
Expanding Gene-Editing Potential in Crop Improvement with Pangenomes

 , , , ,
, , , ,  ,
,  and
and 
Abstract
:1. Introduction
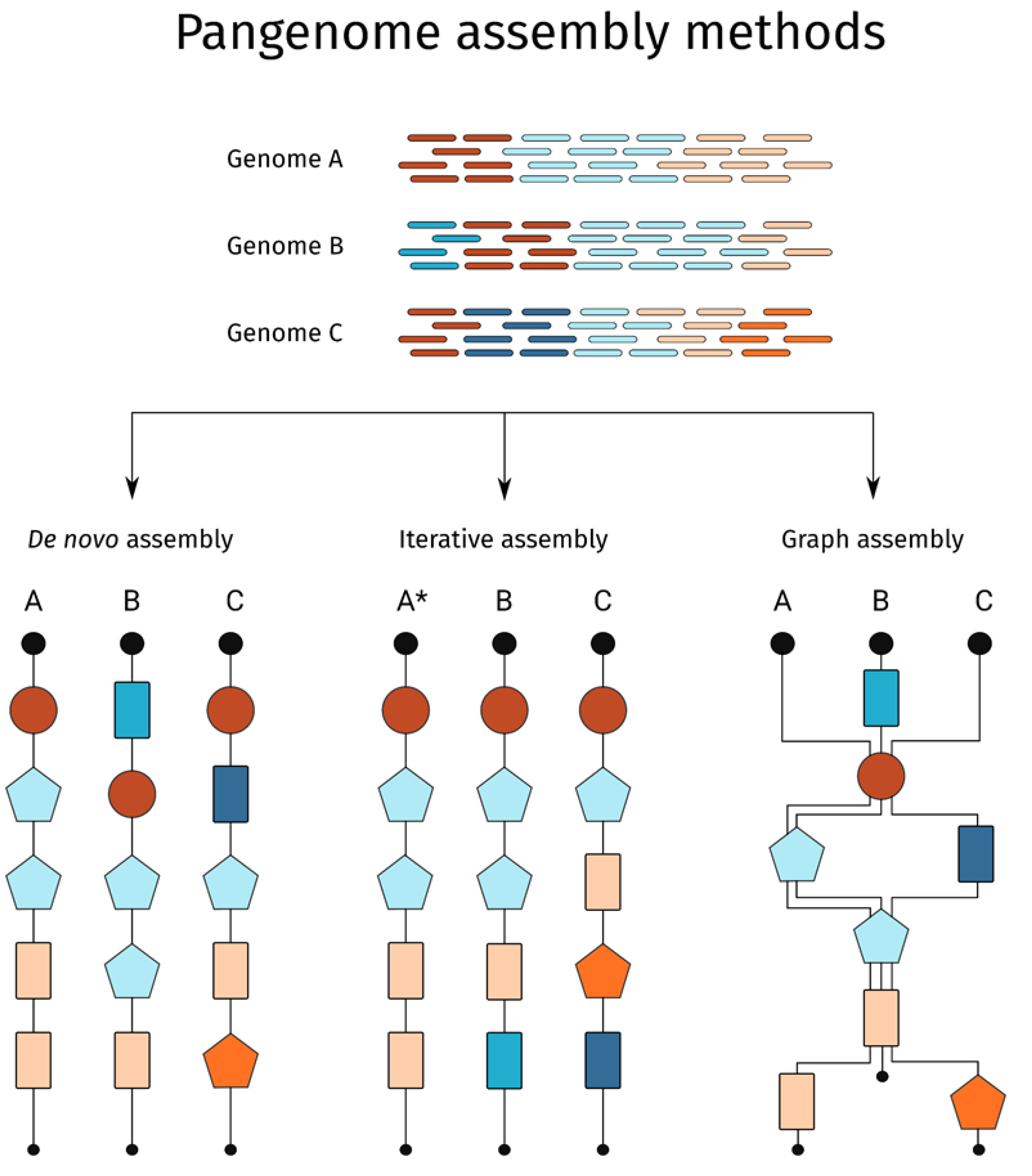
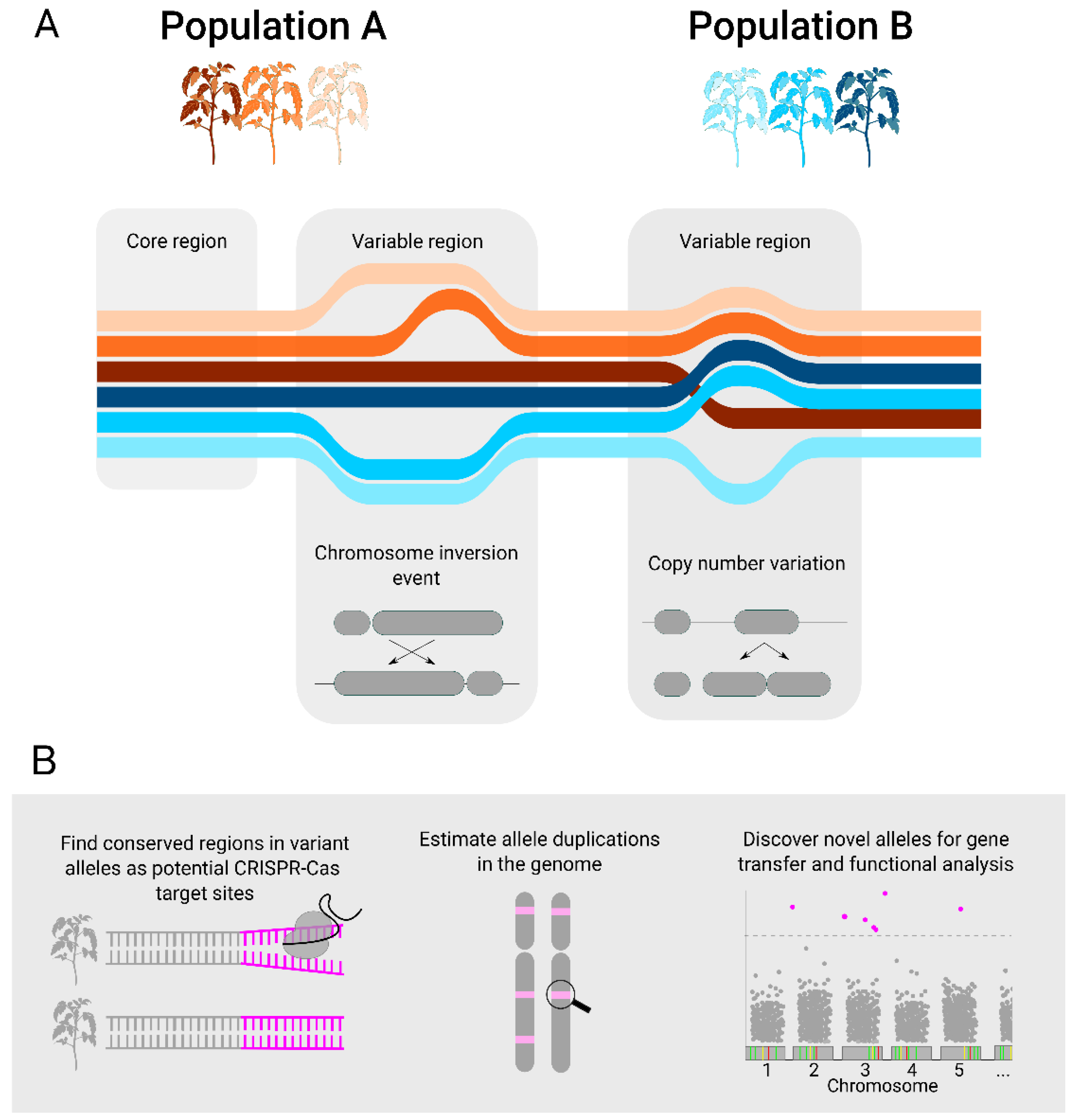
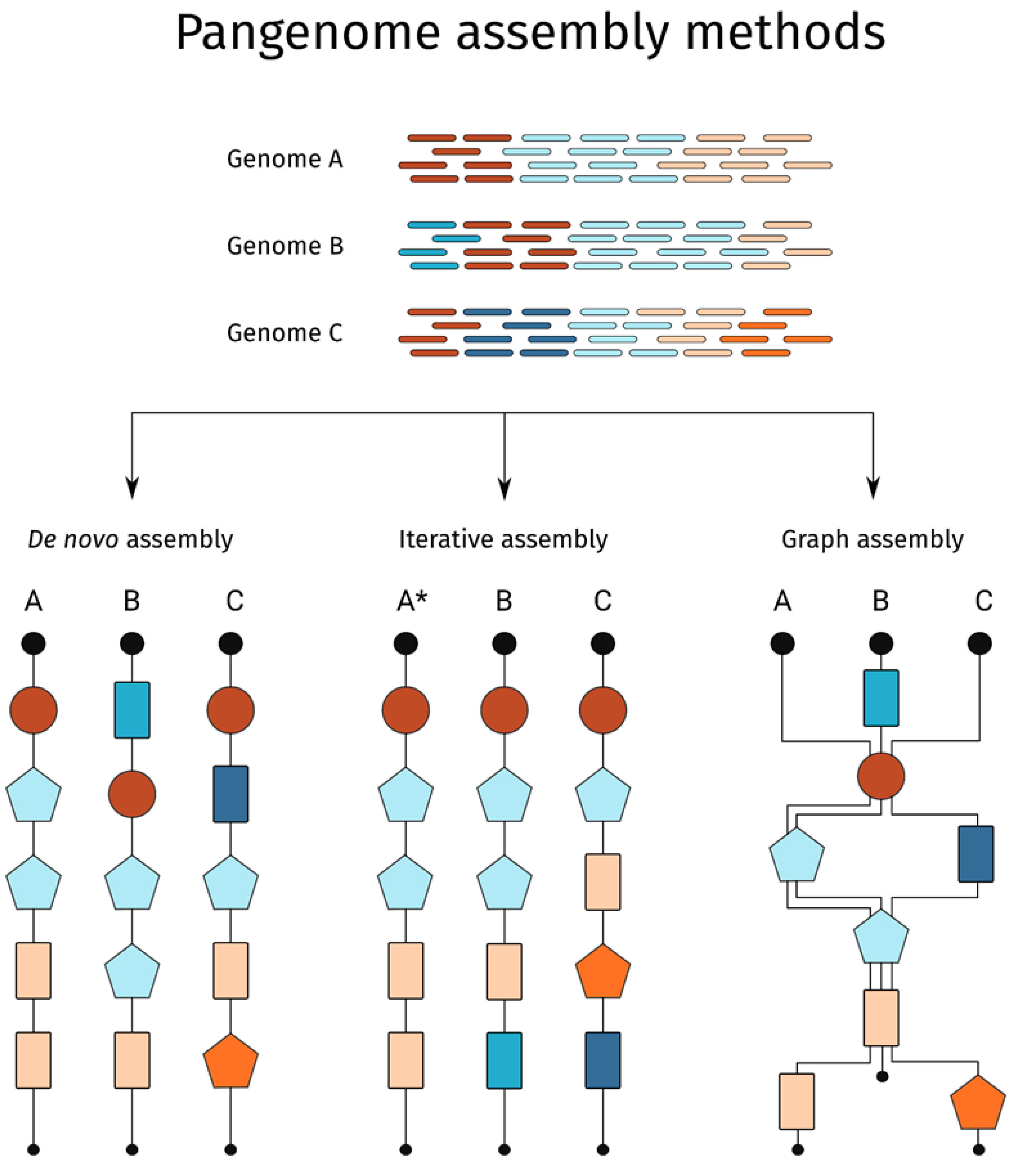
2. Pangenomes
3. Association Analysis Using Pangenomes Can Reveal Valuable Sites for Genome Editing
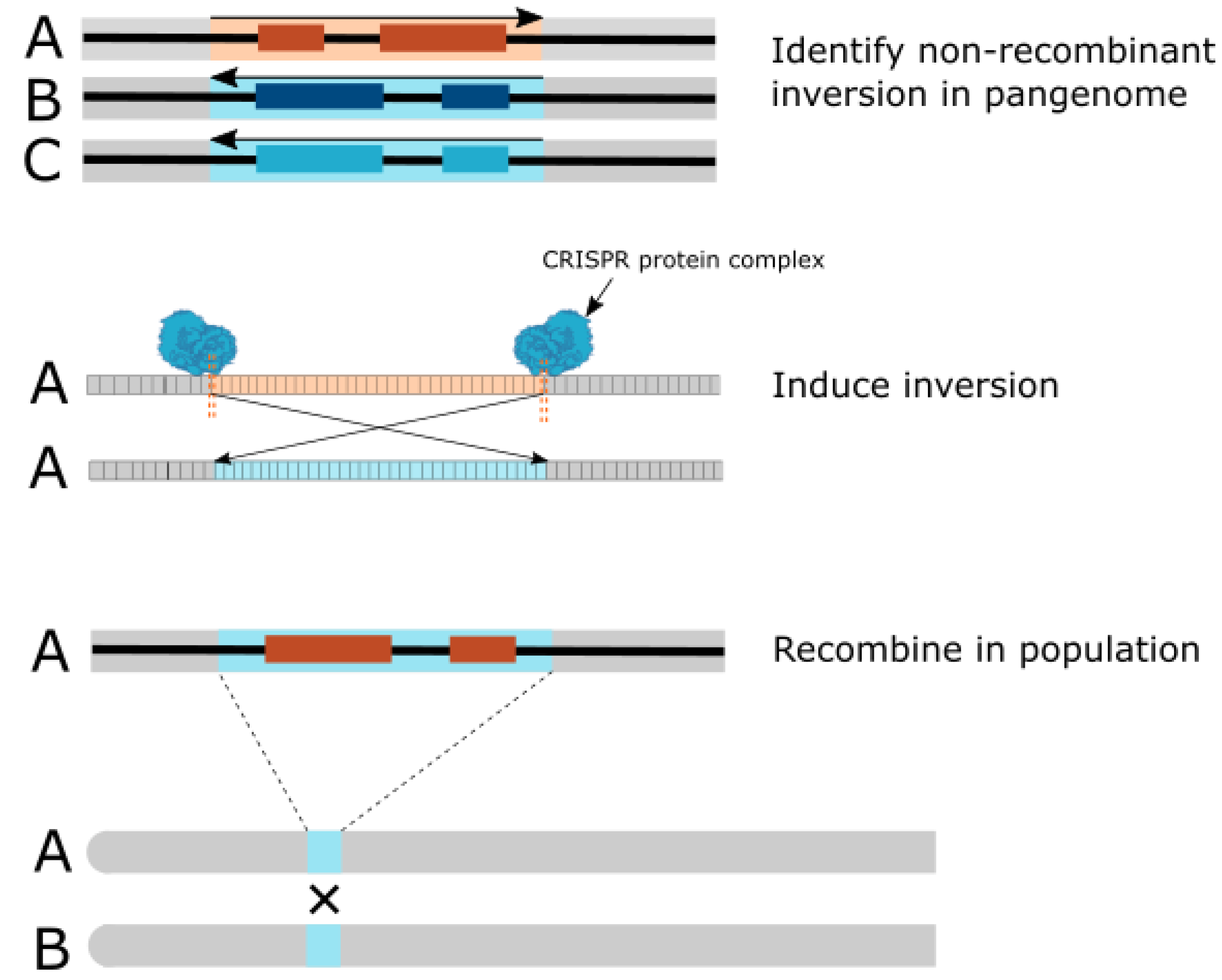
4. Targeted Mutagenesis Guided by Pangenomes
5. Off-Targets Effects in Multiplexed Editing
6. Future Applications of Pangenomes in Genome Editing through Super-Pangenome Guided CRISPR-Cas
7. Challenges and Considerations
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roser, M. Future Population Growth. Available online: https://ourworldindata.org/future-population-growth (accessed on 11 June 2021).
