
Transcriptome and Coexpression Network Analyses Provide In-Sights into the Molecular Mechanisms of Hydrogen Cyanide Synthesis during Seed Development in Common Vetch (Vicia sativa L.)

 ,
,  , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
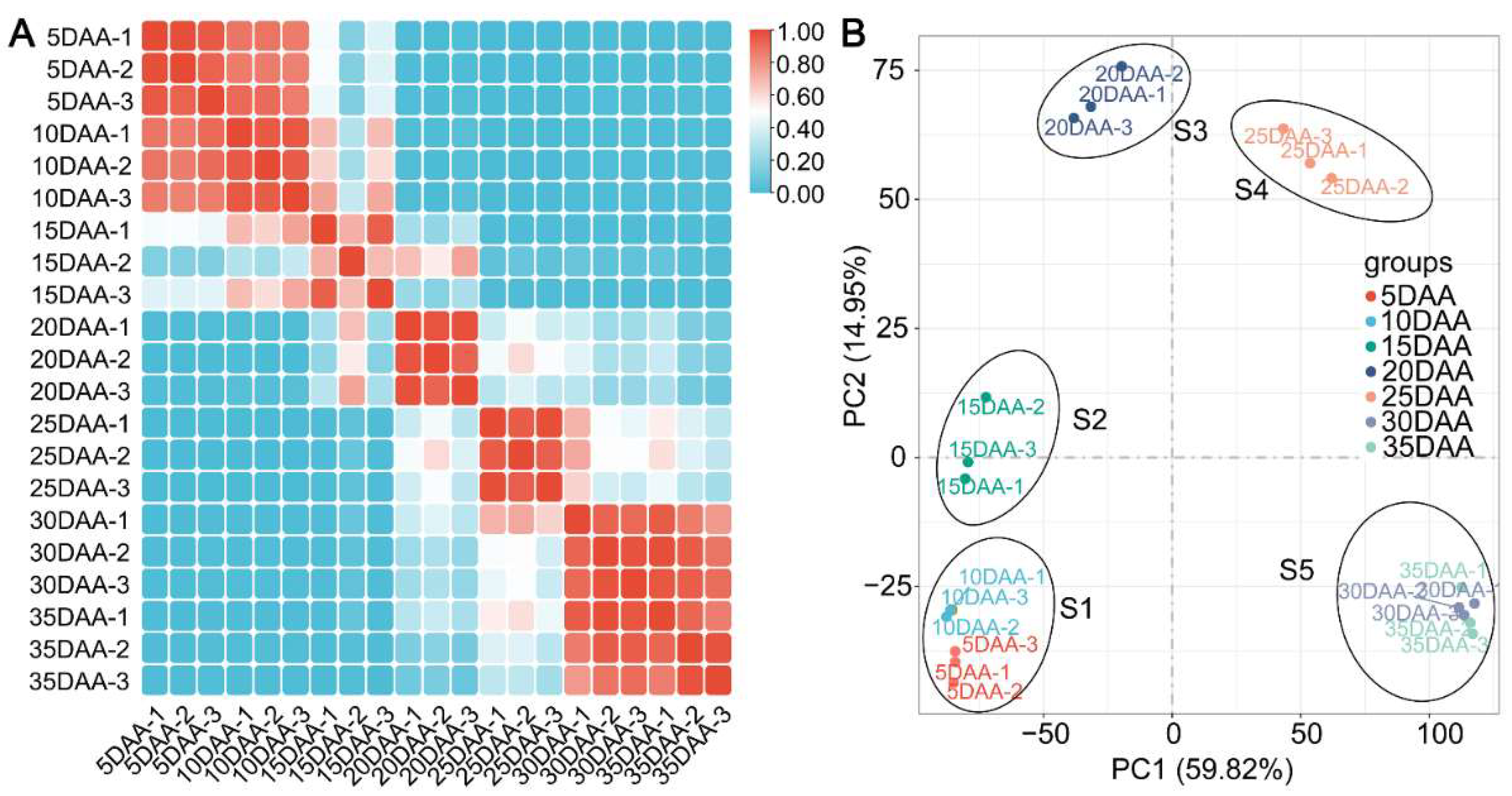
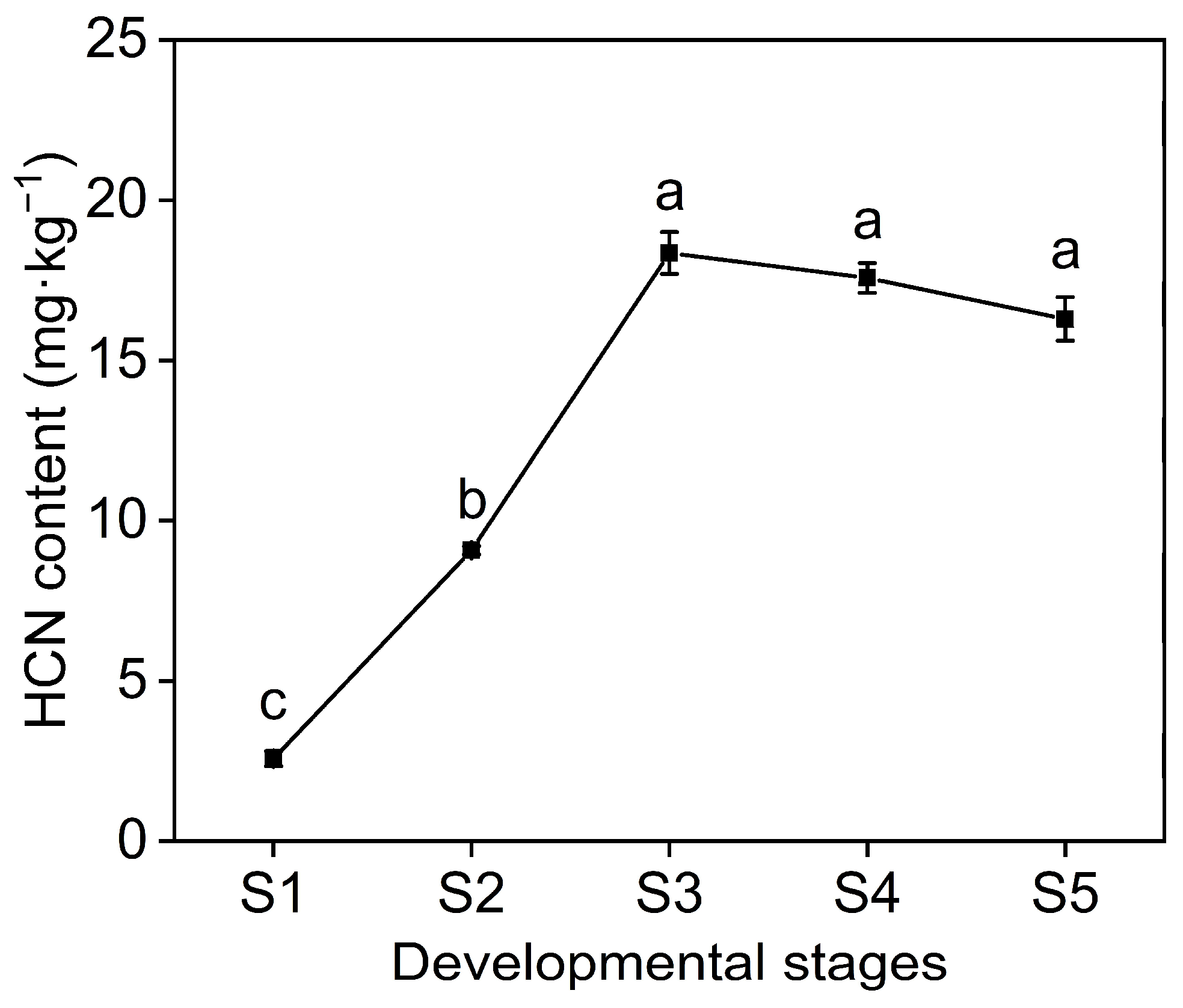
2.1. Morphological and Physiological Changes during Seed Development
2.2. Illumina Sequencing, Assembly and Functional Annotation
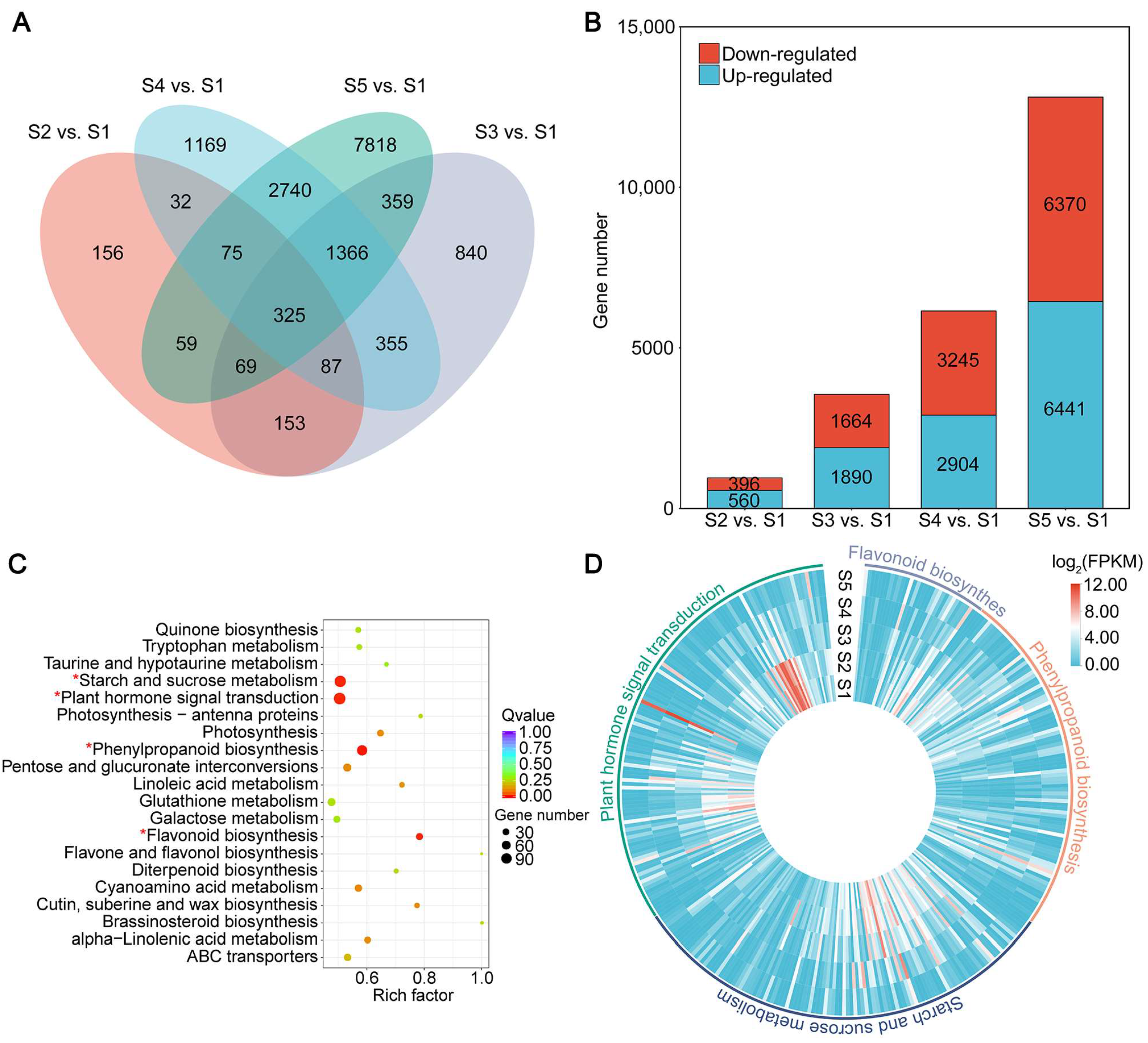
2.3. Differentially Expressed Genes during Seed Development
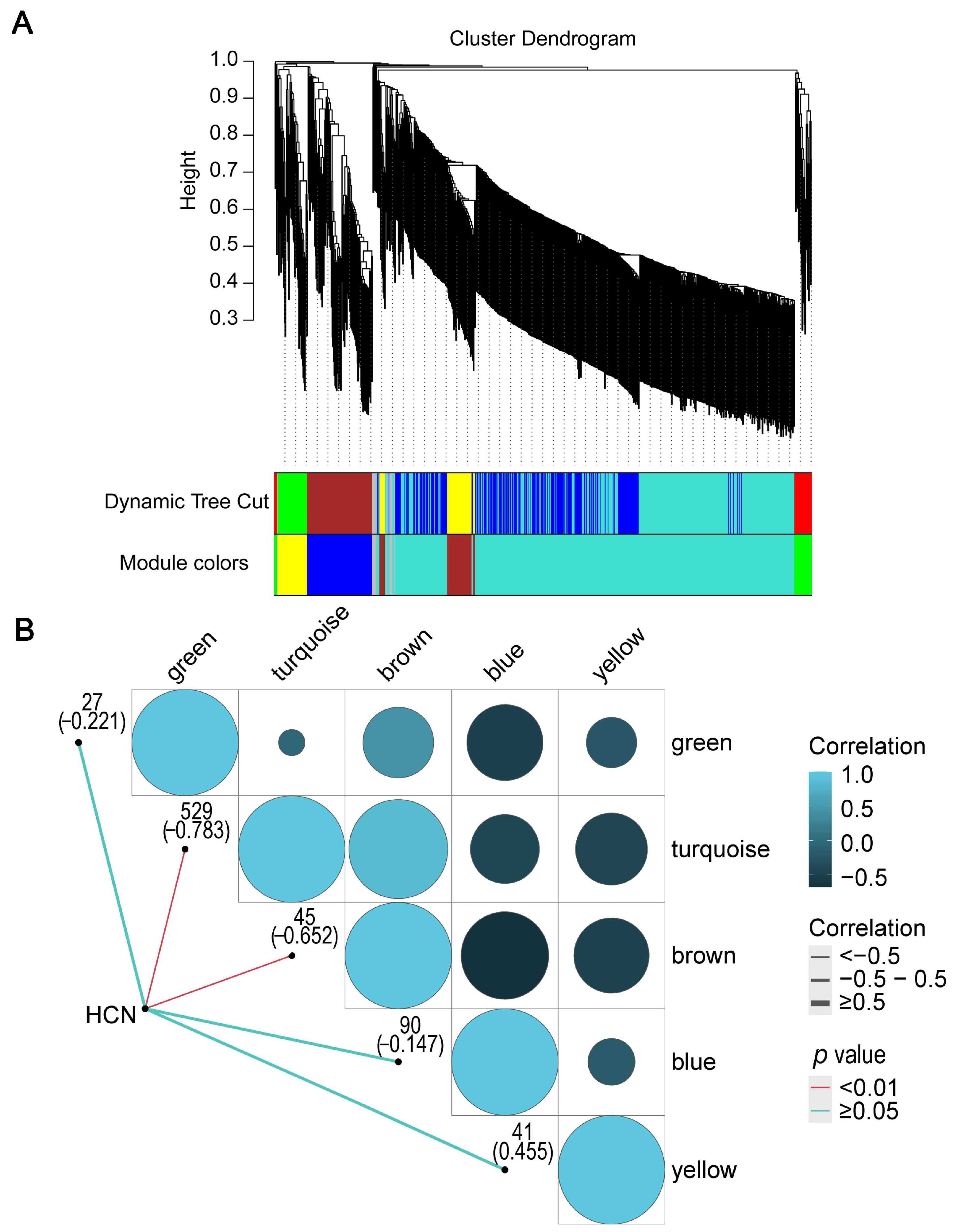
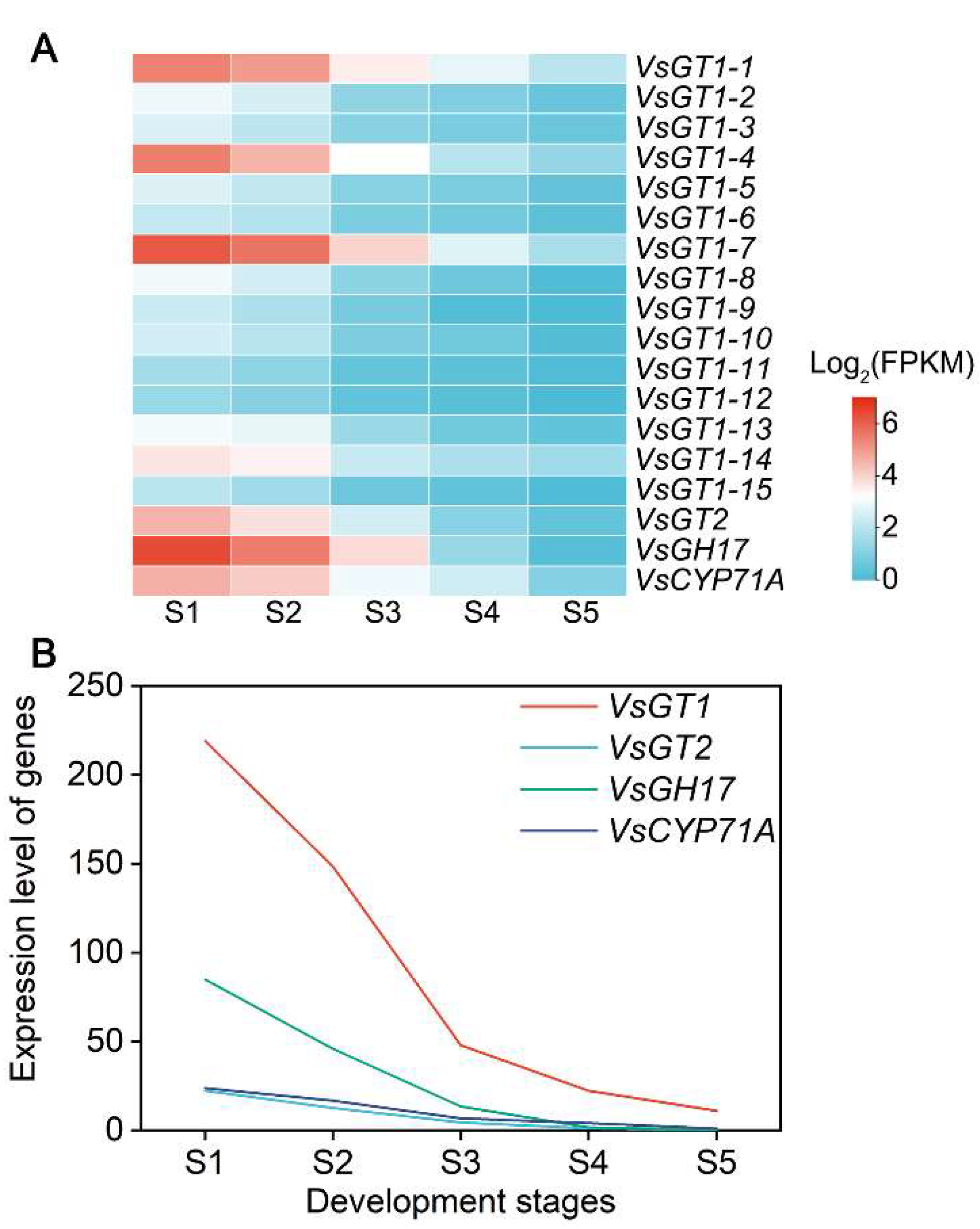
2.4. HCN Content in Seed and WGCNA of HCN Synthesis Genes
2.5. Key Regulatory Pathways for HCN Synthesis
3. Materials and Methods
3.1. Plant Materials
3.2. Morphological and Physiological Characteristics of Seeds
3.3. HCN Content Determination
3.4. RNA Sequencing and Transcriptome Analysis
3.5. Establishment of the Gene Coexpression Network
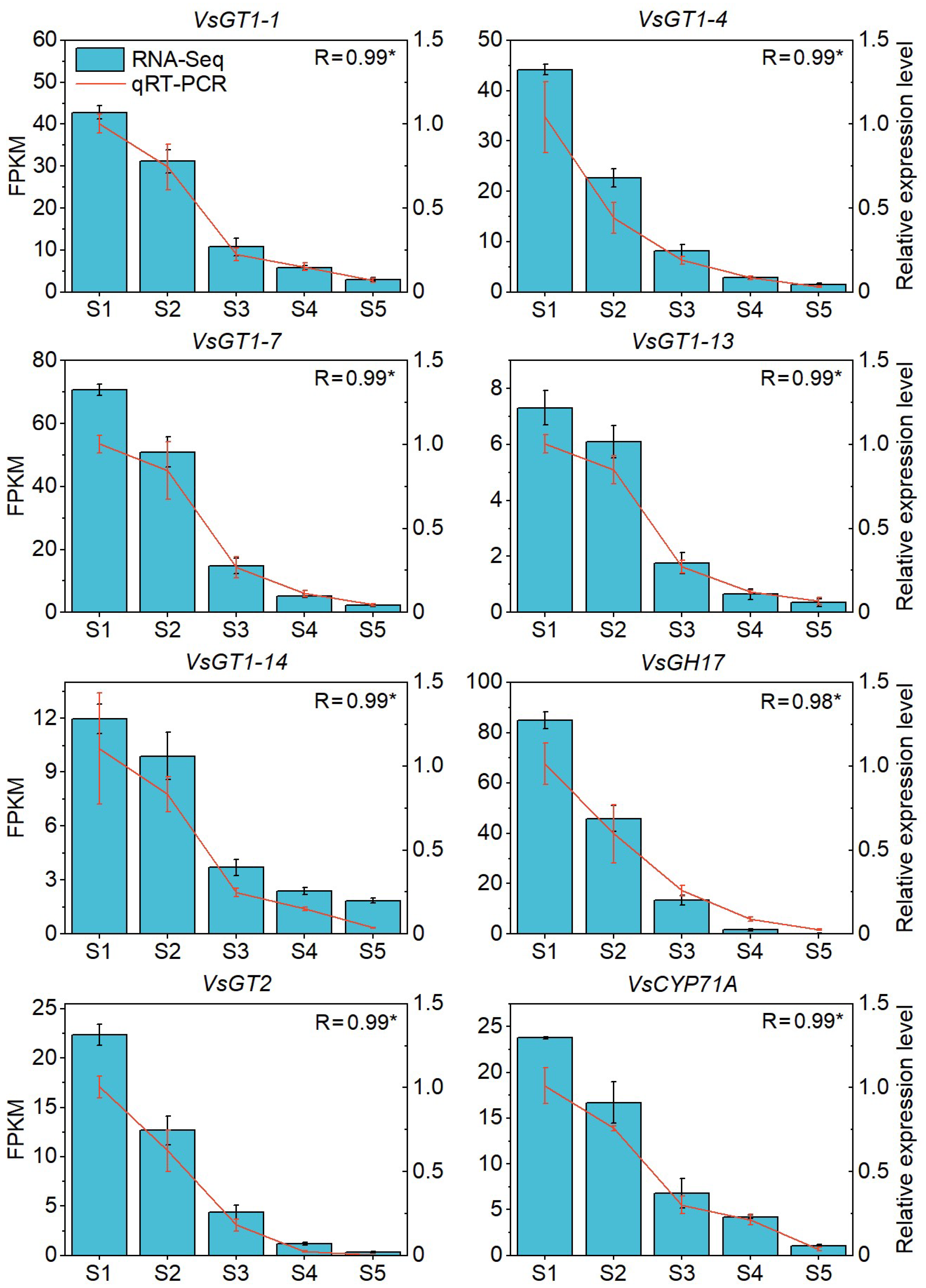
3.6. qRT–PCR Analysis
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Henchion, M.; Hayes, M.; Mullen, A.M.; Fenelon, M.; Tiwari, B. Future protein supply and demand: Strategies and factors influencing a sustainable equilibrium. Foods 2017, 6, 53. [Google Scholar] [CrossRef] [Green Version]
- Francis, C.M.; Enneking, D.; Abd El Moneim, A.M. When and where will vetches have an impact as grain legumes? In Linking Research and Marketing Opportunities for Pulses in the 21st Century; Knight, R., Ed.; Springer: Dordrecht, The Netherlands, 2000; Volume 34, pp. 375–384. [Google Scholar]
- Asgar, M.; Fazilah, A.; Huda, N.; Bhat, R.; Karim, A. Nonmeat protein alternatives as meat extenders and meat analogs. Compr. Rev. Food Sci. 2010, 9, 513–529. [Google Scholar] [CrossRef]
