Lakshmana, M.K. RanBP9 overexpression down-regulates phospho-cofilin, causes early synaptic deficits and impaired learning, and accelerates accumulation of amyloid plaques in the mouse brain. J. Alzheimer’s Dis. 2014, 39, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Filali, M.; Lalonde, R.; Theriault, P.; Julien, C.; Calon, F.; Planel, E. Cognitive and non-cognitive behaviours in the triple transgeinic mouse model of Alzheimer’s Disease expressing mutated APP, PS1 and MAPT (3xTg-AD). Behav. Brain Res. 2012, 234, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Carrera, I.; Novoa, L.; Teijido, O.; Sampedro, C.; Seoane, S.; Lakshamana, M.; Cacabelos, R. Comparative characterization of transgenic mouse models of Alzheimer’s disease. J. Genom. Med. Pharmacogenom. 2017, 2, 331–337. [Google Scholar]
- Urdinguio, R.G.; Sanxhez-Mut, J.V.; Esteller, M. Epigenetic mechanisms in neurological diseases: Genes, syndromes, and therapies. Lancet Neurol. 2009, 8, 1056–1072. [Google Scholar] [CrossRef]
- Sweatt, J.D. The emerging field of neuroepigenetics. Neuron 2013, 80, 624–632. [Google Scholar] [CrossRef]
- Cacabelos, R.; Tellado, I.; Cacabelos, P. The epigenetic machinery in the lyfe cycle and pharmacoepigenetics. In Pharmacoepigenetics; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Delgado-Morales, R.; Esteller, M. Opening up the DNA methylome of dementia. Mol. Psychiatry 2017, 22, 485–496. [Google Scholar] [CrossRef]
- Maloney, B.; Lahiri, D. Epigenetics of dementia: Understanding the disease as a transformation rather than a state. Lancet Neurol. 2016, 15, 760–774. [Google Scholar] [CrossRef]
- Martinez-Iglesias, O.; Carrera, I.; Carril, J.C.; Fernadez-Novoa, L.; Cacabelos, N.; Cacabelos, R. DNA methylation in Neurodegenerative and Cerebrovascular Disorders. Int. J. Mol. Sci. 2020, 21, 2220. [Google Scholar] [CrossRef]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Adwan, L.; Zawia, N.H. Epigenetics: A novel therapeutic approach for the treatment of Alzheimer’s disease. Pharmacol. Ther. 2013, 139, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Teijido, O.; Cacabelos, R. Pharmacoepigenomic interventions as novel potential treatment for Alzheimer’s Disease and Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 3199. [Google Scholar] [CrossRef] [PubMed]
- Tecalco-Cruz, A.C.; Ramirez-Jarquin, J.O.; Alvarez-Sanchez, M.E.; Zepeda-Cervantes, J. Epigenetic basis of Alzheimer disease. World J. Biol. Chem. 2020, 11, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Cadena-del-Castillo, C.; Valdes-Quezada, C.; Carmona-Aldana, F.; Arias, C.; Bermúdez-Rattoni, F.; Recillas-Targa, F. Age-dependent increment of hydroxymethylation in the brain cortex in the triple-trangenic mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 41, 845–854. [Google Scholar] [CrossRef]
- Chouliaras, L.; Mastroeni, D.; Delvaux, E.; Grover, A.; Kenis, G.; Hof, P.R.; Steinbusch, H.W.M.; Coleman, P.D.; Rutten, B.P.F.; van den hove, D. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 2091–2099. [Google Scholar] [CrossRef]