- Masson-Delmotte, V.; Zhai, P.; Pirani, A.; Connors, S.L.; Péan, C.; Berger, S.; Caud, N.; Chen, Y.; Goldfarb, L.; Gomis, M.I.; et al. Climate Change 2021: The Physical Science Basis. In Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, MA, USA, 2021. [Google Scholar]
- Abberton, M.; Batley, J.; Bentley, A.; Bryant, J.; Cai, H.; Cockram, J.; de Oliveira, A.C.; Cseke, L.J.; Dempewolf, H.; De Pace, C.; et al. Global agricultural intensification during climate change: A role for genomics. Plant Biotechnol. J. 2016, 14, 1095–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, R.; Bayer, P.E.; Edwards, D. Climate change and the need for agricultural adaptation. Curr. Opin. Plant Biol. 2020, 56, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.Q.; Doyon, Y.; Ainley, W.M.; Miller, J.C.; Dekelver, R.C.; Moehle, E.A.; Rock, J.M.; Lee, Y.L.; Garrison, R.; Schulenberg, L.; et al. Targeted transgene integration in plant cells using designed zinc finger nucleases. Plant Mol. Biol. 2009, 69, 699–709. [Google Scholar] [CrossRef] [PubMed]
- De Pater, S.; Neuteboom, L.W.; Pinas, J.E.; Hooykaas, P.J.; van der Zaal, B.J. ZFN-induced mutagenesis and gene-targeting in Arabidopsis through Agrobacterium-mediated floral dip transformation. Plant Biotechnol. J. 2009, 7, 821–835. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, A.; Plaisier, C.L.; Carroll, D.; Drews, G.N. Targeted mutagenesis using zinc-finger nucleases in Arabidopsis. Proc. Natl. Acad. Sci. USA 2005, 102, 2232–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhang, F.; Li, X.; Baller, J.A.; Qi, Y.; Starker, C.G.; Bogdanove, A.J.; Voytas, D.F. Transcription activator-like effector nucleases enable efficient plant genome engineering. Plant Physiol. 2013, 161, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Forsyth, A.; Weeks, T.; Richael, C.; Duan, H. Transcription Activator-Like Effector Nucleases (TALEN)-Mediated Targeted DNA Insertion in Potato Plants. Front. Plant Sci. 2016, 7, 1572. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Liu, B.; Spalding, M.H.; Weeks, D.P.; Yang, B. High-efficiency TALEN-based gene editing produces disease-resistant rice. Nat. Biotechnol. 2012, 30, 390–392. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819. [Google Scholar] [CrossRef] [Green Version]
- Scheben, A.; Edwards, D. Genome editors take on crops. Science 2017, 355, 1122. [Google Scholar] [CrossRef]
- Scheben, A.; Wolter, F.; Batley, J.; Puchta, H.; Edwards, D. Towards CRISPR/Cas crops—Bringing together genomics and genome editing. New Phytologist. 2017, 216, 682–698. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Gao, H.; Wang, H.; Lafitte, H.R.; Archibald, R.L.; Yang, M.; Hakimi, S.M.; Mo, H.; Habben, J.E. ARGOS8 variants generated by CRISPR-Cas9 improve maize grain yield under field drought stress conditions. Plant Biotechnol. J. 2017, 15, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Zhang, H.; Zhu, H. CRISPR technology is revolutionizing the improvement of tomato and other fruit crops. Hortic. Res. 2019, 6, 77. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Y.; Wen, J.; Zhao, W.; Wang, Q.; Huang, W. Rational Improvement of Rice Yield and Cold Tolerance by Editing the Three Genes OsPIN5b, GS3, and OsMYB30 With the CRISPR–Cas9 System. Front. Plant Sci. 2020, 10, 1663. [Google Scholar] [CrossRef] [Green Version]
- Bayer, P.E.; Golicz, A.A.; Scheben, A.; Batley, J.; Edwards, D. Plant pan-genomes are the new reference. Nat. Plants 2020, 6, 914–920. [Google Scholar] [CrossRef]
- Danilevicz, M.F.; Tay Fernandez, C.G.; Marsh, J.I.; Bayer, P.E.; Edwards, D. Plant pangenomics: Approaches, applications and advancements. Curr. Opin. Plant Biol. 2020, 54, 18–25. [Google Scholar] [CrossRef]
- Gabriel, R.; von Kalle, C.; Schmidt, M. Mapping the precision of genome editing. Nat. Biotechnol. 2015, 33, 150–152. [Google Scholar] [CrossRef]
- Doebley, J.F.; Gaut, B.S.; Smith, B.D. The Molecular Genetics of Crop Domestication. Cell 2006, 127, 1309–1321. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Zhao, X.; Mace, E.; Henry, R.; Jordan, D. Exploring and Exploiting Pan-genomics for Crop Improvement. Mol. Plant 2019, 12, 156–169. [Google Scholar] [CrossRef] [Green Version]
- The Rice, C.; Sequencing, C. The sequence of rice chromosomes 11 and 12, rich in disease resistance genes and recent gene duplications. BMC Biol. 2005, 3, 20. [Google Scholar] [CrossRef] [Green Version]
- Woodhouse, M.R.; Schnable, J.C.; Pedersen, B.S.; Lyons, E.; Lisch, D.; Subramaniam, S.; Freeling, M. Following tetraploidy in maize, a short deletion mechanism removed genes preferentially from one of the two homologs. PLoS Biol. 2010, 8, e1000409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, D.; Ferguson, A.A.; Jiang, N. What makes up plant genomes: The vanishing line between transposable elements and genes. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2016, 1859, 366–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, L.; Li, D.; Lv, J.; Chen, W.; Zhang, Z.; Li, X.; Yang, B.; Zhou, S.; Yang, S.; Li, W.; et al. Pan-genome of cultivated pepper (Capsicum) and its use in gene presence–absence variation analyses. New Phytol. 2018, 220, 360–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schatz, M.C.; Maron, L.G.; Stein, J.C.; Wences, A.H.; Gurtowski, J.; Biggers, E.; Lee, H.; Kramer, M.; Antoniou, E.; Ghiban, E.; et al. Whole genome de novo assemblies of three divergent strains of rice, Oryza sativa, document novel gene space of aus and indica. Genome Biol. 2014, 15, 506. [Google Scholar] [CrossRef] [Green Version]
- Song, J.-M.; Guan, Z.; Hu, J.; Guo, C.; Yang, Z.; Wang, S.; Liu, D.; Wang, B.; Lu, S.; Zhou, R.; et al. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 2020, 6, 34–45. [Google Scholar] [CrossRef]
- Hurgobin, B.; Golicz, A.A.; Bayer, P.E.; Chan, C.-K.K.; Tirnaz, S.; Dolatabadian, A.; Schiessl, S.V.; Samans, B.; Montenegro, J.D.; Parkin, I.A.P.; et al. Homoeologous exchange is a major cause of gene presence/absence variation in the amphidiploid Brassica napus. Plant Biotechnol. J. 2018, 16, 1265–1274. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Bayer, P.E.; Ruperao, P.; Saxena, R.K.; Khan, A.W.; Golicz, A.A.; Nguyen, H.T.; Batley, J.; Edwards, D.; Varshney, R.K. Trait associations in the pangenome of pigeon pea (Cajanus cajan). Plant Biotechnol. J. 2020, 18, 1946–1954. [Google Scholar] [CrossRef] [Green Version]
- Malzahn, A.; Lowder, L.; Qi, Y. Plant genome editing with TALEN and CRISPR. Cell Biosci. 2017, 7, 21. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, A.; Nain, V. TALENs—an indispensable tool in the era of CRISPR: A mini review. J. Genet. Eng. Biotechnol. 2021, 19, 125. [Google Scholar] [CrossRef]