- Harper, J.; Arscott, G. Toxicity of common and hairy vetch seed for poults and chicks. Poult. Sci. 1962, 41, 1968–1974. [Google Scholar] [CrossRef]
- Cakmakci, S.; Aydinoglu, B.; Karaca, M.; Bilgen, M. Heritability of yield components in common vetch (Vicia sativa L.). Acta Agric. Scand. Sect. B-Soil Plant Sci. 2006, 56, 54–59. [Google Scholar]
- Shirasawa, K.; Kosugi, S.; Sasaki, K.; Ghelfi, A.; Okazaki, K.; Toyoda, A.; Hirakawa, H.; Isobe, S. Genome features of common vetch (Vicia sativa) in natural habitats. Plant Direct 2021, 5, e352. [Google Scholar] [CrossRef]
- Ribeiro, A.C.; Teixeira, A.R.; Ferreira, R.B. Characterization of globulins from common vetch (Vicia sativa L.). J. Agr. Food Chem. 2004, 52, 4913–4920. [Google Scholar] [CrossRef]
- Megías, C.; Pastor-Cavada, E.; Torres-Fuentes, C.; Girón-Calle, J.; Alaiz, M.; Juan, R.; Pastor, J.; Vioque, J. Chelating, antioxidant and antiproliferative activity of Vicia sativa polyphenol extracts. Eur. Food Res. Technol. 2009, 230, 353–359. [Google Scholar] [CrossRef]
- Uzun, A.; Gücer, S.; Acikgoz, E. Common vetch (Vicia sativa L.) germplasm: Correlations of crude protein and mineral content to seed traits. Plant Food Hum. Nutr. 2011, 66, 254–260. [Google Scholar] [CrossRef]
- Mao, Z.; Fu, H.; Nan, Z.; Wan, C. Fatty acid, amino acid, and mineral composition of four common vetch seeds on Qinghai-Tibetan plateau. Food Chem. 2015, 171, 13–18. [Google Scholar] [CrossRef]
- Saleem, M.; Karim, M.; Qadir, M.I.; Ahmed, B.; Rafiq, M.; Ahmad, B. In vitro antibacterial activity and phytochemical analysis of hexane extract of Vicia sativa. Bangl. J. Pharmacol. 2014, 9, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Ock, A.Y.; Hiromichi, S.; Masaharu, M.; Bun-Ichi, S.; Kanzo, S. Vicianin hydrolase is a novel cyanogenic β-glycosidase specific to β-vicianoside (6-O-α-l-arabinopyranosyl-β-d-glucopyranoside) in seeds of Vicia angustifolia. Plant Cell Physiol. 2007, 48, 938–947. [Google Scholar]