- Conliffe, D.; Wong, A.; Troakes, C.; Proitsi, P.; Patel, Y.; Chouliaras, L.; Fernandes, C.; Cooper, J.; Lovestone, S.; Schalkwyk, L. Cross-region reduction in 5-hydroxymethylcytosine in Alzheimer’s disease brain. Neurobiol. Aging 2014, 31, 1850–1854. [Google Scholar] [CrossRef]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic changes in Alzheimer’s disease: Decrements in DNA methylation. Neurobiol. Aging 2010, 31, 2025–2037. [Google Scholar] [CrossRef]
- Mastroeni, D.; McKee, A.; Grover, A.; Rogers, J.; Coleman, P.D. Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer’s disease. PLoS ONE 2009, 4, e6617. [Google Scholar] [CrossRef]
- Janczura, K.J.; Volmar, C.-H.; Sartor, G.C.; Rao, S.J.; Ricciardi, N.R.; Lambet, G.; Brothers, S.; Wahlestedt, C. Inhibition of HDAC3 reverses Alzheimer’s disease-realted pathologies in vitro and in the 3xTg-AD mouse model. Proc. Natl. Acad. Sci. USA 2018, 115, E11148–E11157. [Google Scholar] [CrossRef]
- McQuown, S.C.; Barrett, R.M.; Matheos, D.P.; Post, R.J.; Rogge, G.A.; Alenghat, T.; Mullican, S.E.; Jones, S.; Rusche, J.R.; Lazar, M.A. HDAC3 is a critical negative regulator of long-term memory formation. J. Neurosci. 2011, 31, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wang, S.; Yu, L.; Jin, J.; Ye, X.; Liu, Y.; Xu, Y. HDAC3 negatively regulates spatial memory in a mouse model of Alzheimer’s disease. Aging Cell 2017, 16, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.J.; Guo, J.L.; Huratdo, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M.Y. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011, 2, 252. [Google Scholar] [CrossRef] [PubMed]
- Julien, C.; Tremblay, C.; Emond, V.; Lebbadi, M.; Salem, N., Jr.; Bennett, D.A.; Calon, F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimerñs disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef]
- Di Battista, A.M.; Heinsinger, N.M.; Rebeck, G.W. Alzheimer’s Disease Genetic Risk Factor APOE-ε4 Also Affects Normal Brain Function. Curr. Alzheimer Res. 2016, 13, 1200–1207. [Google Scholar] [CrossRef]
- Lambert, J.-C.; Mann, D.; Richard, F.; Tian, J.; Shi, J.; Thaker, U.; Merrot, S.; Harris, J.; Frigard, B.; Iwatsubo, T. Is there a relation between APOE expression and brain amyloid load in Alzheimer’s disease? J. Neurol. Neurosurg. Psychiatry 2005, 76, 928–933. [Google Scholar] [CrossRef][Green Version]
- de la Monte, S.M.; Jhaveri, A.; Maron, B.A.; Wands, J.R. Nitric oxide synthase 3-mediated neurodegeneration after intracerebral gene delivery. J. Neuropath. Exp. Neurol. 2007, 66, 272–283. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Chiche, J.-D.; von dem Bussche, A.; Sanyal, S.; Lahousse, S.A.; Janssens, S.P.; Bloch, K.D. Nitric oxide synthase-3 overexpression causes apoptosis and impairs neuronal mitochondrial function: Relevance to alxheimer’s type degeneration. Lab. Investig. 2003, 83, 287–298. [Google Scholar] [CrossRef]
- Choi, J.Y.; Yeo, I.J.; Kim, K.C.; Choi, W.R.; Jung, J.-K.; Han, S.-B.; Hong, J.T. COX-2 expression in brains of patients with familiar Alzheimer’s disease. Int. Congress Ser. 2003, 1252, 363–372. [Google Scholar]
- Hoozemans, J.J.M.; O’Banion, M.K. The role of COX-1 and COX-2 in Alzheimer’s Disease Patholgy and the therapeutic potentials of non-steroidal anti-inflammatory drugs. Curr. Drugs Targets-CNS Neurol. Dis. 2005, 4, 307–315. [Google Scholar] [CrossRef]
