
The Value of Whole-Genome Sequencing for Mitochondrial DNA Population Studies: Strategies and Criteria for Extracting High-Quality Mitogenome Haplotypes

 ,
,  , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results
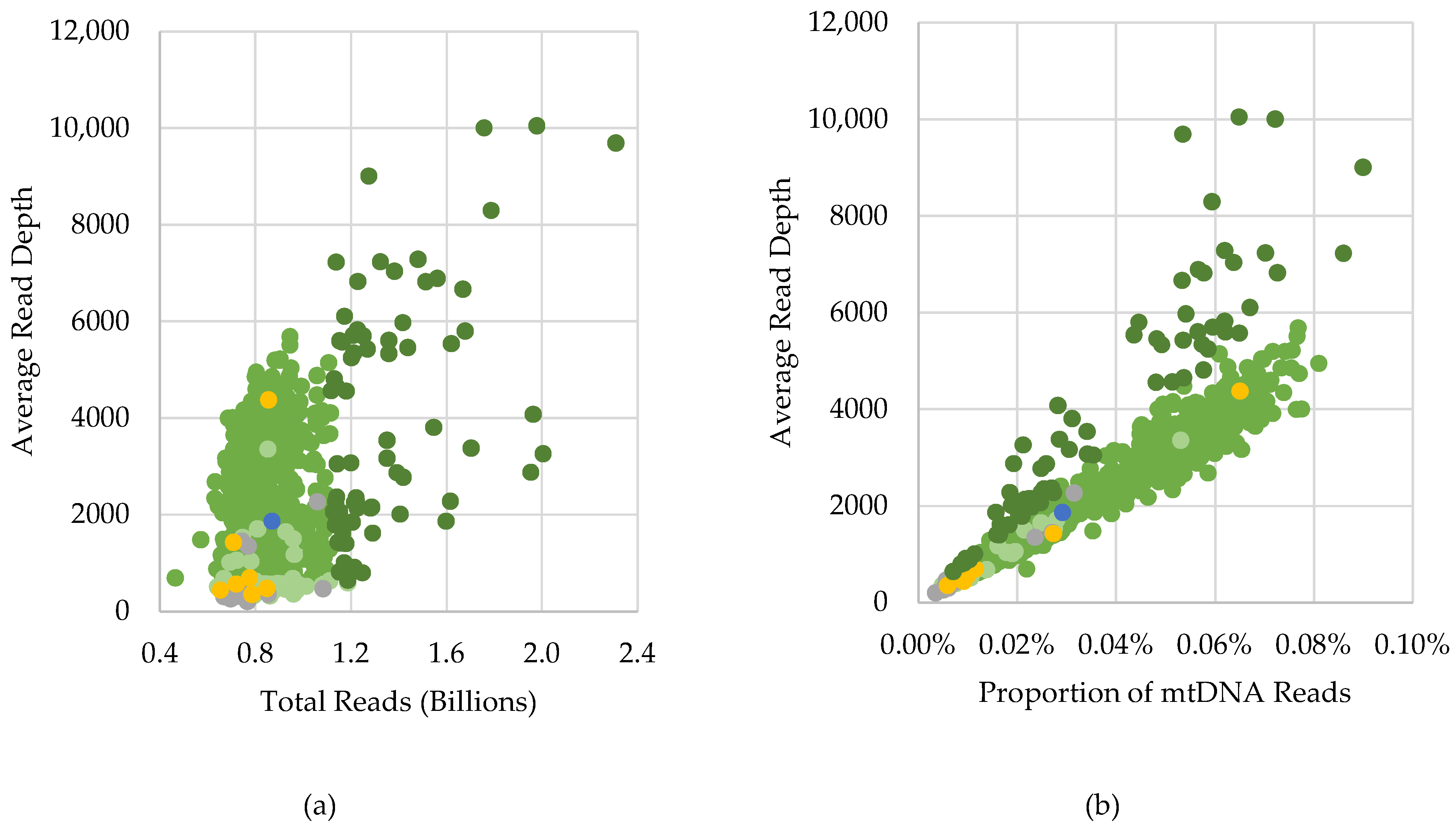
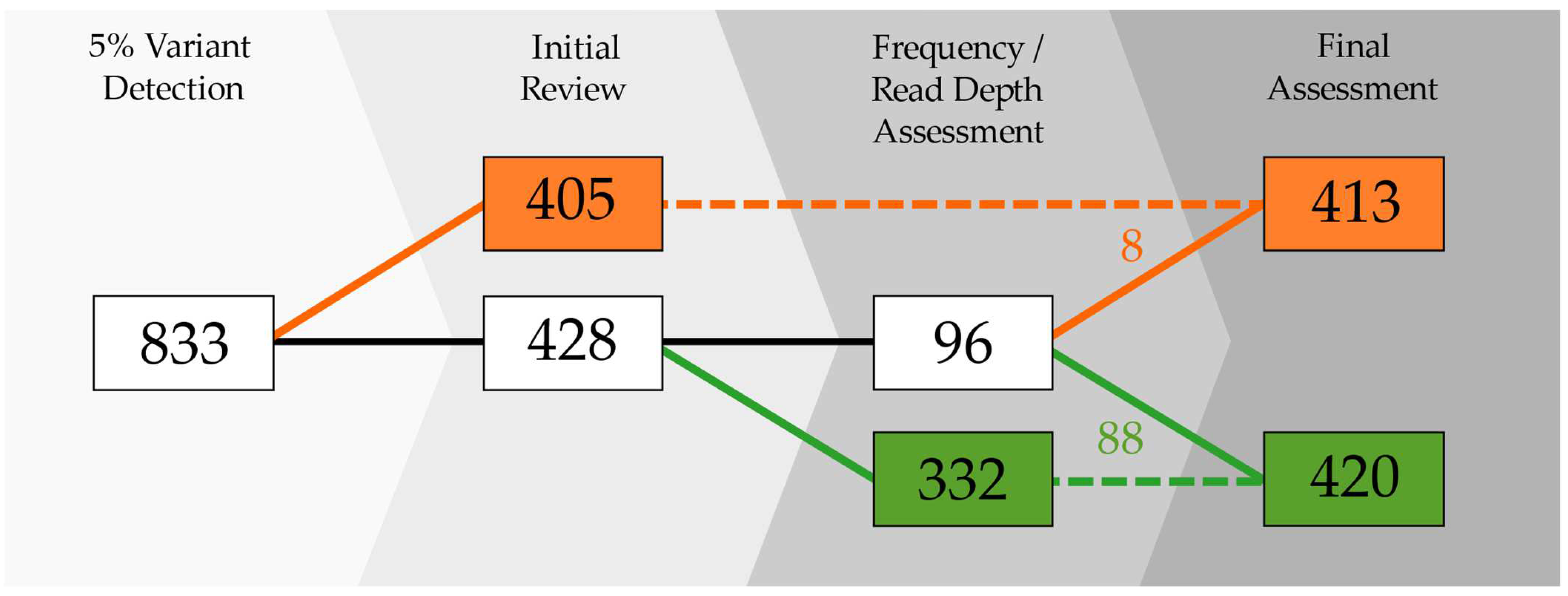
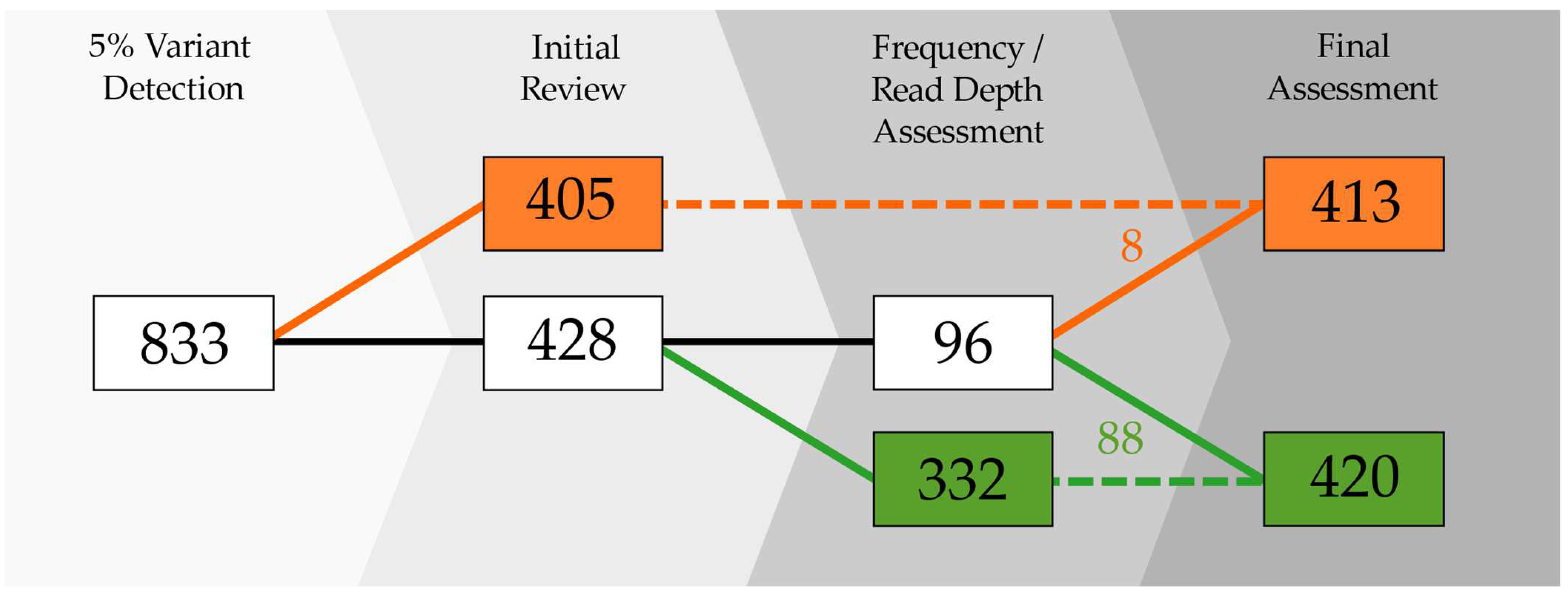
2.1. Overall Performance