- Seigler, D.; Brinker, A. Characterisation of cyanogenic glycosides, cyanolipids, nitroglycosides, organic nitro compounds and nitrile glucosides from plants. In Methods in Plant Biochemistry; Waterman, P.G., Ed.; Academic Press: New York, NY, USA, 1993; Volume 8, p. 51. [Google Scholar]
- Lai, D.; Pičmanová, M.; Abou Hachem, M.; Motawia, M.S.; Olsen, C.E.; Møller, B.L.; Rook, F.; Takos, A.M. Lotus japonicus flowers are defended by a cyanogenic β-glucosidase with highly restricted expression to essential reproductive organs. Plant Mol. Biol. 2015, 89, 21–34. [Google Scholar] [CrossRef] [Green Version]
- Møller, B.; Seigler, D. Biosynthesis of cyanogenic glycosides, cyanolipids and related compounds. In Plant Amino Acids Biochemistry and Biotechnology; Singh, B.K., Ed.; CRC Press: New York, NY, USA, 1999; Volume 21, pp. 563–609. [Google Scholar]
- Adewusi, S.R. Turnover of dhurrin in green sorghum seedlings. Plant Physiol. 1990, 94, 1219–1224. [Google Scholar] [CrossRef] [Green Version]
- Cutler, A.J.; Conn, E.E. The biosynthesis of cyanogenic glucosides in Linum usitatissimum (linen flax) in vitro. Arch. Biochem. Biophys. 1981, 212, 468–474. [Google Scholar] [CrossRef]
- Sinding Hansen, K.; Kristensen, C.; Tattersall, D.B.; Jones, P.R.; Olsen, C.E. The in vitro substrate regiospecificity of recombinant UGT85B1, the cyanohydrin glucosyltransferase from Sorghum bicolor. Phytochemistry 2003, 64, 143–151. [Google Scholar] [CrossRef]