- Alok, A.; Sandhya, D.; Jogam, P.; Rodrigues, V.; Bhati, K.K.; Sharma, H.; Kumar, J. The Rise of the CRISPR/Cpf1 System for Efficient Genome Editing in Plants. Front. Plant Sci. 2020, 11, 264. [Google Scholar] [CrossRef]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Gonda, I.; Sun, H.; Ma, Q.; Bao, K.; Tieman, D.M.; Burzynski-Chang, E.A.; Fish, T.L.; Stromberg, K.A.; Sacks, G.L.; et al. The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet. 2019, 51, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Ruperao, P.; Thirunavukkarasu, N.; Gandham, P.; Selvanayagam, S.; Govindaraj, M.; Nebie, B.; Manyasa, E.; Gupta, R.; Das, R.R.; Gandhi, H.; et al. Sorghum pan-genome explores the functional utility to accelerate the genetic gain. Front. Plant Sci. 2021, 12, 963. [Google Scholar] [CrossRef]
- Liu, H.-J.; Jian, L.; Xu, J.; Zhang, Q.; Zhang, M.; Jin, M.; Peng, Y.; Yan, J.; Han, B.; Liu, J.; et al. High-Throughput CRISPR/Cas9 Mutagenesis Streamlines Trait Gene Identification in Maize[OPEN]. Plant Cell 2020, 32, 1397–1413. [Google Scholar] [CrossRef] [Green Version]
- Ossowski, S.; Schneeberger, K.; Clark, R.M.; Lanz, C.; Warthmann, N.; Weigel, D. Sequencing of natural strains of Arabidopsis thaliana with short reads. Genome Res. 2008, 18, 2024–2033. [Google Scholar] [CrossRef] [Green Version]
- Golicz, A.A.; Bayer, P.E.; Barker, G.C.; Edger, P.P.; Kim, H.; Martinez, P.A.; Chan, C.K.K.; Severn-Ellis, A.; McCombie, W.R.; Parkin, I.A.P.; et al. The pangenome of an agronomically important crop plant Brassica oleracea. Nat. Commun. 2016, 7, 13390. [Google Scholar] [CrossRef]
- Della Coletta, R.; Qiu, Y.; Ou, S.; Hufford, M.B.; Hirsch, C.N. How the pan-genome is changing crop genomics and improvement. Genome Biol. 2021, 22, 3. [Google Scholar] [CrossRef]
- Sirén, J.; Garrison, E.; Novak, A.M.; Paten, B.; Durbin, R. Haplotype-aware graph indexes. Bioinformatics 2020, 36, 400–407. [Google Scholar] [CrossRef]
- Garrison, E.; Sirén, J.; Novak, A.M.; Hickey, G.; Eizenga, J.M.; Dawson, E.T.; Jones, W.; Garg, S.; Markello, C.; Lin, M.F.; et al. Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nat. Biotechnol. 2018, 36, 875–879. [Google Scholar] [CrossRef]
- Rakocevic, G.; Semenyuk, V.; Lee, W.-P.; Spencer, J.; Browning, J.; Johnson, I.J.; Arsenijevic, V.; Nadj, J.; Ghose, K.; Suciu, M.C.; et al. Fast and accurate genomic analyses using genome graphs. Nat. Genet. 2019, 51, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Hu, H.; Tian, Y.; Li, J.; Scheben, A.; Zhang, C.; Li, Y.; Wu, J.; Yang, L.; Fan, X.; et al. The Chicken Pan-Genome Reveals Gene Content Variation and a Promoter Region Deletion in IGF2BP1 Affecting Body Size. Mol. Biol. Evol. 2021, 38, 5066–5081. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [Green Version]
- Gerdol, M.; Moreira, R.; Cruz, F.; Gómez-Garrido, J.; Vlasova, A.; Rosani, U.; Venier, P.; Naranjo-Ortiz, M.A.; Murgarella, M.; Greco, S.; et al. Massive gene presence-absence variation shapes an open pan-genome in the Mediterranean mussel. Genome Biol. 2020, 21, 275. [Google Scholar] [CrossRef]
- Li, R.; Li, Y.; Zheng, H.; Luo, R.; Zhu, H.; Li, Q.; Qian, W.; Ren, Y.; Tian, G.; Li, J.; et al. Building the sequence map of the human pan-genome. Nat. Biotechnol. 2010, 28, 57–63. [Google Scholar] [CrossRef]
- Morgante, M.; De Paoli, E.; Radovic, S. Transposable elements and the plant pan-genomes. Curr. Opin. Plant Biol. 2007, 10, 149–155. [Google Scholar] [CrossRef]
- Li, Y.-h.; Zhou, G.; Ma, J.; Jiang, W.; Jin, L.-g.; Zhang, Z.; Guo, Y.; Zhang, J.; Sui, Y.; Zheng, L.; et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat. Biotechnol. 2014, 32, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Du, H.; Li, P.; Shen, Y.; Peng, H.; Liu, S.; Zhou, G.-A.; Zhang, H.; Liu, Z.; Shi, M.; et al. Pan-Genome of Wild and Cultivated Soybeans. Cell 2020, 182, 162–176.e13. [Google Scholar] [CrossRef]
- Lu, F.; Romay, M.C.; Glaubitz, J.C.; Bradbury, P.J.; Elshire, R.J.; Wang, T.; Li, Y.; Li, Y.; Semagn, K.; Zhang, X.; et al. High-resolution genetic mapping of maize pan-genome sequence anchors. Nat. Commun. 2015, 6, 6914. [Google Scholar] [CrossRef] [Green Version]
- Dolatabadian, A.; Bayer, P.E.; Tirnaz, S.; Hurgobin, B.; Edwards, D.; Batley, J. Characterization of disease resistance genes in the Brassica napus pangenome reveals significant structural variation. Plant Biotechnol. J. 2020, 18, 969–982. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.P.; Contreras-Moreira, B.; Woods, D.P.; Des Marais, D.L.; Burgess, D.; Shu, S.; Stritt, C.; Roulin, A.C.; Schackwitz, W.; Tyler, L.; et al. Extensive gene content variation in the Brachypodium distachyon pan-genome correlates with population structure. Nat. Commun. 2017, 8, 2184. [Google Scholar] [CrossRef]
- Jayakodi, M.; Padmarasu, S.; Haberer, G.; Bonthala, V.S.; Gundlach, H.; Monat, C.; Lux, T.; Kamal, N.; Lang, D.; Himmelbach, A.; et al. The barley pan-genome reveals the hidden legacy of mutation breeding. Nature 2020, 588, 284–289. [Google Scholar] [CrossRef]
- Zhao, Q.; Feng, Q.; Lu, H.; Li, Y.; Wang, A.; Tian, Q.; Zhan, Q.; Lu, Y.; Zhang, L.; Huang, T.; et al. Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat. Genet. 2018, 50, 278–284. [Google Scholar] [CrossRef] [Green Version]
- Varshney, R.K.; Roorkiwal, M.; Sun, S.; Bajaj, P.; Chitikineni, A.; Thudi, M.; Singh, N.P.; Du, X.; Upadhyaya, H.D.; Khan, A.W.; et al. A chickpea genetic variation map based on the sequencing of 3366 genomes. Nature 2021, 599, 622–627. [Google Scholar] [CrossRef]
- Sun, X.; Jiao, C.; Schwaninger, H.; Chao, C.T.; Ma, Y.; Duan, N.; Khan, A.; Ban, S.; Xu, K.; Cheng, L.; et al. Phased diploid genome assemblies and pan-genomes provide insights into the genetic history of apple domestication. Nat. Genet. 2020, 52, 1423–1432. [Google Scholar] [CrossRef]
- Yu, J.; Golicz, A.A.; Lu, K.; Dossa, K.; Zhang, Y.; Chen, J.; Wang, L.; You, J.; Fan, D.; Edwards, D.; et al. Insight into the evolution and functional characteristics of the pan-genome assembly from sesame landraces and modern cultivars. Plant Biotechnol. J. 2019, 17, 881–892. [Google Scholar] [CrossRef] [Green Version]
- Hübner, S.; Bercovich, N.; Todesco, M.; Mandel, J.R.; Odenheimer, J.; Ziegler, E.; Lee, J.S.; Baute, G.J.; Owens, G.L.; Grassa, C.J.; et al. Sunflower pan-genome analysis shows that hybridization altered gene content and disease resistance. Nat. Plants 2019, 5, 54–62. [Google Scholar] [CrossRef]
- Long, E.; Bradbury, P.; Romay, M.; Buckler, E.; Robbins, K. Genome-wide Imputation Using the Practical Haplotype Graph in the Heterozygous Crop Cassava. Genes Genomes Genet. 2021, 12, jkab383. [Google Scholar] [CrossRef]