- Zhao, L.; Cao, K.; Hu, K.; He, X.; Yun, D.; Tong, T.; Han, L. Sirtuins and their biological relevance in aging and age-related disorders. Aging Dis. 2020, 11, 927–945. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Tekwani, B.L. Histone deacetylases inhibitors in Neurodegenerative Diseases, Neuroprotection and Neuronals Differentiation. Front. Pharmacol. 2020, 11, 537. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Iglesias, O.; Cacabelos, R. Epigenetic treatment of neurodegenerative disorders. In Histone Modifications in Therapy; Castelo-Branco, P., Jeronimo, C., Eds.; Elsevier: London, UK, 2020; pp. 311–335. [Google Scholar]
- De Rossi, P.; Andrew, R.J.; Musial, T.F.; Buggia-Prevot, V.; Xu, G.; Ponnusamy, M.; Ly, H.; Krause, S.V.; Rice, R.C.; De L’Estoile, V.; et al. Aberrant accrual of BIN1 near Alzheimer’s disease amyloid deposits in transgenic models. Brain Pathol. 2019, 29, 485–501. [Google Scholar] [CrossRef]
- Wang, R.; Wang, H.; Carrera, I.; Xu, S.; Lakshmana, M.K. COPS5 protein overexpression increases amyloid plaque burden, decreases spinophilin-immuroreactive puncta, and exarcebates learning and memory deficits in the mouse brain. J. Biol. Chem. 2015, 290, 9299–9309. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.-S.; Shen, L.-L.; Zhu, C.; Bu, X.-L.; Liu, Y.-H.; Liu, C.-H.; Yao, X.-Q.; Zhang, L.-L.; Zhou, H.-D.; Walker, D.G.; et al. Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl. Psych. 2016, 6, e907. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekimschtein, P. Brain-Derived neurotrophic factor: A key molecule for memory in the healthy and pathological brain. Front. Cell Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef]
- Mori, Y.; Tsuji, M.; Oguchi, T.; Kasuga, K.; Kimura, A.; Futamura, A.; Sugimoto, A.; Kasai, H.; Kuroda, T.; Yano, S.; et al. Serum BDNF as a potential biomarker of Alzheimer’s Disease: Verification through assessment of serum, cerebrospinal fluid, and medial temporal lobe atrophy. Front. Neurol. 2021, 12, 653267. [Google Scholar] [CrossRef]
- Walker, M.P.; LaFerla, F.M.; Oddo, S.S.; Brewer, G.J. Reversible epigenetic histone modifications and Bdnf expression in neurons with aging and from a mouse model of Alzheimer’s disease. Age 2012, 35, 519–531. [Google Scholar] [CrossRef]
- Martinez-Iglesias, O.; Naidoo, V.; Cacabelos, N.; Cacabelos, R. Epigenetic Biomarkers as diagnostic tools for Neurodegenerative Disorders. Int. J. Mol. Sci. 2022, 23, 13. [Google Scholar] [CrossRef]
- Delabio, R.; Rasmussen, L.; Mizumoto, I.; Viani, G.-A.; Chen, E.; Villares, J.; Costa, I.-B.; Turecki, G.; Aparecido Linde, S.; Cardoso Smith, M.; et al. PSEN1 and PSEN2 gene expression in Alzheimer’s disease brain a new approach. J. Alzheimer’s Dis. 2014, 42, 757–760. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Painter, M.M.; Bu, G.; Kanekiyo, T. Apoliporotein E as a therapeutic target in Alzheimer’s disease: A review of basic research and clinical evicende. CNS Drugs 2017, 30, 773–789. [Google Scholar] [CrossRef]
- Cramer, P.E.; Cirrito, J.R.; Wesson, D.W.; Lee, C.Y.D.; Karlo, J.C.; Zinn, A.E.; Casali, B.T.; Restivo, J.J.; Goebel, W.D.; James, M.J.; et al. ApoE-directed therapeutics rapidly clear betaámyloid and reverse deficits in AD mouse models. Science 2012, 335, 1503–1506. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Zhong, K.; Kinney, J.W.; Heaney, C.; Moll-Tudla, J.; Joshi, A.; Pontecorvo, M.; Vevous, M.; Tang, A.; Bena, J. Double-blind, placebo-cpmtrolled, proof-of-concept trial of bexarotene Xin moderate Alzheimer’s disease. Alzheimer’s Res. Ther. 2016, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Carrera, I.; Martinez-Iglesias, O.; Cacabelos, N.; Naidoo, V. What is the gold standard model for Alzheimer’s disease drug discovery and development? Exp. Opin. Drug Discov. 2021, 16, 1415–1440. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Sinclair, D.A.; Ellis, J.; Steegborn, C. Sirtuin activators and inhibitors:promises, achievements and challenges. Pharmacol. Ther. 2019, 188, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-Y.; Hsu, J.-L.; Wang, H.-C.; Wu, S.-J.; Hong, C.-J.; Cheng, I.H.-J. Alterations of the neuroinflammatory markers IL-6 and TRAIL in Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. Extra 2015, 5, 424–434. [Google Scholar] [CrossRef]
- Park, J.C.; Han, S.H.; Mook-Jung, I. Peripheral inflammatory biomarkers in Alzheimer’s disease: A brief review. BMB Rep. 2020, 53, 10–19. [Google Scholar] [CrossRef]
- Quintanilla, R.-A.; Orellana, D.I.; Gonzalez-Billault, C.; Maccioni, R.B. Interleukin-6 induces Alzheimer-type phosporylation of tau protein by deregulating the cdk5/p35 pathway. Exp. Cell Res. 2004, 295, 245–257. [Google Scholar] [CrossRef]
- Zhao, M.; Cribbd, D.H.; Anderson, A.J.; Cummings, B.J.; Su, J.H.; Wasserman, A.J.; Cotman, C.W. The induction of the TNFalpha death domain signaling patwhway in Alzheimer’s disease brain. Neurochem. Res. 2003, 28, 307–318. [Google Scholar] [CrossRef]
- Janeksins, M.C.; Mastrangelo, M.A.; Park, K.M.; Sudol, K.L.; Narrow, W.C.; Oddo, S.; LaFerla, F.; Callahan, L.M.; Federoff, H.J.; Bowers, W.J. Chronic neuron-specific tumor necrosis factor alpha expression enhances the local inflammatory environment ultimately leading to neuronal death in 3xTg-AD mice. Am. J. Pathol. 2008, 173, 1768–1782. [Google Scholar] [CrossRef]
- Aisen, P.S. Evaluation of selective COX-2 inhibitors for the treatment of Alzheimer’s disease. J. Pain Symptom Manag. 2002, 23, S35–S40. [Google Scholar] [CrossRef]
- Woodling, N.S.; Colas, D.; Wang, Q.; Minhas, P.; Panchal, M.; Liang, X.; Mhatre, S.D.; Brown, H.; Ko, N.; Zagol-Ikapitte, I.; et al. Cyclooxygenase inhibition targets neurons to prevent early behavioural decline in Alzheimer’s disease model mice. Brain 2016, 139, 2063–2081. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Rathod, S.S.S.; Ghoneim, M.M.; Alshehri, S.; Ahmad, J.; Mishra, A.; Alhakamay, N.A. DNA methylation: A promising approach in manegement of Alzheimer’s Disease and other Neurodegenerative Disorder. Biology 2022, 11, 90. [Google Scholar] [CrossRef] [PubMed]
- Esposito, M.; Sherr, G.L. Epigenetic modifications in Alzheimer’s neuropathology and therapeutics. Front. Neurosc. 2019, 13, 476. [Google Scholar] [CrossRef]
- Francesco, A.D.; Arosio, B.; Falconi, A.; Bonaventura, M.M.D.; Karimi, M.; Mari, D.; Casati, M.; Maccarrone, M.; D’Addario, C. Global changes in DNA methylation in Alzheimer’s disease peripheral blood mononuclear cells. Brain Behav. Immun. Mar. 2015, 45, 139–144. [Google Scholar] [CrossRef]
- Martinez-Iglesias, O.; Naidoo, V.; Carril, J.C.; Carrera, I.; Corzo, L.; Rodriguez, S.; Alejo, R.; Cacabelos, N.; Cacabelos, R. AtreMorine treatment regulates DNA methylation in Neurodegenerative Disorders: Epigenetic and pharmacogenetic studies. Curr. Pharm. Pers. Med. 2020, 17, 159–171. [Google Scholar] [CrossRef]
- Lu, X.; Wang, L.; Yu, C.; Yu, D.; Yu, G. Histone acetylation modifiers in the pathogenesis of Alzheimer’s Disease. Front Cell Neurosci. 2015, 9, 226. [Google Scholar] [CrossRef]
- Wood, I.C. The contribution and therapeutic potential of epigenetic modifications in Alzheimer’s Disease. Front Neurosci. 2018, 12, 649. [Google Scholar] [CrossRef]
- Rizzi, L.; Roriz-Cruz, M. Sirtuin 1 and Alzheimer’s disease: An up-to-date review. Neuropeptides 2018, 71, 54–60. [Google Scholar] [CrossRef]
- Donmez, G. The effects of SIRT1 on Alzheimer’s Disease models. Int. J. Alzheimer’s Dis. 2012, 2012, 509529. [Google Scholar]
- Min, S.W.; Sohn, P.D.; Li, Y.; Devidze, N.; Johnson, J.R.; Krogan, N.J.; Masliah, E.; Mok, S.A.; Gestwicki, J.E.; Gan, L. SIRT1 Deacetylates Tau and Reduces Pathogenic Tau Spread in a Mouse Model of Tauopathy. J. Neurosci. 2018, 38, 3680–3688. [Google Scholar] [CrossRef]
- Carrera, I.; Martinez, O. Cacabelos Neuroprotection with natural antioxidants and nutraceuticals in the context of brain cell degeneration: The epigenetic connection. Curr. Top. Med. Chem. 2019, 19, 2999–3011. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Baur, J.A.; Chen, A.; Miller, C.; Adams, J.K.; Kisielewski, A.; Howitz, K.T.; Zipkin, R.E.; Sinclair, D.A. Design and synthesis of compounds that extend yeast replicative lifespan. Aging Cell 2007, 6, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Pukhalskaia, A.E.; Dyatlova, A.S.; Linkova, N.S.; Kozlov, K.L.; Kvetnaia, T.V.; Koroleva, M.V.; Kvetnoy, I.M. Sirtuins as possible predictors of aging and Alzheimer’s Disease development: Verification in the hippocampus and saliva. Bull. Exp. Biol. Med. 2020, 169, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Chaterjee, P.; Sharma, P.K.; Singh, A.K.; Gupta, A.; Gill, K.; Tripathi, M.; Dey, A.B.; Dey, S. Sirtuin1: A primising serum protein marker for early detection of Alzheimers disease. PLoS ONE 2013, 8, e61560. [Google Scholar] [CrossRef]
- Pradhan, R.; Singh, A.K.; Kumar, P.; Bajpai, S.; Pathak, M.; Chatterjee, P.; Dwivedi, S.; Dey, A.B.; Dey, S. Blood circulatory level of seven sirtuins in Alzheimer’s Disease: Potent biomarker based on translational reseach. Mol. Neurobiol. 2022. [Google Scholar] [CrossRef]
- Cacabelos, R.; Carril, J.C.; Cacabelos, N.; Zazantsev, A.; Vostrov, A.V.; Corzo, L.; Cacabelos, P.; Goldgaber, D. Sirtuins in Alzheimer’s Disease-SIRT2-related GenoPhenotypes and implication in Pharmacoepigenetics. Int. J. Mol. Sci. 2019, 20, 1249. [Google Scholar] [CrossRef]
- Xu, K.; Dai, X.L.; Huang, H.C.; Jiang, Z.F. Targeting HDACs: A promising therapy for Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2011, 2011, 143269. [Google Scholar] [CrossRef]
- Fransquet, P.D.; Ryan, J. The current status of blood epigenetic biomarkers for dementia. Crit. Rev. Clin. Lab. Sci. 2019, 56, 435–457. [Google Scholar] [CrossRef]