- Le, B.H.; Cheng, C.; Bui, A.Q.; Wagmaister, J.A.; Henry, K.F.; Pelletier, J.; Kwong, L.; Belmonte, M.; Kirkbride, R.; Horvath, S. Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors. Proc. Natl. Acad. Sci. USA 2010, 107, 8063–8070. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Wang, S.; He, C.; Zhou, B.; Ruan, Y.-L.; Shou, H. Identification of regulatory networks and hub genes controlling soybean seed set and size using RNA sequencing analysis. J. Exp. Bot. 2017, 68, 1955–1972. [Google Scholar] [CrossRef] [Green Version]
- Garg, R.; Singh, V.K.; Rajkumar, M.S.; Kumar, V.; Jain, M. Global transcriptome and coexpression network analyses reveal cultivar-specific molecular signatures associated with seed development and seed size/weight determination in chickpea. Plant J. 2017, 91, 1088–1107. [Google Scholar] [CrossRef] [Green Version]
- Steinbrecher, T.; Leubner-Metzger, G. The biomechanics of seed germination. J. Exp. Bot. 2017, 68, 765–783. [Google Scholar] [CrossRef]
- Fang, L.; Xu, X.; Li, J.; Zheng, F.; Li, M.; Yan, J.; Li, Y.; Zhang, X.; Li, L.; Ma, G. Transcriptome analysis provides insights into the non-methylated lignin synthesis in Paphiopedilum armeniacum seed. BMC Genom. 2020, 21, 524. [Google Scholar] [CrossRef]
- Zhang, X.-M.; Zhao, L.; Larson-Rabin, Z.; Li, D.-Z.; Guo, Z.-H. De novo sequencing and characterization of the floral transcriptome of Dendrocalamus latiflorus (Poaceae: Bambusoideae). PLoS ONE 2012, 7, e42082. [Google Scholar] [CrossRef]
- Tsanakas, G.F.; Manioudaki, M.E.; Economou, A.S.; Kalaitzis, P. De novo transcriptome analysis of petal senescence in Gardenia jasminoides Ellis. BMC Genom. 2014, 15, 554. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Deng, J.; Shi, T.; Chen, Q.; Liang, C.; Meng, Z.; Zhu, L.; Wang, Y.; Zhao, F.; Yu, S. Global transcriptome analysis and identification of genes involved in nutrients accumulation during seed development of rice tartary buckwheat (Fagopyrum Tararicum). Sci. Rep. 2017, 7, 11792. [Google Scholar] [CrossRef]