- Jensen, S.; Charles, J.R.; Muleta, K.; Bradbury, P.; Casstevens, T.; Deshpande, S.P.; Gore, M.A.; Gupta, R.; Ilut, D.C.; Johnson, L.; et al. A sorghum Practical Haplotype Graph facilitates genome-wide imputation and cost-effective genomic prediction. Plant Genome 2020, 13, e20009. [Google Scholar] [CrossRef] [Green Version]
- Montenegro, J.D.; Golicz, A.A.; Bayer, P.E.; Hurgobin, B.; Lee, H.; Chan, C.-K.K.; Visendi, P.; Lai, K.; Doležel, J.; Batley, J.; et al. The pangenome of hexaploid bread wheat. Plant J. 2017, 90, 1007–1013. [Google Scholar] [CrossRef] [Green Version]
- Jiao, W.-B.; Schneeberger, K. Chromosome-level assemblies of multiple Arabidopsis genomes reveal hotspots of rearrangements with altered evolutionary dynamics. Nat. Commun. 2020, 11, 989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Scheben, A.; Verpaalen, B.; Tirnaz, S.; Bayer, P.E.; Hodel, R.G.J.; Batley, J.; Soltis, D.E.; Soltis, P.S.; Edwards, D. Amborella gene presence/absence variation is associated with abiotic stress responses that may contribute to environmental adaptation. New Phytol. 2021, 233, 1548–1555. [Google Scholar] [CrossRef]
- Li, J.; Yuan, D.; Wang, P.; Wang, Q.; Sun, M.; Liu, Z.; Si, H.; Xu, Z.; Ma, Y.; Zhang, B.; et al. Cotton pan-genome retrieves the lost sequences and genes during domestication and selection. Genome Biol. 2021, 22, 119. [Google Scholar] [CrossRef]
- Zhou, P.; Silverstein, K.A.T.; Ramaraj, T.; Guhlin, J.; Denny, R.; Liu, J.; Farmer, A.D.; Steele, K.P.; Stupar, R.M.; Miller, J.R.; et al. Exploring structural variation and gene family architecture with De Novo assemblies of 15 Medicago genomes. BMC Genom. 2017, 18, 261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayer, P.E.; Golicz, A.A.; Tirnaz, S.; Chan, C.-K.K.; Edwards, D.; Batley, J. Variation in abundance of predicted resistance genes in the Brassica oleracea pangenome. Plant Biotechnol. J. 2019, 17, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Bayer, P.E.; Valliyodan, B.; Hu, H.; Marsh, J.I.; Yuan, Y.; Vuong, T.D.; Patil, G.; Song, Q.; Batley, J.; Varshney, R.K.; et al. Sequencing the USDA core soybean collection reveals gene loss during domestication and breeding. Plant Genome 2021, e20109. [Google Scholar] [CrossRef] [PubMed]
- Ali, Z.; Mahfouz, M.M.; Mansoor, S. CRISPR-TSKO: A Tool for Tissue-Specific Genome Editing in Plants. Trends Plant Sci. 2020, 25, 123–126. [Google Scholar] [CrossRef]
- Li, Q.; Wu, G.; Zhao, Y.; Wang, B.; Zhao, B.; Kong, D.; Wei, H.; Chen, C.; Wang, H. CRISPR/Cas9-mediated knockout and overexpression studies reveal a role of maize phytochrome C in regulating flowering time and plant height. Plant Biotechnol. J. 2020, 18, 2520–2532. [Google Scholar] [CrossRef]
- Ueta, R.; Abe, C.; Watanabe, T.; Sugano, S.S.; Ishihara, R.; Ezura, H.; Osakabe, Y.; Osakabe, K. Rapid breeding of parthenocarpic tomato plants using CRISPR/Cas9. Sci. Rep. 2017, 7, 507. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, Y.; Chen, S.; Tian, H.; Fu, D.; Zhu, B.; Luo, Y.; Zhu, H. Lycopene Is Enriched in Tomato Fruit by CRISPR/Cas9-Mediated Multiplex Genome Editing. Front. Plant Sci. 2018, 9, 559. [Google Scholar] [CrossRef]
- Yu, Q.-h.; Wang, B.; Li, N.; Tang, Y.; Yang, S.; Yang, T.; Xu, J.; Guo, C.; Yan, P.; Wang, Q.; et al. CRISPR/Cas9-induced Targeted Mutagenesis and Gene Replacement to Generate Long-shelf Life Tomato Lines. Sci. Rep. 2017, 7, 11874. [Google Scholar] [CrossRef]
- Zhang, N.; Roberts, H.M.; Van Eck, J.; Martin, G.B. Generation and Molecular Characterization of CRISPR/Cas9-Induced Mutations in 63 Immunity-Associated Genes in Tomato Reveals Specificity and a Range of Gene Modifications. Front. Plant Sci. 2020, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decaestecker, W.; Buono, R.A.; Pfeiffer, M.L.; Vangheluwe, N.; Jourquin, J.; Karimi, M.; Van Isterdael, G.; Beeckman, T.; Nowack, M.K.; Jacobs, T.B. CRISPR-TSKO: A Technique for Efficient Mutagenesis in Specific Cell Types, Tissues, or Organs in Arabidopsis. Plant Cell 2019, 31, 2868–2887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Zhang, Q.; Zhu, Q.; Liu, W.; Chen, Y.; Qiu, R.; Wang, B.; Yang, Z.; Li, H.; Lin, Y.; et al. A Robust CRISPR/Cas9 System for Convenient, High-Efficiency Multiplex Genome Editing in Monocot and Dicot Plants. Mol. Plant 2015, 8, 1274–1284. [Google Scholar] [CrossRef]
- Borrill, P.; Harrington, S.A.; Uauy, C. Applying the latest advances in genomics and phenomics for trait discovery in polyploid wheat. Plant J. 2019, 97, 56–72. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Sapkota, M.; van der Knaap, E. Perspectives of CRISPR/Cas-mediated cis-engineering in horticulture: Unlocking the neglected potential for crop improvement. Hortic. Res. 2020, 7, 36. [Google Scholar] [CrossRef] [Green Version]
- Gruner, K.; Esser, T.; Acevedo-Garcia, J.; Freh, M.; Habig, M.; Strugala, R.; Stukenbrock, E.; Schaffrath, U.; Panstruga, R. Evidence for Allele-Specific Levels of Enhanced Susceptibility of Wheat mlo Mutants to the Hemibiotrophic Fungal Pathogen Magnaporthe oryzae pv. Triticum. Genes 2020, 11, 517. [Google Scholar] [CrossRef]
- Consonni, C.; Humphry, M.E.; Hartmann, H.A.; Livaja, M.; Durner, J.; Westphal, L.; Vogel, J.; Lipka, V.; Kemmerling, B.; Schulze-Lefert, P.; et al. Conserved requirement for a plant host cell protein in powdery mildew pathogenesis. Nat. Genet. 2006, 38, 716–720. [Google Scholar] [CrossRef]
- Tian, D.; Chen, Z.; Chen, Z.; Zhou, Y.; Wang, Z.; Wang, F.; Chen, S. Allele-specific marker-based assessment revealed that the rice blast resistance genes Pi2 and Pi9 have not been widely deployed in Chinese indica rice cultivars. Rice 2016, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Chai, L.; Zhang, J.; Fernando, W.G.D.; Li, H.; Huang, X.; Cui, C.; Jiang, J.; Zheng, B.; Liu, Y.; Jiang, L. Detection of Blackleg Resistance Gene Rlm1 in Double-Low Rapeseed Accessions from Sichuan Province, by Kompetitive Allele-Specific PCR. Plant Pathol. J. 2021, 37, 194–199. [Google Scholar] [CrossRef]
- Van Bezouw, R.F.H.M.; Janssen, E.M.; Ashrafuzzaman, M.; Ghahramanzadeh, R.; Kilian, B.; Graner, A.; Visser, R.G.F.; van der Linden, C.G. Shoot sodium exclusion in salt stressed barley (Hordeum vulgare L.) is determined by allele specific increased expression of HKT1;5. J. Plant Physiol. 2019, 241, 153029. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Lin, J.; Li, Z.; Lin, Z.; Ma, Y.; Wang, Y.; Ming, R. Allele specific expression of Dof genes responding to hormones and abiotic stresses in sugarcane. PLoS ONE 2020, 15, e0227716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacomini, D.A.; Patterson, E.L.; Küpper, A.; Beffa, R.; Gaines, T.A.; Tranel, P.J. Coexpression Clusters and Allele-Specific Expression in Metabolism-Based Herbicide Resistance. Genome Biol. Evol. 2020, 12, 2267–2278. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wen, W.; Liu, J.; Zhang, Y.; Cao, S.; He, Z.; Rasheed, A.; Jin, H.; Zhang, C.; Yan, J.; et al. Genetic architecture of grain yield in bread wheat based on genome-wide association studies. BMC Plant Biol. 2019, 19, 168. [Google Scholar] [CrossRef]