2.1.1. Passing Samples
2.1.2. Excluded Samples
2.2. Classification of Mixed Positions
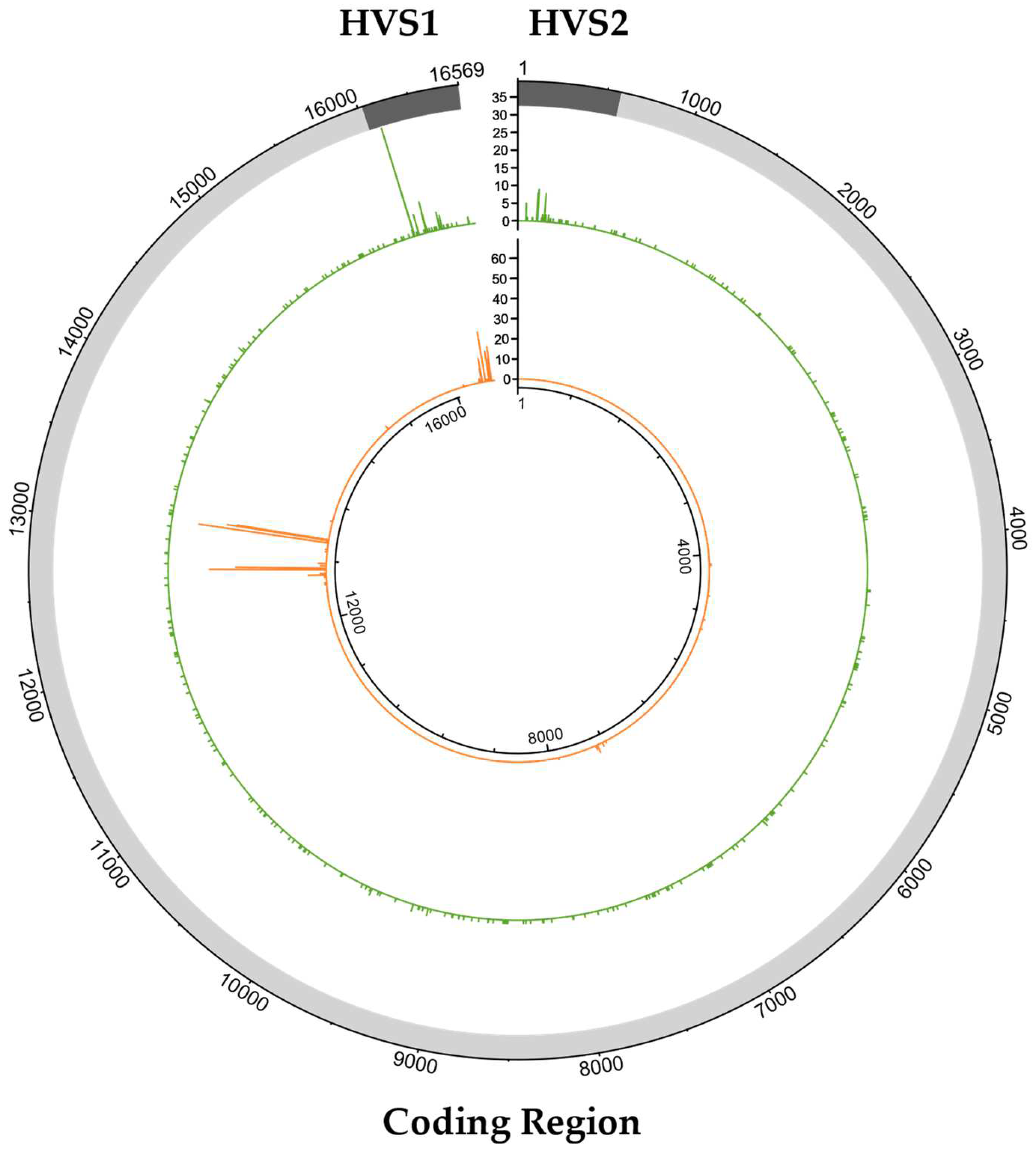
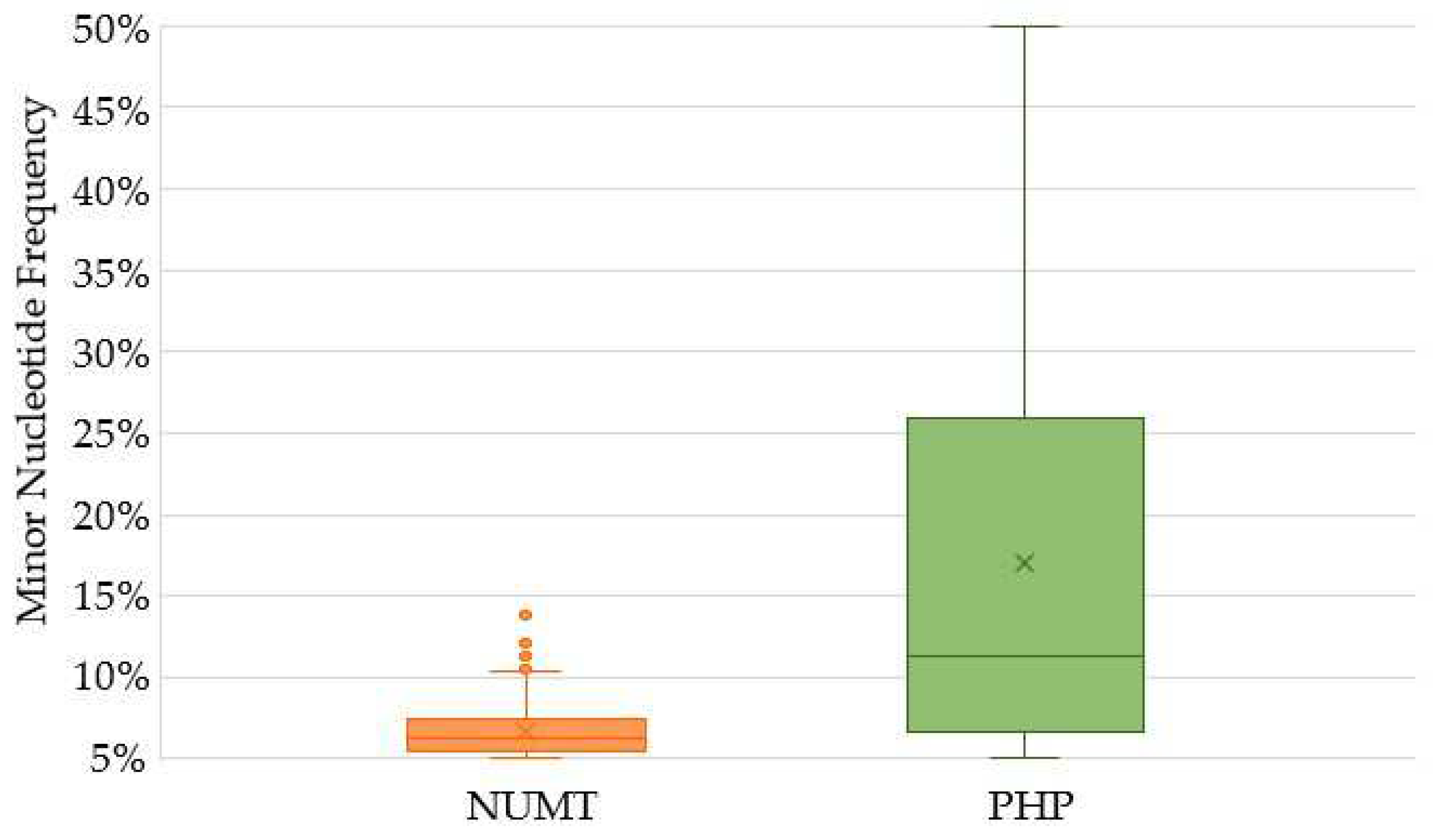
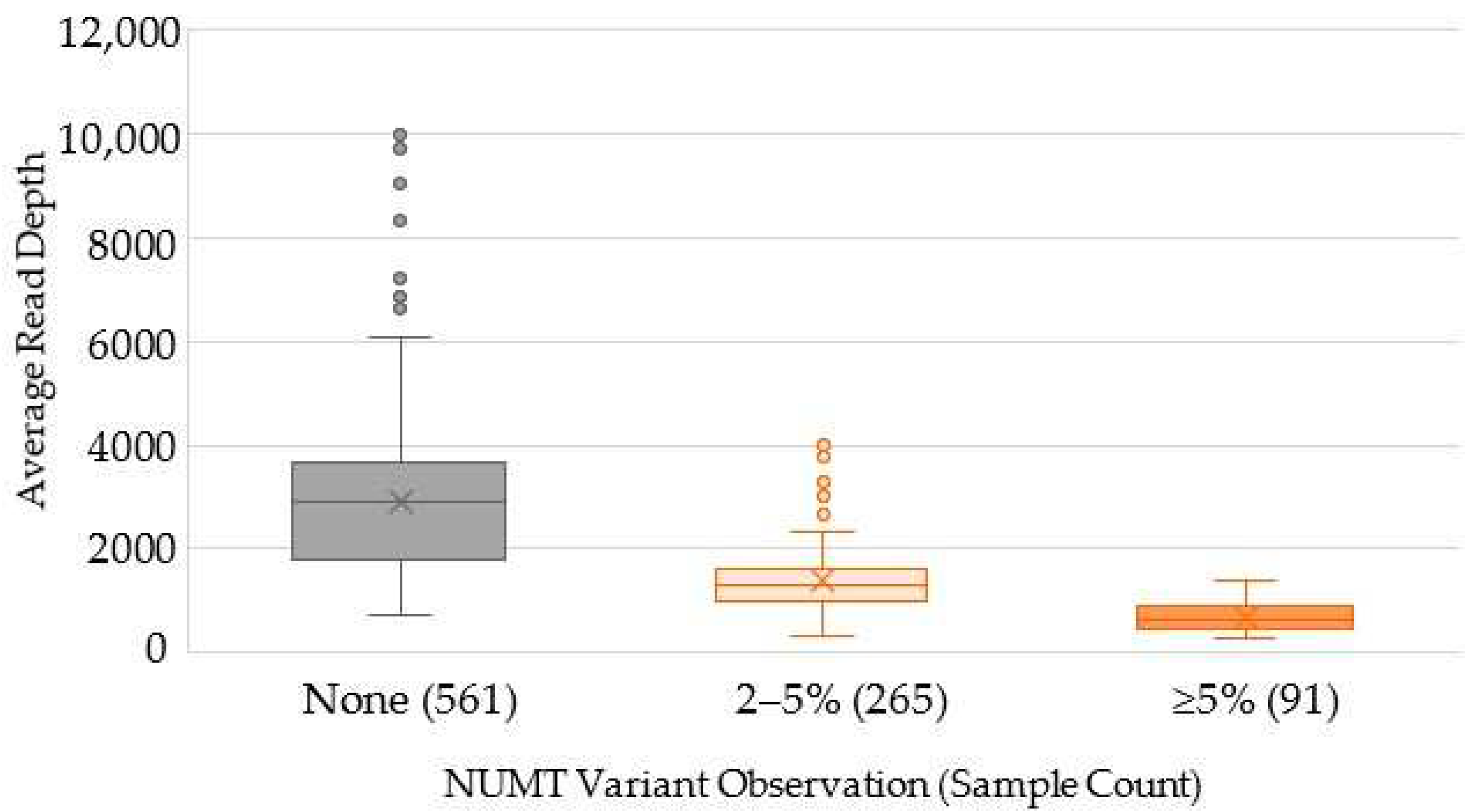
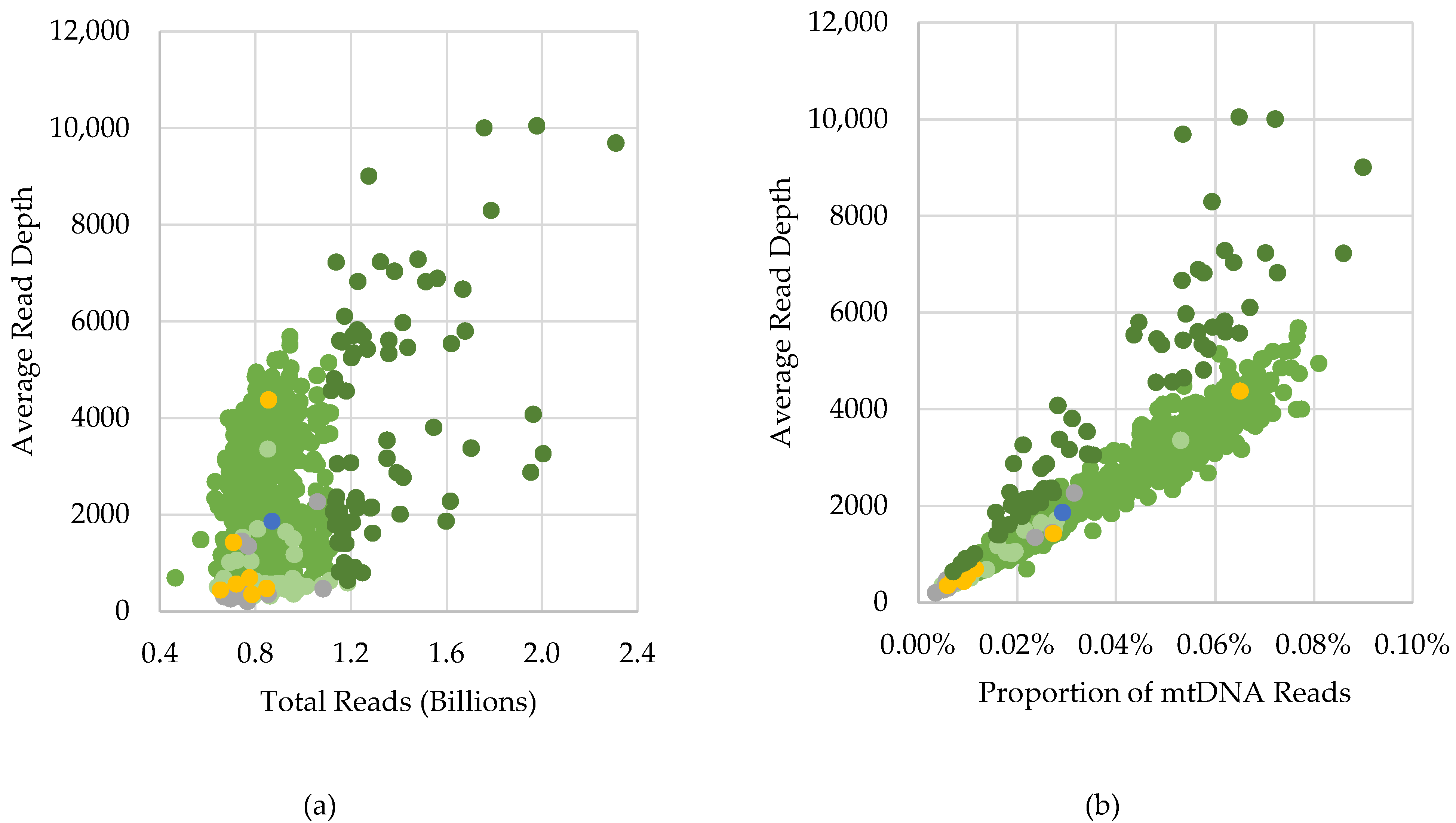
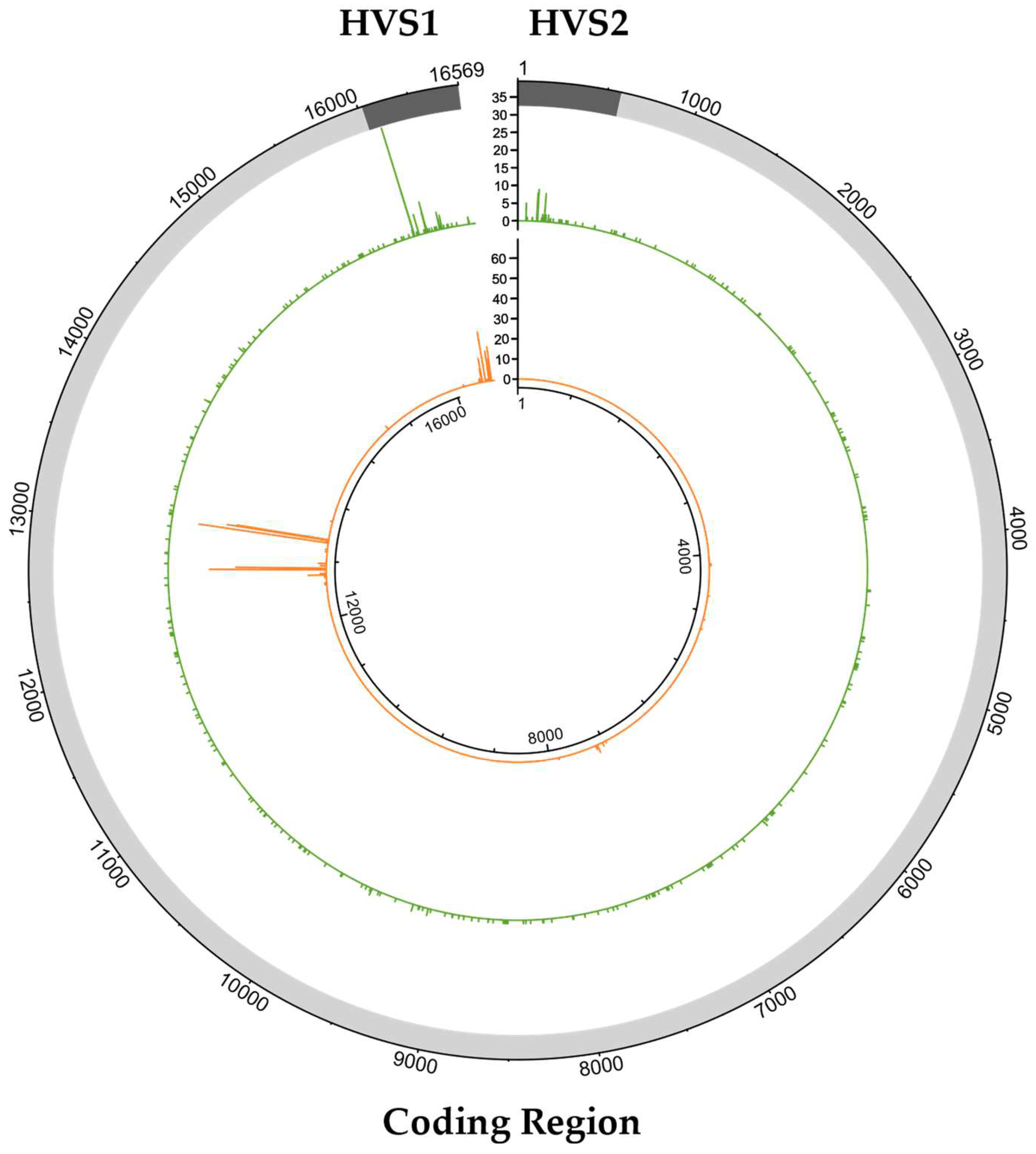
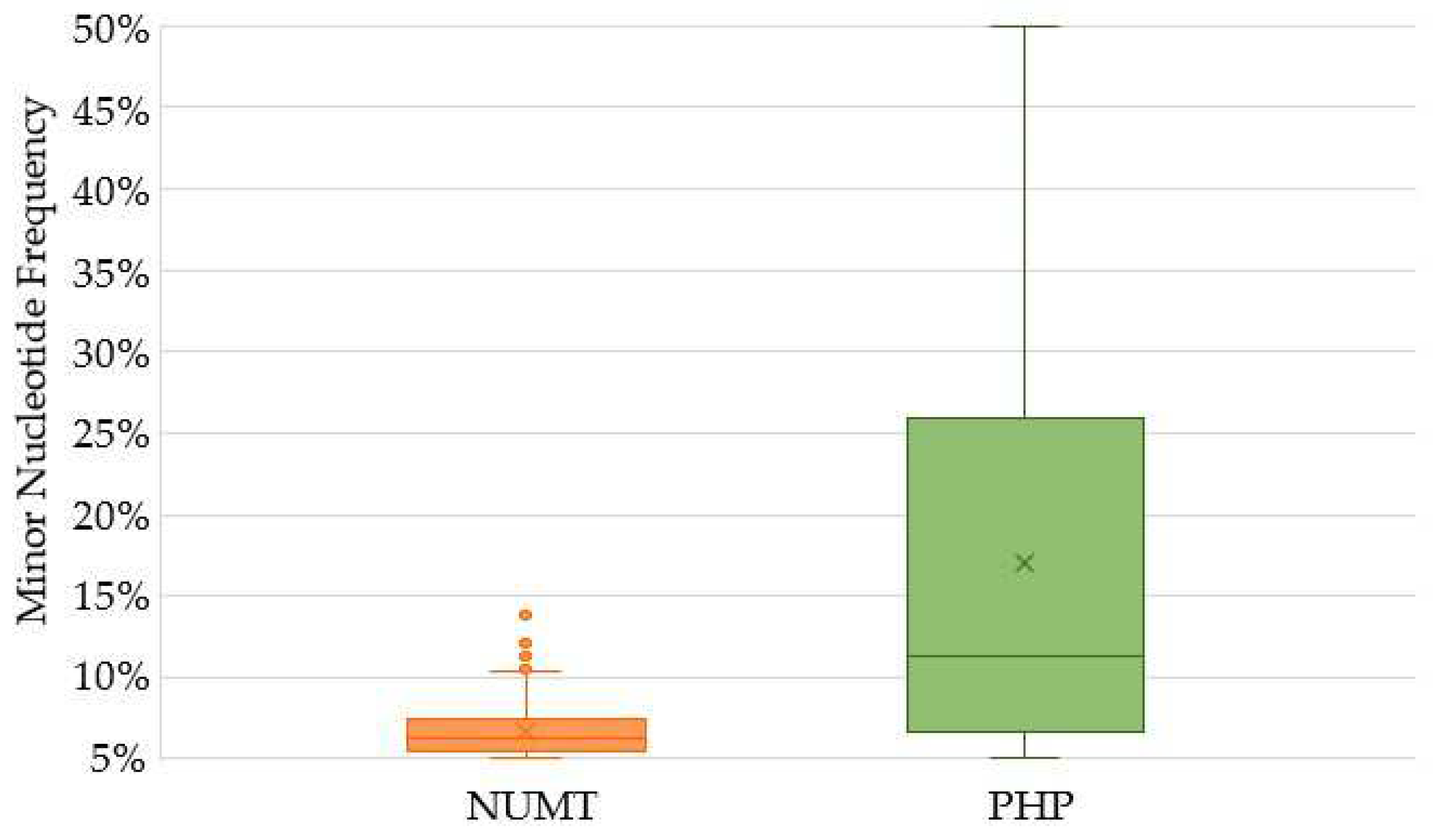