- Han, X.; Zhang, L.; Miao, X.; Hu, X.; Nan, S.; Fu, H. Transcriptome analysis reveals the molecular mechanisms of mucilage biosynthesis during Artemisia sphaerocephala seed development. Ind. Crop. Prod. 2020, 145, 111991. [Google Scholar] [CrossRef]
- Dong, R.; Dong, D.; Luo, D.; Zhou, Q.; Chai, X.; Zhang, J.; Xie, W.; Liu, W.; Dong, Y.; Wang, Y.; et al. Transcriptome analyses reveal candidate pod shattering-associated genes involved in the pod ventral sutures of common vetch (Vicia sativa L.). Front. Plant Sci. 2017, 8, 649. [Google Scholar] [CrossRef]
- Rui, H.; Zhang, X.; Shinwari, K.I.; Zheng, L.; Shen, Z. Comparative transcriptomic analysis of two Vicia sativa L. varieties with contrasting responses to cadmium stress reveals the important role of metal transporters in cadmium tolerance. Plant Soil 2018, 423, 241–255. [Google Scholar] [CrossRef]
- Xue, L.; Zhang, J.; Xue, H. Genome-wide analysis of the complex transcriptional networks of rice developing seeds. PLoS ONE 2012, 7, e31081. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zeevaart, J.A. Expression of ABA 8′-hydroxylases in relation to leaf water relations and seed development in bean. Plant J. 2006, 47, 675–686. [Google Scholar] [CrossRef]
- Finkelstein, R.R.; Gampala, S.S.; Rock, C.D. Abscisic acid signaling in seeds and seedlings. Plant Cell 2002, 14, S15–S45. [Google Scholar] [CrossRef] [Green Version]
- Walton, L.J.; Kurepin, L.V.; Yeung, E.C.; Shah, S.; Emery, R.N.; Reid, D.M.; Pharis, R.P. Ethylene involvement in silique and seed development of canola, Brassica napus L. Plant Physiol. Bioch. 2012, 58, 142–150. [Google Scholar] [CrossRef]
- Bernardi, J.; Li, Q.-B.; Gao, Y.; Zhao, Y.; Battaglia, R.; Marocco, A.; Chourey, P.S. The auxin-deficient defective kernel18 (dek18) mutation alters the expression of seed-specific biosynthetic genes in maize. J. Plant Growth Regul. 2016, 35, 770–777. [Google Scholar] [CrossRef]