- Morineau, C.; Bellec, Y.; Tellier, F.; Gissot, L.; Kelemen, Z.; Nogué, F.; Faure, J.-D. Selective gene dosage by CRISPR-Cas9 genome editing in hexaploid Camelina sativa. Plant Biotechnol. J. 2017, 15, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Schaart, J.G.; van de Wiel, C.C.M.; Smulders, M.J.M. Genome editing of polyploid crops: Prospects, achievements and bottlenecks. Transgenic Res. 2021, 30, 337–351. [Google Scholar] [CrossRef]
- Zaman, Q.U.; Li, C.; Cheng, H.; Hu, Q. Genome editing opens a new era of genetic improvement in polyploid crops. Crop J. 2019, 7, 141–150. [Google Scholar] [CrossRef]
- Schwartz, C.; Lenderts, B.; Feigenbutz, L.; Barone, P.; Llaca, V.; Fengler, K.; Svitashev, S. CRISPR–Cas9-mediated 75.5-Mb inversion in maize. Nat. Plants 2020, 6, 1427–1431. [Google Scholar] [CrossRef]
- Huang, J.; Li, J.; Zhou, J.; Wang, L.; Yang, S.; Hurst, L.D.; Li, W.-H.; Tian, D. Identifying a large number of high-yield genes in rice by pedigree analysis, whole-genome sequencing, and CRISPR-Cas9 gene knockout. Proc. Natl. Acad. Sci. USA 2018, 115, E7559. [Google Scholar] [CrossRef] [Green Version]
- Matchett-Oates, L.; Braich, S.; Spangenberg, G.C.; Rochfort, S.; Cogan, N.O.I. In silico analysis enabling informed design for genome editing in medicinal cannabis; gene families and variant characterisation. PLoS ONE 2021, 16, e0257413. [Google Scholar] [CrossRef]
- Tang, S.; Zhao, H.; Lu, S.; Yu, L.; Zhang, G.; Zhang, Y.; Yang, Q.-Y.; Zhou, Y.; Wang, X.; Ma, W.; et al. Genome- and transcriptome-wide association studies provide insights into the genetic basis of natural variation of seed oil content in Brassica napus. Mol. Plant 2021, 14, 470–487. [Google Scholar] [CrossRef] [PubMed]
- Martina, M.; Tikunov, Y.; Portis, E.; Bovy, A.G. The Genetic Basis of Tomato Aroma. Genes 2021, 12, 226. [Google Scholar] [CrossRef] [PubMed]
- Hirschhorn, J.N.; Daly, M.J. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 2005, 6, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Li, G.; Zhao, H.; Wang, G.; Lian, X.; Xie, W. Exploring the rice dispensable genome using a metagenome-like assembly strategy. Genome Biol. 2015, 16, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, H.-L.; Dong, L.; Wang, Z.-P.; Zhang, H.-Y.; Han, C.-Y.; Liu, B.; Wang, X.-C.; Chen, Q.-J. A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol. 2014, 14, 327. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Ye, X.; Guo, R.; Huang, J.; Wang, W.; Tang, J.; Tan, L.; Zhu, J.K.; Chu, C.; Qian, Y. Genome-wide Targeted Mutagenesis in Rice Using the CRISPR/Cas9 System. Mol. Plant 2017, 10, 1242–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.; Yu, H.; Zhang, Y.; Zhuang, F.; Song, X.; Gao, S.; Gao, C.; Li, J. Construction of a Genome-Wide Mutant Library in Rice Using CRISPR/Cas9. Mol. Plant 2017, 10, 1238–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Hao, M.; Wang, W.; Wang, H.; Chen, F.; Chu, W.; Zhang, B.; Mei, D.; Cheng, H.; Hu, Q. An Efficient CRISPR/Cas9 Platform for Rapidly Generating Simultaneous Mutagenesis of Multiple Gene Homoeologs in Allotetraploid Oilseed Rape. Front. Plant Sci. 2018, 9, 442. [Google Scholar] [CrossRef]
- Bai, M.; Yuan, J.; Kuang, H.; Gong, P.; Li, S.; Zhang, Z.; Liu, B.; Sun, J.; Yang, M.; Yang, L.; et al. Generation of a multiplex mutagenesis population via pooled CRISPR-Cas9 in soya bean. Plant Biotechnol. J. 2020, 18, 721–731. [Google Scholar] [CrossRef] [Green Version]
- Findlay, G.M.; Daza, R.M.; Martin, B.; Zhang, M.D.; Leith, A.P.; Gasperini, M.; Janizek, J.D.; Huang, X.; Starita, L.M.; Shendure, J. Accurate classification of BRCA1 variants with saturation genome editing. Nature 2018, 562, 217–222. [Google Scholar] [CrossRef]
- Hashimoto, R.; Ueta, R.; Abe, C.; Osakabe, Y.; Osakabe, K. Efficient Multiplex Genome Editing Induces Precise, and Self-Ligated Type Mutations in Tomato Plants. Front. Plant Sci. 2018, 9, 916. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Huang, H.-H.; Jimenez, J.; Del Vecchio, D. Resource Competition Shapes the Response of Genetic Circuits. ACS Synth. Biol. 2017, 6, 1263–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Voigt, C.A. Engineered dCas9 with reduced toxicity in bacteria: Implications for genetic circuit design. Nucleic Acids Res. 2018, 46, 11115–11125. [Google Scholar] [CrossRef] [Green Version]
- McCarty, N.S.; Graham, A.E.; Studená, L.; Ledesma-Amaro, R. Multiplexed CRISPR technologies for gene editing and transcriptional regulation. Nat. Commun. 2020, 11, 1281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-H.; Tee, L.Y.; Wang, X.-G.; Huang, Q.-S.; Yang, S.-H. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef]
- Xu, K.; Xu, X.; Fukao, T.; Canlas, P.; Maghirang-Rodriguez, R.; Heuer, S.; Ismail, A.M.; Bailey-Serres, J.; Ronald, P.C.; Mackill, D.J. Sub1A is an ethylene-response-factor-like gene that confers submergence tolerance to rice. Nature 2006, 442, 705–708. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Tu, M.; Wang, Y.; Yin, W.; Zhang, Y.; Wu, H.; Gu, Y.; Li, Z.; Xi, Z.; Wang, X. Whole-genome sequencing reveals rare off-target mutations in CRISPR/Cas9-edited grapevine. Hortic. Res. 2021, 8, 114. [Google Scholar] [CrossRef]
- Li, J.; Manghwar, H.; Sun, L.; Wang, P.; Wang, G.; Sheng, H.; Zhang, J.; Liu, H.; Qin, L.; Rui, H.; et al. Whole genome sequencing reveals rare off-target mutations and considerable inherent genetic or/and somaclonal variations in CRISPR/Cas9-edited cotton plants. Plant Biotechnol. J. 2019, 17, 858–868. [Google Scholar] [CrossRef]
- Grohmann, L.; Keilwagen, J.; Duensing, N.; Dagand, E.; Hartung, F.; Wilhelm, R.; Bendiek, J.; Sprink, T. Detection and Identification of Genome Editing in Plants: Challenges and Opportunities. Front. Plant Sci. 2019, 10, 236. [Google Scholar] [CrossRef] [Green Version]
- Golicz, A.A.; Batley, J.; Edwards, D. Towards plant pangenomics. Plant Biotechnol. J. 2016, 14, 1099–1105. [Google Scholar] [CrossRef]