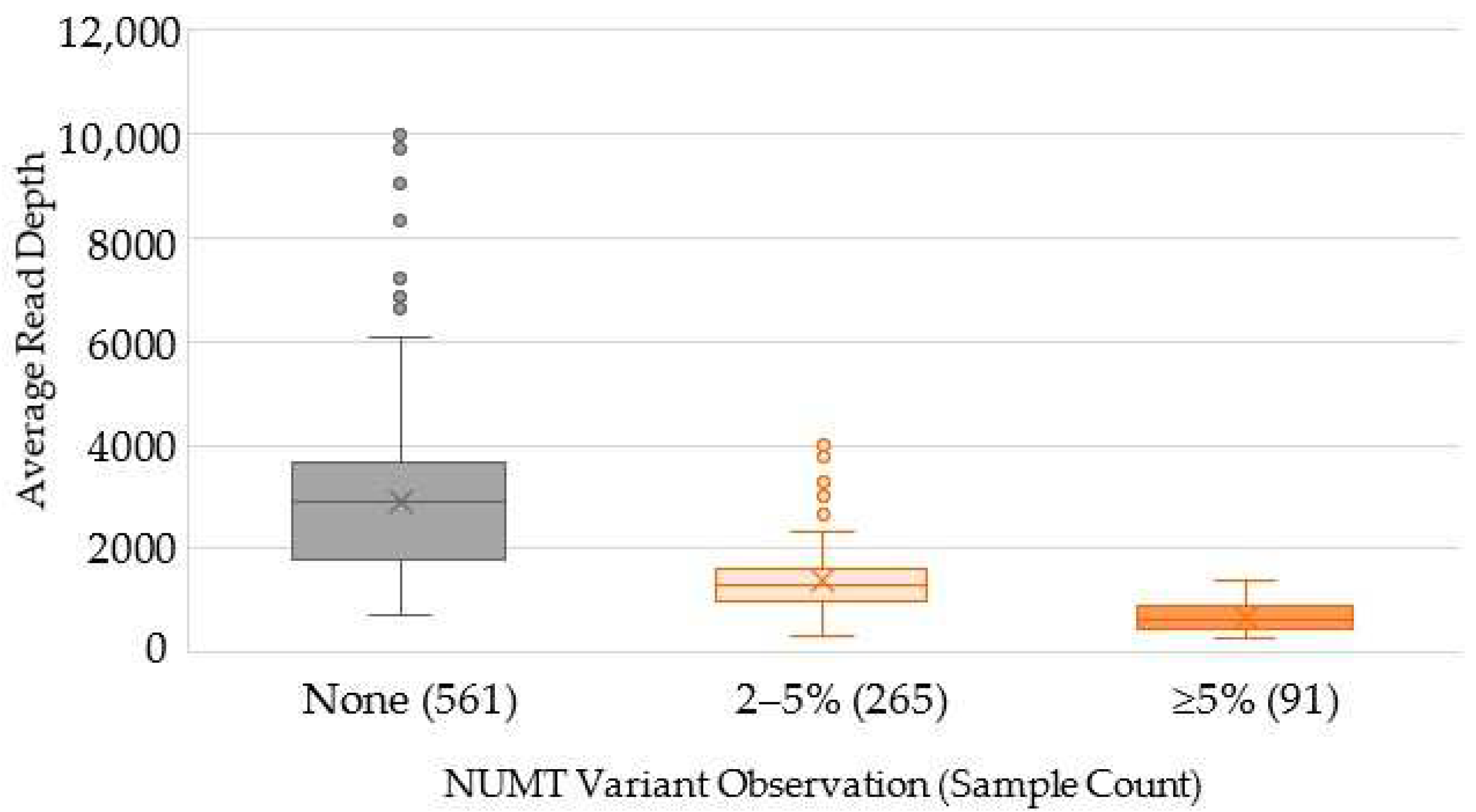
2.3. Characterization of Variants Associated with NUMTs
2.4. Heteroplasmy
2.5. Comparison with High-Quality Mitogenome Datasets
3. Discussion
4. Materials and Methods
4.1. Samples and Whole-Genome Sequencing
4.2. Mitochondrial Genome Haplotype Generation
4.3. Classification of Mixed Positions
4.4. Additional Quality Control Measures