- Schruff, M.C.; Spielman, M.; Tiwari, S.; Adams, S.; Fenby, N.; Scott, R.J. The AUXIN RESPONSE FACTOR 2 gene of Arabidopsis links auxin signalling, cell division, and the size of seeds and other organs. Development 2006, 133, 251–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, A.; Park, S.; Zheng-Jun, X.; Nakajima, M.; Yamaguchi, I. Immunohistochemistry of active gibberellins and gibberellin-inducible α-amylase in developing seeds of morning glory. Plant Physiol. 2002, 129, 1045–1053. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.P.; Jermakow, A.M.; Swain, S.M. Gibberellins are required for seed development and pollen tube growth in Arabidopsis. Plant Cell. 2002, 14, 3133–3147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartrina, I.; Otto, E.; Strnad, M.; Werner, T.; Schmülling, T. Cytokinin regulates the activity of reproductive meristems, flower organ size, ovule formation, and thus seed yield in Arabidopsis thaliana. Plant Cell 2011, 23, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Nougué, O.; Corbi, J.; Ball, S.G.; Manicacci, D.; Tenaillon, M.I. Molecular evolution accompanying functional divergence of duplicated genes along the plant starch biosynthesis pathway. BMC Evol. Biol. 2014, 14, 103. [Google Scholar] [CrossRef] [Green Version]
- Vogt, T. Phenylpropanoid biosynthesis. Mol. Plant. 2010, 3, 2–20. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, X.; Li, M.; Wang, N.; Qu, X.; Fan, S. Transcriptome analysis of elm (Ulmus pumila) fruit to identify phytonutrients associated genes and pathways. Forests 2019, 10, 738. [Google Scholar] [CrossRef] [Green Version]
- Nambisan, B. Strategies for elimination of cyanogens from cassava for reducing toxicity and improving food safety. Food Chem. Toxicol. 2011, 49, 690–693. [Google Scholar] [CrossRef]
- Han, X.; Zhang, L.; Niu, D.; Nan, S.; Miao, X.; Hu, X.; Li, C.; Fu, H. Transcriptome and co-expression network analysis reveal molecular mechanisms of mucilage formation during seed development in Artemisia sphaerocephala. Carbohyd. Polym. 2021, 251, 117044. [Google Scholar] [CrossRef] [PubMed]
- Bak, S.; Paquette, S.M.; Morant, M.; Morant, A.V.; Saito, S.; Bjarnholt, N.; Zagrobelny, M.; Jørgensen, K.; Osmani, S.; Simonsen, H.T. Cyanogenic glycosides: A case study for evolution and application of cytochromes P450. Phytochem. Rev. 2006, 5, 309–329. [Google Scholar] [CrossRef]
- Møller, B.L. Functional diversifications of cyanogenic glucosides. Curr. Opin. Plant Biol. 2010, 13, 337–346. [Google Scholar] [CrossRef]
- Kautz, S.; Trisel, J.A.; Ballhorn, D.J. Jasmonic acid enhances plant cyanogenesis and resistance to herbivory in lima bean. J. Chem. Ecol. 2014, 40, 1186–1196. [Google Scholar] [CrossRef]
- Sun, M.; Li, J.; Li, D.; Huang, F.; Wang, D.; Li, H.; Xing, Q.; Zhu, H.; Shi, L. Full-length transcriptome sequencing and modular organization analysis of the naringin/neoeriocitrin-related gene expression pattern in Drynaria roosii. Plant Cell Physiol. 2018, 59, 1398–1414. [Google Scholar] [CrossRef]
- Feng, D.; Shen, Y.; Chavez, E.R. Effectiveness of different processing methods in reducing hydrogen cyanide content of flaxseed. J. Sci. Food Agr. 2003, 83, 836–841. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Ko Pathway | Gene Number |
|---|---|---|
| Phenylpropanoid biosynthesis | ko00940 | 20 |
| Starch and sucrose metabolism | ko00500 | 19 |
| Cyanoamino acid metabolism | ko00460 | 12 |
| Flavonoid biosynthesis | ko00941 | 6 |