- Schouten, H.J.; Tikunov, Y.; Verkerke, W.; Finkers, R.; Bovy, A.; Bai, Y.; Visser, R.G.F. Breeding Has Increased the Diversity of Cultivated Tomato in The Netherlands. Front. Plant Sci. 2019, 10, 1606. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Jiang, Y.; Wang, Z.; Gou, Z.; Lyu, J.; Li, W.; Yu, Y.; Shu, L.; Zhao, Y.; Ma, Y.; et al. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 2015, 33, 408–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warschefsky, E.; Penmetsa, R.V.; Cook, D.R.; von Wettberg, E.J.B. Back to the wilds: Tapping evolutionary adaptations for resilient crops through systematic hybridization with crop wild relatives. Am. J. Bot. 2014, 101, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Krattinger, S.G.; Keller, B. Molecular genetics and evolution of disease resistance in cereals. New Phytol. 2016, 212, 320–332. [Google Scholar] [CrossRef] [Green Version]
- Sakai, H.; Itoh, T. Massive gene losses in Asian cultivated rice unveiled by comparative genome analysis. BMC Genom. 2010, 11, 121. [Google Scholar] [CrossRef] [Green Version]
- Mbuvi, D.; Masiga, C.; Kuria, E.; Masanga, J.; Wamalwa, M.; Mohamed, A.; Odeny, D.; Hamza, N.; Timko, M.; Runo, S. Novel Sources of Witchweed (Striga) Resistance from Wild Sorghum Accessions. Front. Plant Sci. 2017, 8, 116. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Mezmouk, S.; Baumgarten, A.; Buckler, E.S.; Guill, K.E.; McMullen, M.D.; Mumm, R.H.; Ross-Ibarra, J. Incomplete dominance of deleterious alleles contributes substantially to trait variation and heterosis in maize. PLoS Genet. 2017, 13, e1007019. [Google Scholar] [CrossRef] [Green Version]
- Soyk, S.; Müller, N.A.; Park, S.J.; Schmalenbach, I.; Jiang, K.; Hayama, R.; Zhang, L.; Van Eck, J.; Jiménez-Gómez, J.M.; Lippman, Z.B. Variation in the flowering gene SELF PRUNING 5G promotes day-neutrality and early yield in tomato. Nat. Genet. 2017, 49, 162–168. [Google Scholar] [CrossRef]
- Khan, A.W.; Garg, V.; Roorkiwal, M.; Golicz, A.A.; Edwards, D.; Varshney, R.K. Super-Pangenome by Integrating the Wild Side of a Species for Accelerated Crop Improvement. Trends Plant. Sci. 2020, 25, 148–158. [Google Scholar] [CrossRef] [Green Version]
- Scheben, A.; Edwards, D. Bottlenecks for genome-edited crops on the road from lab to farm. Genome Biol. 2018, 19, 178. [Google Scholar] [CrossRef] [Green Version]
- Østerberg, J.T.; Xiang, W.; Olsen, L.I.; Edenbrandt, A.K.; Vedel, S.E.; Christiansen, A.; Landes, X.; Andersen, M.M.; Pagh, P.; Sandøe, P.; et al. Accelerating the Domestication of New Crops: Feasibility and Approaches. Trends Plant Sci. 2017, 22, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Komatsuda, T.; Pourkheirandish, M.; He, C.; Azhaguvel, P.; Kanamori, H.; Perovic, D.; Stein, N.; Graner, A.; Wicker, T.; Tagiri, A.; et al. Six-rowed barley originated from a mutation in a homeodomain-leucine zipper I-class homeobox gene. Proc. Natl. Acad. Sci. USA 2007, 104, 1424–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Nussbaum-Wagler, T.; Li, B.; Zhao, Q.; Vigouroux, Y.; Faller, M.; Bomblies, K.; Lukens, L.; Doebley, J.F. The origin of the naked grains of maize. Nature 2005, 436, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Huang, W.; Gao, J.-P.; Yang, J.; Shi, M.; Zhu, M.-Z.; Luo, D.; Lin, H.-X. Genetic control of rice plant architecture under domestication. Nat. Genet. 2008, 40, 1365–1369. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Li, X.; Shannon, L.M.; Yeh, C.-T.; Wang, M.L.; Bai, G.; Peng, Z.; Li, J.; Trick, H.N.; Clemente, T.E.; et al. Parallel domestication of the Shattering1 genes in cereals. Nat. Genet. 2012, 44, 720–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.-Y.; Cong, B.; Wing, R.; Vrebalov, J.; Tanksley, S.D. Changes in Regulation of a Transcription Factor lead to Autogamy in Cultivated Tomatoes. Science 2007, 318, 643. [Google Scholar] [CrossRef]
- Zsögön, A.; Cermak, T.; Voytas, D.; Peres, L.E.P. Genome editing as a tool to achieve the crop ideotype and de novo domestication of wild relatives: Case study in tomato. Plant Sci. 2017, 256, 120–130. [Google Scholar] [CrossRef]
- Khan, M.Z.; Zaidi, S.S.-e.-A.; Amin, I.; Mansoor, S. A CRISPR Way for Fast-Forward Crop Domestication. Trends Plant Sci. 2019, 24, 293–296. [Google Scholar] [CrossRef]
- Zsögön, A.; Čermák, T.; Naves, E.R.; Notini, M.M.; Edel, K.H.; Weinl, S.; Freschi, L.; Voytas, D.F.; Kudla, J.; Peres, L.E.P. De novo domestication of wild tomato using genome editing. Nat. Biotechnol. 2018, 36, 1211–1216. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Yang, X.; Yu, Y.; Si, X.; Zhai, X.; Zhang, H.; Dong, W.; Gao, C.; Xu, C. Domestication of wild tomato is accelerated by genome editing. Nat. Biotechnol. 2018, 36, 1160–1163. [Google Scholar] [CrossRef]
- Sedbrook, J.C.; Phippen, W.B.; Marks, M.D. New approaches to facilitate rapid domestication of a wild plant to an oilseed crop: Example pennycress (Thlaspi arvense L.). Plant Sci. 2014, 227, 122–132. [Google Scholar] [CrossRef]
- Shapter, F.M.; Cross, M.; Ablett, G.; Malory, S.; Chivers, I.H.; King, G.J.; Henry, R.J. High-Throughput Sequencing and Mutagenesis to Accelerate the Domestication of Microlaena stipoides as a New Food Crop. PLoS ONE 2013, 8, e82641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Merrick, P.; Zhang, Z.; Ji, C.; Yang, B.; Fei, S.-Z. Targeted mutagenesis in tetraploid switchgrass (Panicum virgatum L.) using CRISPR/Cas9. Plant Biotechnol. J. 2018, 16, 381–393. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Lin, Y.; Chen, S.; Liu, H.; Chen, Z.; Fan, M.; Hu, T.; Mei, F.; Chen, J.; Chen, L.; et al. CRISPR/Cas9-mediated functional recovery of the recessive rc allele to develop red rice. Plant Biotechnol. J. 2019, 17, 2096–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Wang, K.; Tang, H.; Gong, Q.; Du, L.; Pei, X.; Ye, X. CRISPR/Cas9 editing of wheat TaQ genes alters spike morphogenesis and grain threshability. J. Genet. Genom. 2020, 47, 563–575. [Google Scholar] [CrossRef]
- Zhang, Y.; Thomas, W.; Bayer, P.E.; Edwards, D.; Batley, J. Frontiers in Dissecting and Managing Brassica Diseases: From Reference-Based RGA Candidate Identification to Building Pan-RGAomes. Int. J. Mol. Sci. 2020, 21, 8964. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Y.; Zhu, J.-K. Developing naturally stress-resistant crops for a sustainable agriculture. Nat. Plants 2018, 4, 989–996. [Google Scholar] [CrossRef]
- Kole, C. Wild Crop Relatives: Genomic and Breeding Resources: Cereals; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2011; Volume 1. [Google Scholar]
- Smýkal, P.; Nelson, M.N.; Berger, J.D.; Von Wettberg, E.J. The impact of genetic changes during crop domestication. Agronomy 2018, 8, 119. [Google Scholar] [CrossRef] [Green Version]
- Pinosio, S.; Giacomello, S.; Faivre-Rampant, P.; Taylor, G.; Jorge, V.; Le Paslier, M.C.; Zaina, G.; Bastien, C.; Cattonaro, F.; Marroni, F.; et al. Characterization of the Poplar Pan-Genome by Genome-Wide Identification of Structural Variation. Mol. Biol. Evol. 2016, 33, 2706–2719. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Zhu, W.; Diao, S.; Wu, X.; Lu, J.; Ding, C.; Su, X. The poplar pangenome provides insights into the evolutionary history of the genus. Commun. Biol. 2019, 2, 215. [Google Scholar] [CrossRef]