4.5. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Ostell, J.; Pruitt, K.D.; Sayers, E.W. GenBank. Nucleic Acids Res. 2018, 46, D41–D47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayama, G.; Emery, S.B.; Kidd, J.M.; Mills, R.E. The genomic landscape of polymorphic human nuclear mitochondrial insertions. Nucleic Acids Res. 2014, 42, 12640–12649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woerner, A.E.; Cihlar, J.C.; Smart, U.; Budowle, B. Numt identification and removal with RtN! Bioinformatics 2020, 36, 5115–5116. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.; Parson, W. Interpreting NUMTs in forensic genetics: Seeing the forest for the trees. Forensic Sci. Int. Genet. 2021, 53, 102497. [Google Scholar] [CrossRef] [PubMed]
- Balciuniene, J.; Balciunas, D. A Nuclear mtDNA Concatemer (Mega-NUMT) Could Mimic Paternal Inheritance of Mitochondrial Genome. Front. Genet. 2019, 10, 518. [Google Scholar] [CrossRef] [PubMed]
- Lutz-Bonengel, S.; Niederstatter, H.; Naue, J.; Koziel, R.; Yang, F.; Sanger, T.; Huber, G.; Berger, C.; Pflugradt, R.; Strobl, C.; et al. Evidence for multi-copy Mega-NUMTs in the human genome. Nucleic Acids Res. 2021, 49, 1517–1531. [Google Scholar] [CrossRef] [PubMed]
- Wachsmuth, M.; Hubner, A.; Li, M.; Madea, B.; Stoneking, M. Age-Related and Heteroplasmy-Related Variation in Human mtDNA Copy Number. PLoS Genet. 2016, 12, e1005939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, C.R.; Kiesler, K.M.; Sturk-Andreaggi, K.; Ring, J.D.; Parson, W.; Schanfield, M.; Vallone, P.M.; Marshall, C. Platinum-Quality Mitogenome Haplotypes from United States Populations. Genes 2020, 11, 1290. [Google Scholar] [CrossRef]
- Just, R.S.; Scheible, M.K.; Fast, S.A.; Sturk-Andreaggi, K.; Rock, A.W.; Bush, J.M.; Higginbotham, J.L.; Peck, M.A.; