
Discovery of SARS-CoV-2 3CLPro Peptidomimetic Inhibitors through the Catalytic Dyad Histidine-Specific Protein–Ligand Interactions

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
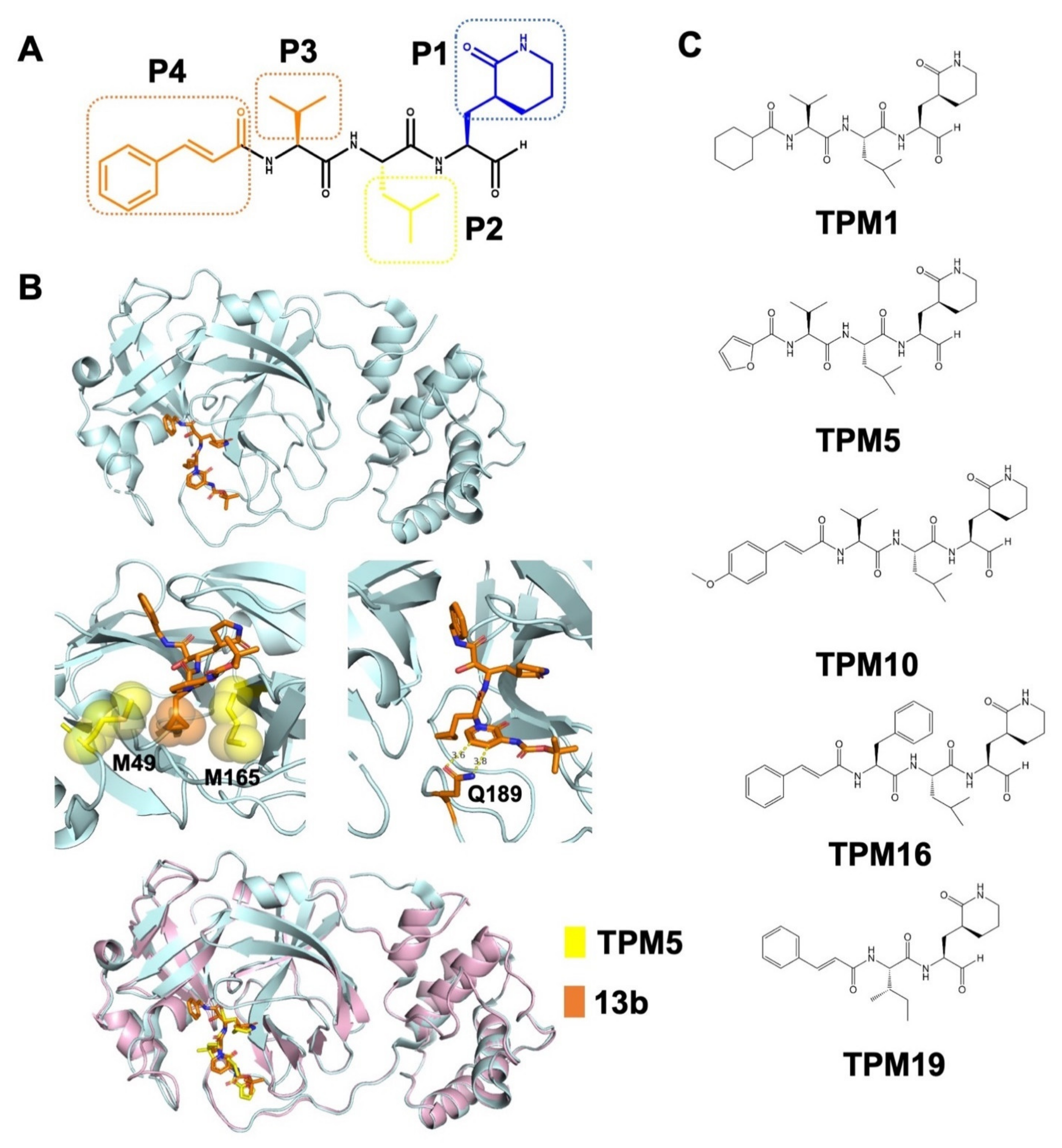
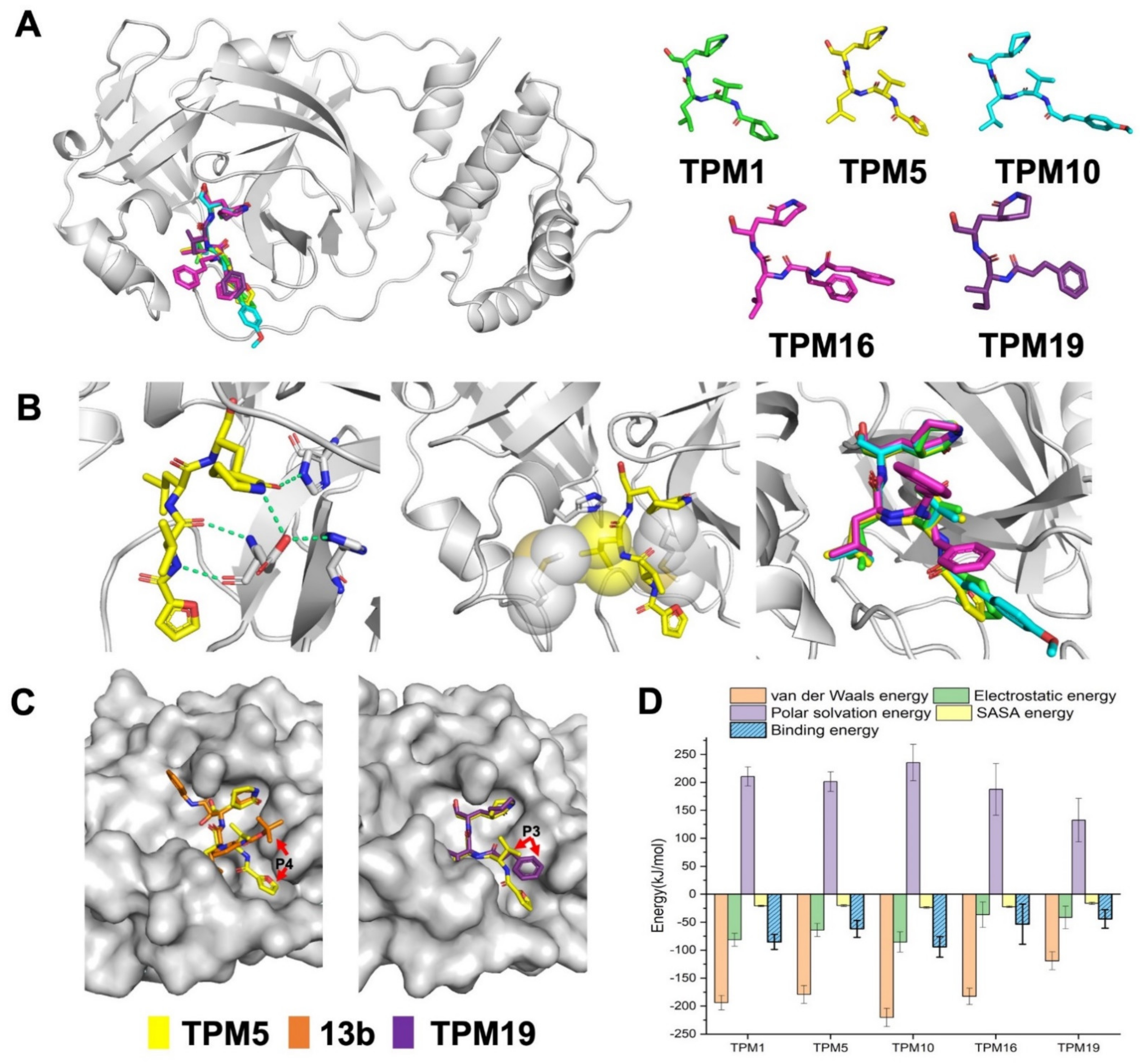