| Brassinosteroid biosynthesis | ko00905 | 5 |
| Flavone and flavonol biosynthesis | ko00944 | 4 |
| Isoflavonoid biosynthesis | ko00943 | 3 |
| Cutin, suberine and wax biosynthesis | ko00073 | 3 |
| Zeatin biosynthesis | ko00908 | 2 |
| Stilbenoid, diarylheptanoid and gingerol biosynthesis | ko00945 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Zhao, L.; Zhou, Q.; Fang, L.; Luo, D.; Liu, W.; Searle, I.R.; Liu, Z. Transcriptome and Coexpression Network Analyses Provide In-Sights into the Molecular Mechanisms of Hydrogen Cyanide Synthesis during Seed Development in Common Vetch (Vicia sativa L.). Int. J. Mol. Sci. 2022, 23, 2275. https://doi.org/10.3390/ijms23042275
Li M, Zhao L, Zhou Q, Fang L, Luo D, Liu W, Searle IR, Liu Z. Transcriptome and Coexpression Network Analyses Provide In-Sights into the Molecular Mechanisms of Hydrogen Cyanide Synthesis during Seed Development in Common Vetch (Vicia sativa L.). International Journal of Molecular Sciences. 2022; 23(4):2275. https://doi.org/10.3390/ijms23042275
Chicago/Turabian StyleLi, Mingyu, Lu Zhao, Qiang Zhou, Longfa Fang, Dong Luo, Wenxian Liu, Iain Robert Searle, and Zhipeng Liu. 2022. "Transcriptome and Coexpression Network Analyses Provide In-Sights into the Molecular Mechanisms of Hydrogen Cyanide Synthesis during Seed Development in Common Vetch (Vicia sativa L.)" International Journal of Molecular Sciences 23, no. 4: 2275. https://doi.org/10.3390/ijms23042275
APA StyleLi, M., Zhao, L., Zhou, Q., Fang, L., Luo, D., Liu, W., Searle, I. R., & Liu, Z. (2022). Transcriptome and Coexpression Network Analyses Provide In-Sights into the Molecular Mechanisms of Hydrogen Cyanide Synthesis during Seed Development in Common Vetch (Vicia sativa L.). International Journal of Molecular Sciences, 23(4), 2275. https://doi.org/10.3390/ijms23042275