- Rijzaani, H.; Bayer, P.E.; Rouard, M.; Doležel, J.; Batley, J.; Edwards, D. The pangenome of banana highlights differences between genera and genomes. Plant Genome 2021, 11, e20100. [Google Scholar] [CrossRef]
- Sharwood, R.E. Engineering chloroplasts to improve Rubisco catalysis: Prospects for translating improvements into food and fiber crops. New Phytol. 2017, 213, 494–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franks, S.J.; Hoffmann, A.A. Genetics of Climate Change Adaptation. Annu. Rev. Genet. 2012, 46, 185–208. [Google Scholar] [CrossRef] [PubMed]
- Lambers, H.; Martinoia, E.; Renton, M. Plant adaptations to severely phosphorus-impoverished soils. Curr. Opin. Plant Biol. 2015, 25, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Cole, M.B.; Augustin, M.A.; Robertson, M.J.; Manners, J.M. The science of food security. Npj Sci. Food 2018, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Hess, G.T.; Frésard, L.; Han, K.; Lee, C.H.; Li, A.; Cimprich, K.A.; Montgomery, S.B.; Bassik, M.C. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat. Methods 2016, 13, 1036–1042. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.; Joshi, R.K.; Zhao, K. Base editing in crops: Current advances, limitations and future implications. Plant Biotechnol. J. 2020, 18, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Dong, O.X.; Yu, S.; Jain, R.; Zhang, N.; Duong, P.Q.; Butler, C.; Li, Y.; Lipzen, A.; Martin, J.A.; Barry, K.W.; et al. Marker-free carotenoid-enriched rice generated through targeted gene insertion using CRISPR-Cas9. Nat. Commun. 2020, 11, 1178. [Google Scholar] [CrossRef] [Green Version]
- Hoppe, C.; Ashe, H.L. CRISPR-Cas9 strategies to insert MS2 stem-loops into endogenous loci in Drosophila embryos. STAR Protoc. 2021, 2, 100380. [Google Scholar] [CrossRef]
- Lu, Y.; Tian, Y.; Shen, R.; Yao, Q.; Wang, M.; Chen, M.; Dong, J.; Zhang, T.; Li, F.; Lei, M.; et al. Targeted, efficient sequence insertion and replacement in rice. Nat. Biotechnol. 2020, 38, 1402–1407. [Google Scholar] [CrossRef] [PubMed]
- Poernbacher, I.; Crossman, S.; Kurth, J.; Nojima, H.; Baena-Lopez, A.; Alexandre, C.; Vincent, J.-P. Lessons in genome engineering: Opportunities, tools and pitfalls. bioRxiv 2019, 710871. [Google Scholar] [CrossRef]
- Hendriks, D.; Clevers, H.; Artegiani, B. CRISPR-Cas Tools and Their Application in Genetic Engineering of Human Stem Cells and Organoids. Cell Stem Cell 2020, 27, 705–731. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
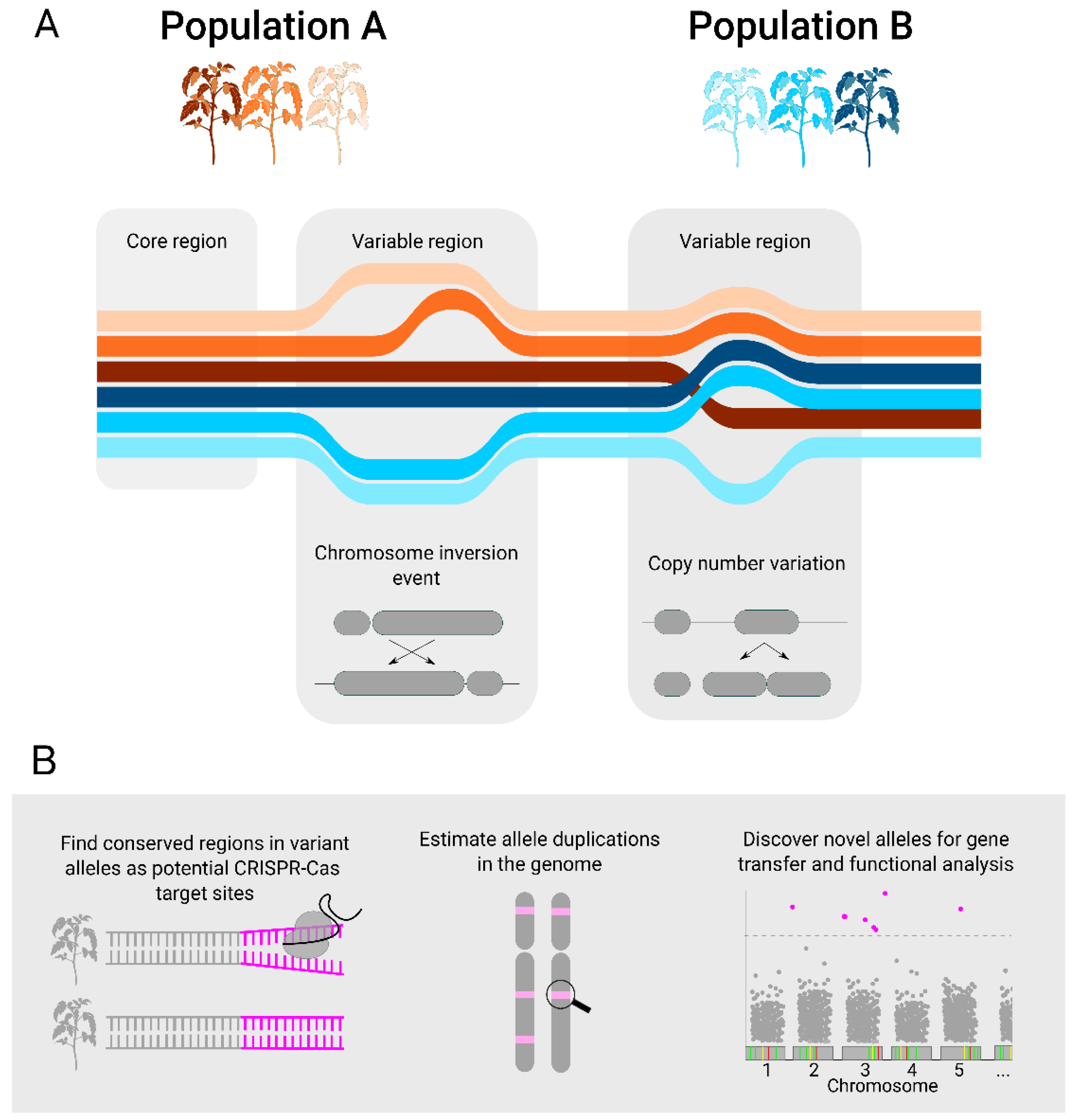{kind=link}
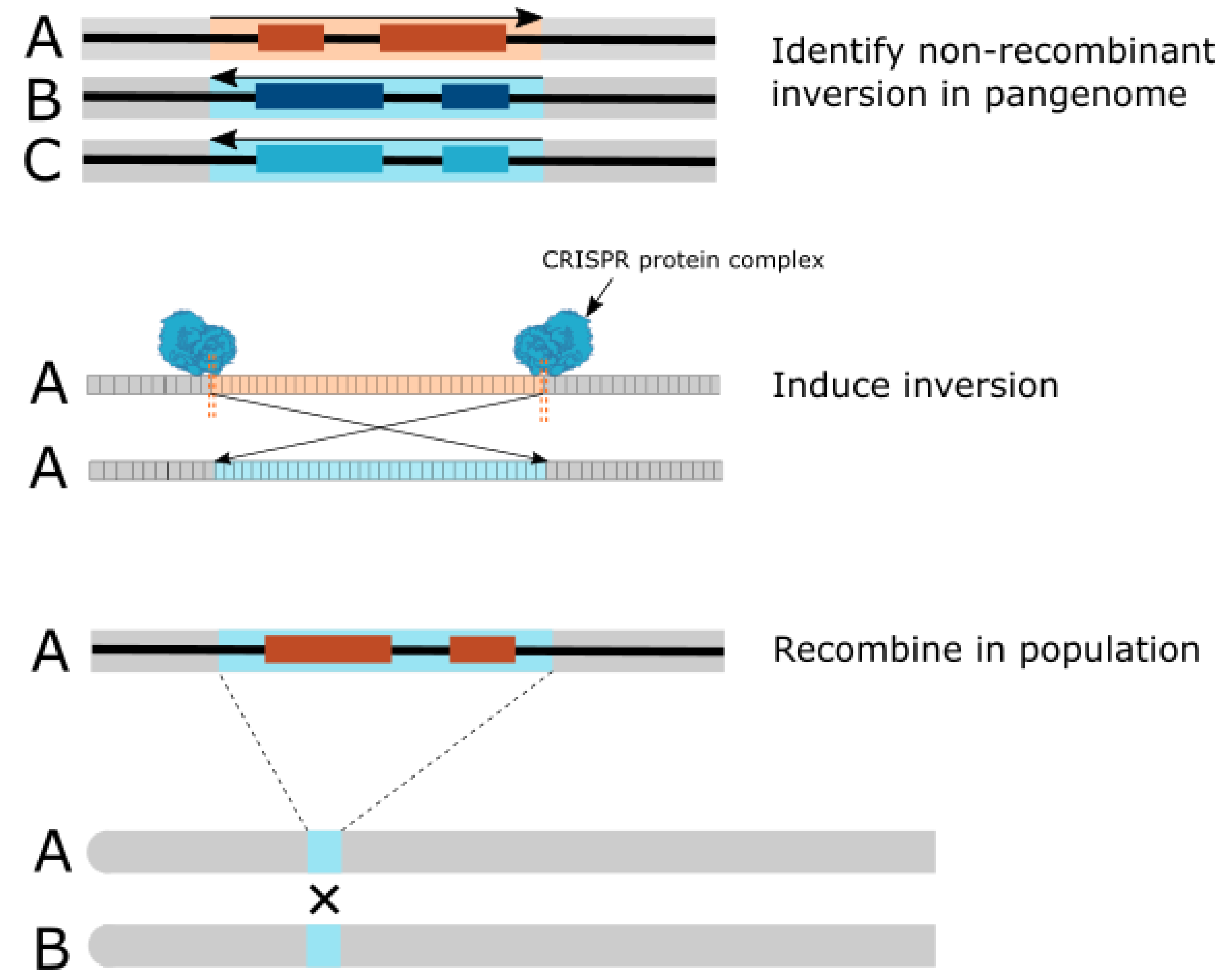{kind=link}
| Species Name | Accessions | Size (Individuals) | Analysis | Reference |
|---|---|---|---|---|
| Glycine max | USDA collection | 1110 | PAV, GO, SNP discovery and population genetics analysis | [68] |
| Chinese population | 26 | Synteny, SV, genetic variation and gene expression analysis | [50] | |
| Zea mays | USDA Collection | 14,129 | GBS tagging, GWAS mapping, and PAV analysis | [51] |
| Solanum lycopersicum | NCBI SRA database | 725 | SNP calling, QTL mapping, expression analysis, and PAV analysis | [35] |
| Brassica oleracea | Chinese Kale/TO100 | 10 | Gene clustering, TE annotation, SNP calling, phylogenetic, PAV, and GO analysis | [39] |
| Brassica napus | Diversity set | 8 | Phylogenetic, SNP, InDels, SV, PAV, population analysis | [27] |
| Diversity set | 53 | Candidate identification, QTL, and SNP analysis | [52] | |
| Brassica distachyon | Diversity set | 54 | Pan-gene clustering, variant calling, TE, and indel phylogenetic analysis | [53] |
| Hordeum vulgare | Diversity set | 20 | GWAS, inversion calling, SNP calling, QTL mapping, and PAV analysis | [54] |
| Oryza sativa | China National Rice Research Institute andNational Institute of Genetics in Japan | 1529 | Evolutionarily and PAV analysis | [55] |
| Cajanus cajan | ICRISAT | 89 | SNP and PAV analysis | [29] |
| Diversity set | 3366 | SNP, SV, CNV, phylogenetic, GWAS analysis, and genomeprediction | [56] | |
| Malus domestica | Plant Genetic Resources Unit | 91 | Gene prediction, comparative analysis, and PAV/variant calling | [57] |
| Sesamum indicum | Diversity set | 5 | PAV and evolutionary analysis | [58] |
| Helianthus annuus | USDA Collection | 493 | SNP calling, genome positioning, and GO and GWAS analysis | [59] |
| Manihot esculenta | Diversity set | 57 | Haplotype sampling and genomic prediction | [60] |
| Sorghum bicolor | Diversity set | 354 | PAV, SNP, GWAS, diversity, and population analysis | [36] |
| Chibas sorghum breeding program | 24 | Genotype prediction, haplotype sampling, and WGS | [61] | |
| Triticum aestivum | Chinese Spring | 18 | PAV and SNP analysis | [62] |
| Arabidopsis thaliana | MPI for Plant Breeding Research | 7 | Pangenomic, CNV, and synteny analysis | [63] |
| Amborella trichopoda | Ambroella Genome Project | 10 | PAV, GO, candidate gene, phylogenetic, and SNP analysis | [64] |
| Medicago truncatula | Diversity set | 15 | Comparative genomic analysis, protein orthlog, diversity, and SV analysis | [66] |
| Gossypium | NCBI database | 1961 | InDel, population structure, LD, SV, CNV, PAV, and metagenome association analysis | [65] |
| Capsicum | Diversity set | 5 | PAV and GWAS analysis | [25] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tay Fernandez, C.G.; Nestor, B.J.; Danilevicz, M.F.; Marsh, J.I.; Petereit, J.; Bayer, P.E.; Batley, J.; Edwards, D. Expanding Gene-Editing Potential in Crop Improvement with Pangenomes. Int. J. Mol. Sci. 2022, 23, 2276. https://doi.org/10.3390/ijms23042276
Tay Fernandez CG, Nestor BJ, Danilevicz MF, Marsh JI, Petereit J, Bayer PE, Batley J, Edwards D. Expanding Gene-Editing Potential in Crop Improvement with Pangenomes. International Journal of Molecular Sciences. 2022; 23(4):2276. https://doi.org/10.3390/ijms23042276
Chicago/Turabian StyleTay Fernandez, Cassandria G., Benjamin J. Nestor, Monica F. Danilevicz, Jacob I. Marsh, Jakob Petereit, Philipp E. Bayer, Jacqueline Batley, and David Edwards. 2022. "Expanding Gene-Editing Potential in Crop Improvement with Pangenomes" International Journal of Molecular Sciences 23, no. 4: 2276. https://doi.org/10.3390/ijms23042276
APA StyleTay Fernandez, C. G., Nestor, B. J., Danilevicz, M. F., Marsh, J. I., Petereit, J., Bayer, P. E., Batley, J., & Edwards, D. (2022). Expanding Gene-Editing Potential in Crop Improvement with Pangenomes. International Journal of Molecular Sciences, 23(4), 2276. https://doi.org/10.3390/ijms23042276