2.1. Structural-Based Rational Design of TPM Inhibitors
2.2. Biophysical Characterization of TPM–3CLpro Interactions
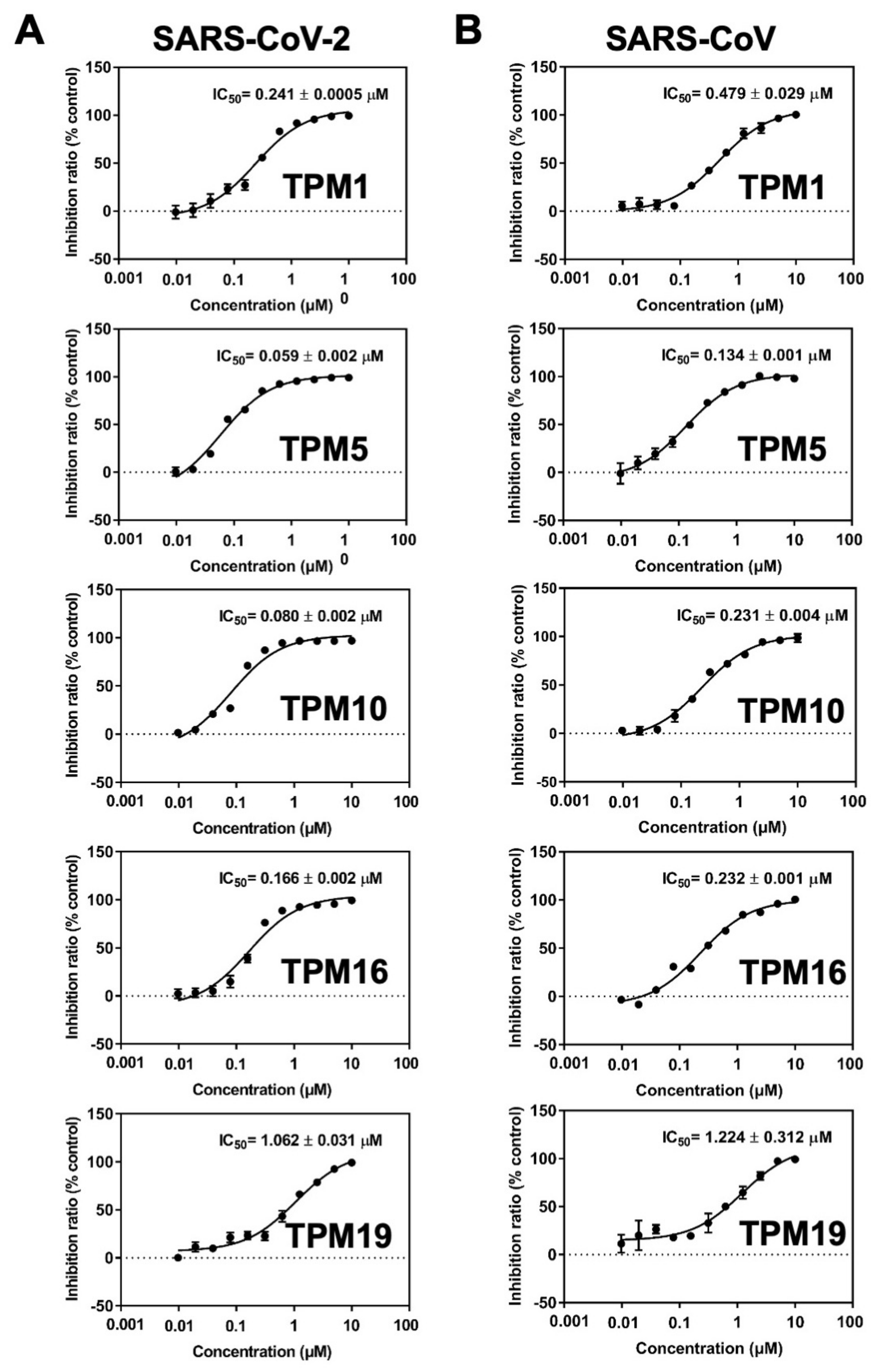
2.3. Inhibitory Activity of TPMs against SARS-CoV-2 3CLPro
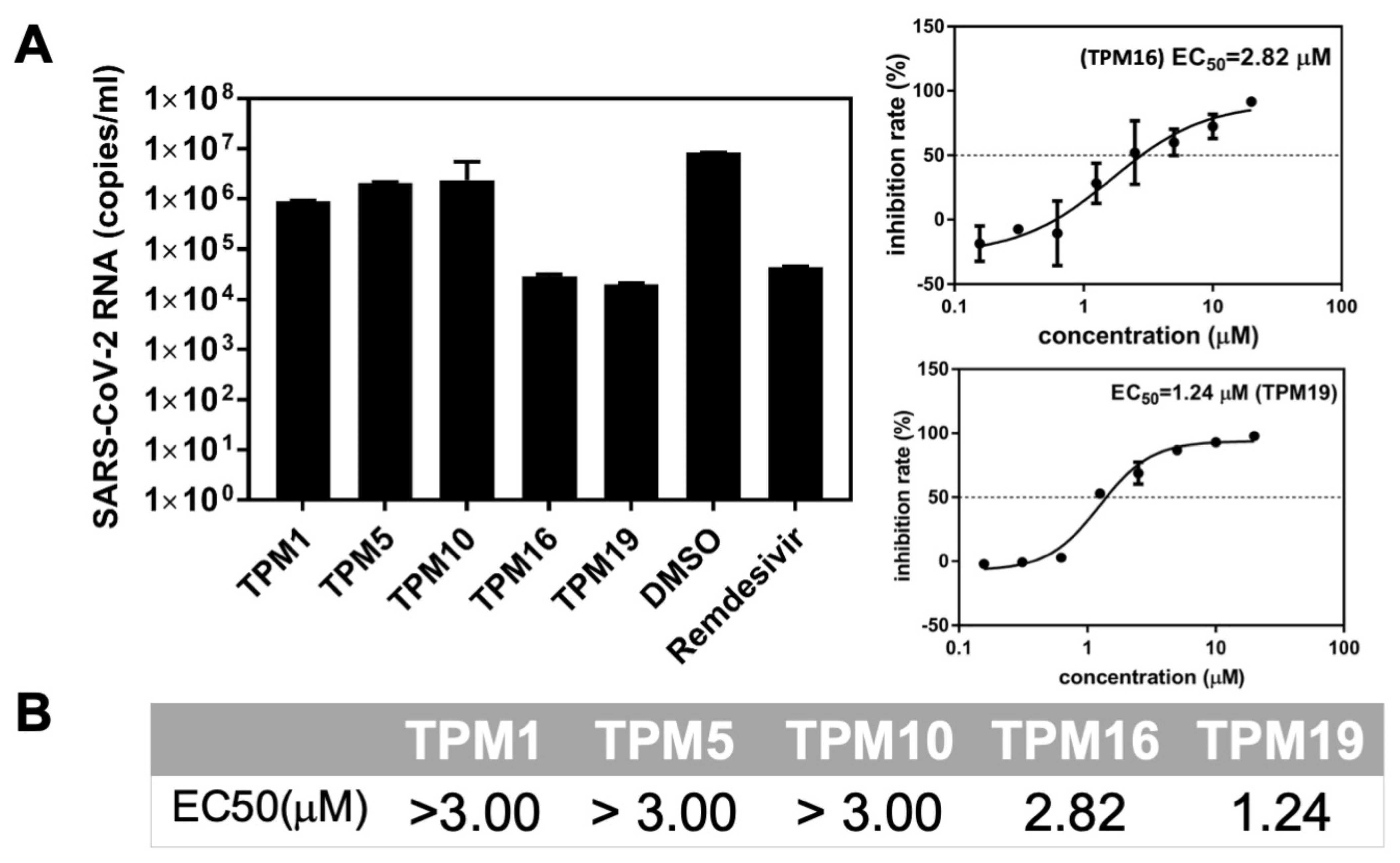
2.4. The Ex-Vivo Anti-Viral Potencies of TPMs against SARS-CoV-2
3. Discussion and Conclusions
4. Materials and Methods
4.1. Expression and Purification of the Recombinant Protein
4.2. Synthesis of TPM Inhibitors
4.3. In Vitro Enzymatic and Inhibition Assay
4.4. Cytotoxicity
4.5. Crystallization, Data Collection, and Structure Determination
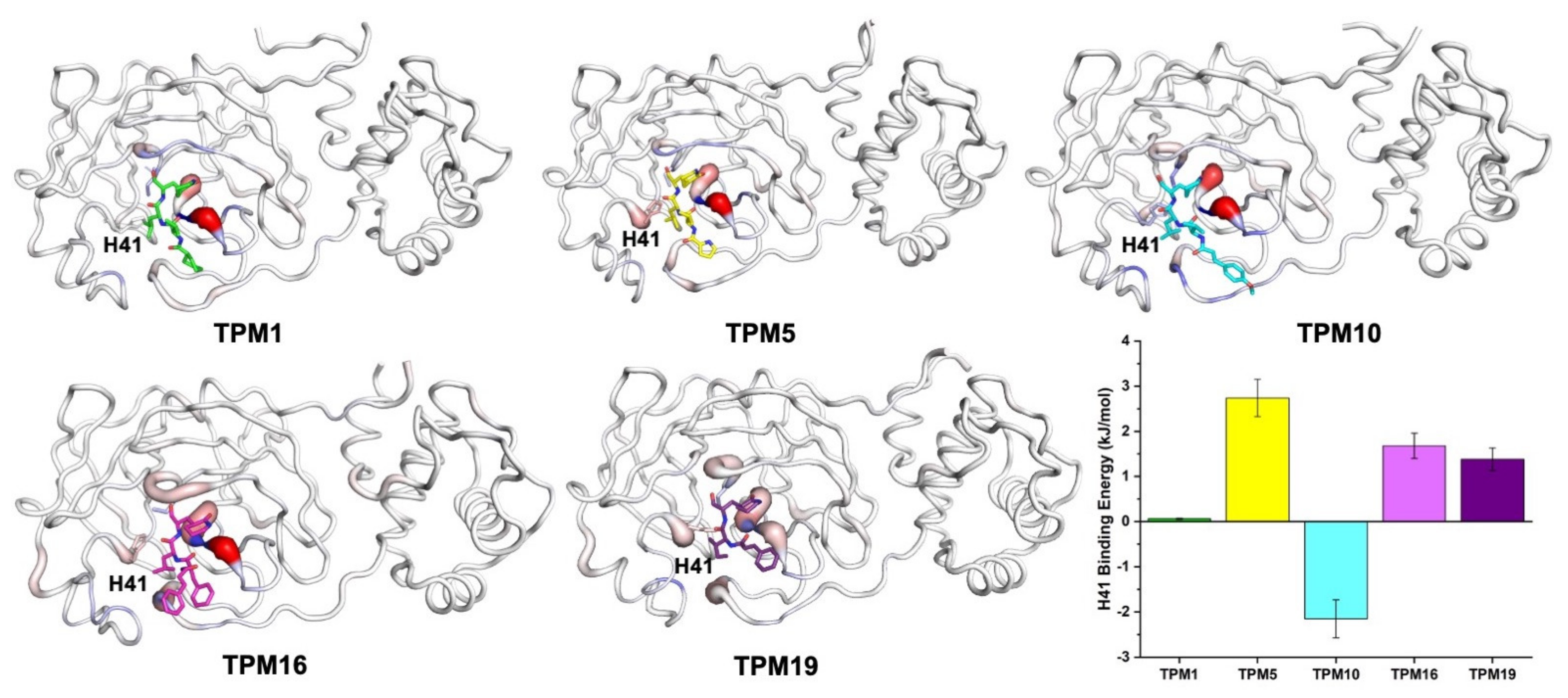
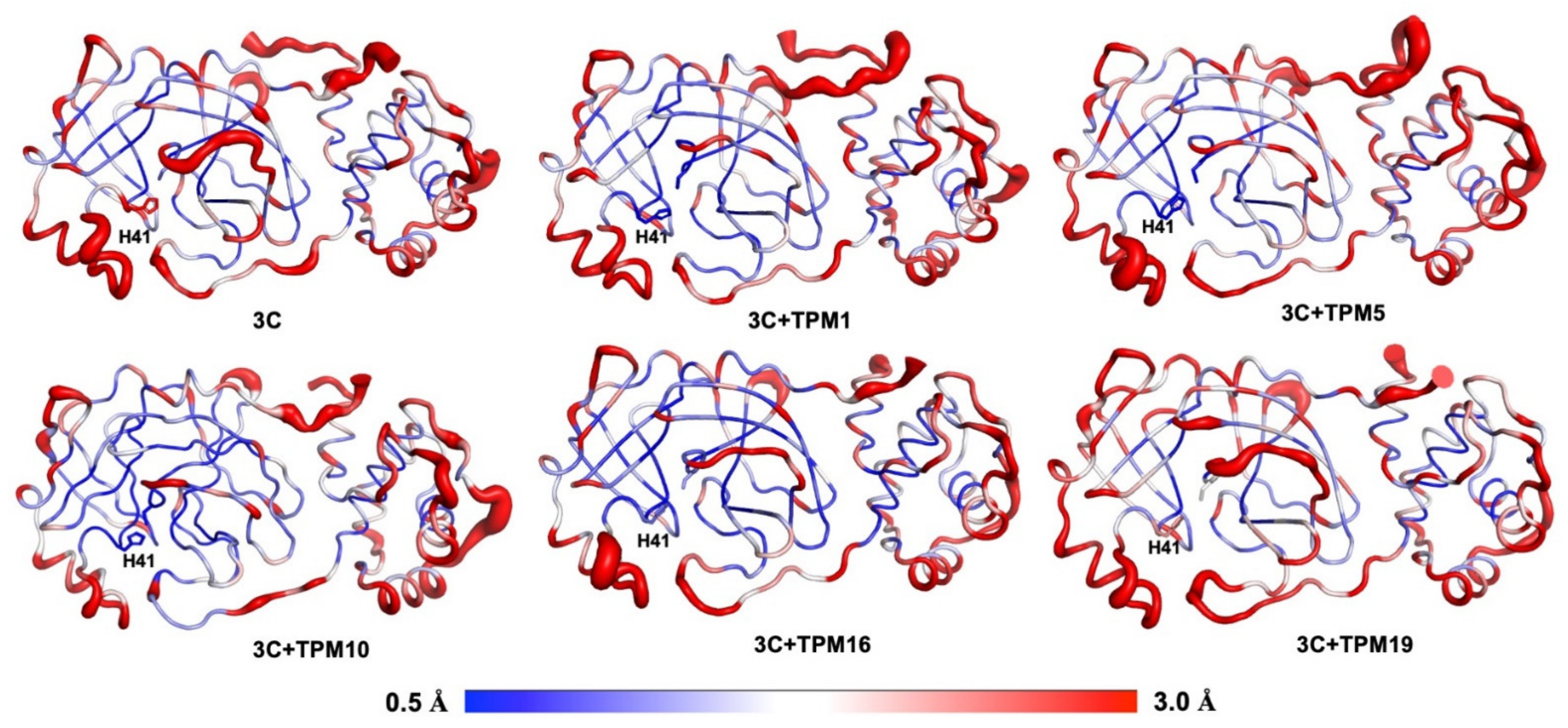
4.6. Molecular Dynamics Simulations
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Makela, M.J.; Puhakka, T.; Ruuskanen, O.; Leinonen, M.; Saikku, P.; Kimpimaki, M.; Blomqvist, S.; Hyypia, T.; Arstila, P. Viruses and bacteria in the etiology of the common cold. J. Clin. Microbiol. 1998, 36, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 2249. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Worldometer, Coronavirus Update (Live), 424,012,619 Cases and 5,902,222 Deaths from COVID-19 Virus Pandemic. Available online: www.worldometers.info/coronavirus/ (accessed on 31 January 2022).
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef]
- Wang, H.; He, S.; Deng, W.; Zhang, Y.; Li, G.; Sun, J.; Zhao, W.; Guo, Y.; Yin, Z.; Li, D.; et al. Comprehensive Insights into the Catalytic Mechanism of Middle East Respiratory Syndrome 3C-Like Protease and Severe Acute Respiratory Syndrome 3C-Like Protease. ACS Catal. 2020, 10, 5871–5890. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liang, C.; Xin, L.; Ren, X.; Tian, L.; Ju, X.; Li, H.; Wang, Y.; Zhao, Q.; Liu, H.; et al. The development of Coronavirus 3C-Like protease (3CL(pro)) inhibitors from 2010 to 2020. Eur. J. Med. Chem. 2020, 206, 112711. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.H. An Overview of Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, R.L.; Kania, R.S.; Brothers, M.A.; Davies, J.F.; Ferre, R.A.; Gajiwala, K.S.; He, M.; Hogan, R.J.; Kozminski, K.; Li, L.Y.; et al. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. [Google Scholar] [CrossRef]
- Su, H.; Yao, S.; Zhao, W.; Zhang, Y.; Liu, J.; Shao, Q.; Wang, Q.; Li, M.; Xie, H.; Shang, W.; et al. Identification of pyrogallol as a warhead in design of covalent inhibitors for the SARS-CoV-2 3CL protease. Nat. Commun. 2021, 12, 3623. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Li, Y.S.; Zeng, R.; Liu, F.L.; Luo, R.H.; Huang, C.; Wang, Y.F.; Zhang, J.; Quan, B.; Shen, C.; et al. SARS-CoV-2 M(pro) inhibitors with antiviral activity in a transgenic mouse model. Science 2021, 371, 1374–1378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.H.; Stone, E.A.; Deshmukh, M.; Ippolito, J.A.; Ghahremanpour, M.M.; Tirado-Rives, J.; Spasov, K.A.; Zhang, S.; Takeo, Y.; Kudalkar, S.N.; et al. Potent Noncovalent Inhibitors of the Main Protease of SARS-CoV-2 from Molecular Sculpting of the Drug Perampanel Guided by Free Energy Perturbation Calculations. ACS Cent. Sci. 2021, 7, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Osipiuk, J.; Azizi, S.A.; Dvorkin, S.; Endres, M.; Jedrzejczak, R.; Jones, K.A.; Kang, S.; Kathayat, R.S.; Kim, Y.; Lisnyak, V.G.; et al. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2021, 12, 743. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Zhao, Y.; Fang, C.; Zhang, Q.; Zhang, R.; Zhao, X.; Duan, Y.; Wang, H.; Zhu, Y.; Feng, L.; Zhao, J.; et al. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332. Protein Cell 2021, 1–5. [Google Scholar] [CrossRef]
- Drayman, N.; DeMarco, J.K.; Jones, K.A.; Azizi, S.A.; Froggatt, H.M.; Tan, K.; Maltseva, N.I.; Chen, S.; Nicolaescu, V.; Dvorkin, S.; et al. Masitinib is a broad coronavirus 3CL inhibitor that blocks replication of SARS-CoV-2. Science 2021, 373, 931–936. [Google Scholar] [CrossRef]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; De Jesus, P.D.; Teriete, P.; Hull, M.V.; et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 2020, 586, 113–119. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Cannalire, R.; Cerchia, C.; Beccari, A.R.; Di Leva, F.S.; Summa, V. Targeting SARS-CoV-2 Proteases and Polymerase for COVID-19 Treatment: State of the Art and Future Opportunities. J. Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.A.; Banerjee, S.; Singh, S.; Qureshi, I.A.; Gayen, S.; Jha, T. First structure-activity relationship analysis of SARS-CoV-2 virus main protease (Mpro) inhibitors: An endeavor on COVID-19 drug discovery. Mol. Divers. 2021, 25, 1827–1838. [Google Scholar] [CrossRef]
- Yin, G.; Lv, G.; Zhang, J.; Jiang, H.; Lai, T.; Yang, Y.; Ren, Y.; Wang, J.; Yi, C.; Chen, H.; et al. Early-stage structure-based drug discovery for small GTPases by NMR spectroscopy. Pharmacol. Ther. 2022, 236, 108110. [Google Scholar] [CrossRef]
- Ma, Y.; Li, L.; He, S.; Shang, C.; Sun, Y.; Liu, N.; Meek, T.D.; Wang, Y.; Shang, L. Application of Dually Activated Michael Acceptor to the Rational Design of Reversible Covalent Inhibitor for Enterovirus 71 3C Protease. J. Med. Chem. 2019, 62, 6146–6162. [Google Scholar] [CrossRef] [PubMed]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Grosse-Kunstleve, R.W.; Hung, L.W.; Ioerger, T.R.; McCoy, A.J.; Moriarty, N.W.; Read, R.J.; Sacchettini, J.C.; Sauter, N.K.; Terwilliger, T.C. PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 1948–1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Bjelkmar, P.; Larsson, P.; Cuendet, M.A.; Hess, B.; Lindahl, E. Implementation of the CHARMM Force Field in GROMACS: Analysis of Protein Stability Effects from Correction Maps, Virtual Interaction Sites, and Water Models. J. Chem. Theory Comput. 2010, 6, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Xu, B.; Ma, S.; Wang, H.; Shang, L.; Zhu, C.; Ye, S. Discovery of SARS-CoV-2 3CLPro Peptidomimetic Inhibitors through the Catalytic Dyad Histidine-Specific Protein–Ligand Interactions. Int. J. Mol. Sci. 2022, 23, 2392. https://doi.org/10.3390/ijms23042392
Wang Y, Xu B, Ma S, Wang H, Shang L, Zhu C, Ye S. Discovery of SARS-CoV-2 3CLPro Peptidomimetic Inhibitors through the Catalytic Dyad Histidine-Specific Protein–Ligand Interactions. International Journal of Molecular Sciences. 2022; 23(4):2392. https://doi.org/10.3390/ijms23042392
Chicago/Turabian StyleWang, Yaxin, Binghong Xu, Sen Ma, Hao Wang, Luqing Shang, Cheng Zhu, and Sheng Ye. 2022. "Discovery of SARS-CoV-2 3CLPro Peptidomimetic Inhibitors through the Catalytic Dyad Histidine-Specific Protein–Ligand Interactions" International Journal of Molecular Sciences 23, no. 4: 2392. https://doi.org/10.3390/ijms23042392
APA StyleWang, Y., Xu, B., Ma, S., Wang, H., Shang, L., Zhu, C., & Ye, S. (2022). Discovery of SARS-CoV-2 3CLPro Peptidomimetic Inhibitors through the Catalytic Dyad Histidine-Specific Protein–Ligand Interactions. International Journal of Molecular Sciences, 23(4), 2392. https://doi.org/10.3390/ijms23042392