- Kawarabayasji, T.; Terakawa, T.; Takahashi, A.; Hasegawa, H.; Narita, S.; Sato, K.; Nakamura, T.; Seino, Y.; Hirohata, M.; Baba, N.; et al. Oral Immunization with Soybean Storage Protein Containing Amyloid-β 4–10 Prevents Spatial Learning Decline. J Alzheimer’s Dis. 2019, 70, 487–503. [Google Scholar] [CrossRef]
- Kratochvill, F.; Gratz, N.; Qualls, J.E.; Van de Velde, L.-A.; Chi, H.; Kovarik, P.; Murray, P.J. Tristetraprolin Limits Inflammatory Cytokine Production in Tumor-Associated Macrophages in an mRNA Decay-Independent Manner. Cancer Res. 2015, 75, 3054–3064. [Google Scholar] [CrossRef] [PubMed]
- Mehla, J.; Lacoursiere, S.G.; Lapointe, V.; McNaughton, B.L.; Suyherland, R.J.; McDonald, R.J.; Mohajerani, M.H. Age-dependent behavioral and biochemical characterization of single APP knock-in mouse (APPNL-G-F/NL-G-F) model of Alzheimer’s disease. Neurobiol. Aging 2019, 75, 25–37. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence |
|---|---|
| APP forward | AGG ACT GAC CAC TCG ACC AG |
| APP reverse | CGG GGG TCT AGT TCT GCA T |
| BIN/COPS5 forward | GAC TAC AAA GAC CAT GAC GGT |
| BIN reverse | CAG GTT AGT TTG AGC TAC GAG |
| COPS5 reverse | CCA CCC GAT TGC ATT TTC AAG |
| IL-2 forward | CTA GGC CAC AGA ATT GAA AGA TCT |
| IL-2 reverse | GTA GGT GGA AAT TCT AGC ATC ATC C |
| Temperature | Time | Cycles | |
|---|---|---|---|
| APP PCR | |||
| Denaturation | 93 °C | 3 min | 1 |
| Denaturation | 93 °C | 15 s | 40 |
| Annealing | 56 °C | 30 s | |
| Extension | 68 °C | 1 min | 1 |
| BIN/COPS5 PCR | |||
| Denaturation | 98 °C | 30 s | 1 |
| Denaturation | 98 °C | 5 s | 40 |
| Annealing | 52 °C | 5 s | |
| Extension | 72 °C | 15 s | 1 |
| Extension | 72 °C | 1 min | 1 |
| Antibody | Species | Clonality | Supplier | Product Number | Ref. |
|---|---|---|---|---|---|
| CD11b | Rat | Monoclonal | ThermoFisher | 14-0112-82 | [83] |
| β-amyloid | Rabbit | Monoclonal | ThermoFisher | MA5-35187 | [82] |
| GFAP | Mouse | Monoclonal | ThermoFisher | MA5-12023 | [84] |
| GENE | ID |
|---|---|
| PSEN1 | Mm05001104_m1 |
| PSEN2 | Mm00440405_m1 |
| NOS3 | Mm0045217_m1 |
| COX-2 | Mm0329438_g1 |
| TNFα | Mm0044258_m1 |
| IL-6 | Mm00446190_m1 |
| SIRT1 | Mm0168521_m1 |
| SIRT2 | Mm01492014_m1 |
| HDAC3 | Mm0515816_m1 |
| S18 | Mm03929990_g1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Iglesias, O.; Naidoo, V.; Carrera, I.; Cacabelos, R. Epigenetic Studies in the Male APP/BIN1/COPS5 Triple-Transgenic Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 2446. https://doi.org/10.3390/ijms23052446
Martínez-Iglesias O, Naidoo V, Carrera I, Cacabelos R. Epigenetic Studies in the Male APP/BIN1/COPS5 Triple-Transgenic Mouse Model of Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(5):2446. https://doi.org/10.3390/ijms23052446
Chicago/Turabian StyleMartínez-Iglesias, Olaia, Vinogran Naidoo, Iván Carrera, and Ramón Cacabelos. 2022. "Epigenetic Studies in the Male APP/BIN1/COPS5 Triple-Transgenic Mouse Model of Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 5: 2446. https://doi.org/10.3390/ijms23052446
APA StyleMartínez-Iglesias, O., Naidoo, V., Carrera, I., & Cacabelos, R. (2022). Epigenetic Studies in the Male APP/BIN1/COPS5 Triple-Transgenic Mouse Model of Alzheimer’s Disease. International Journal of Molecular Sciences, 23(5), 2446. https://doi.org/10.3390/ijms23052446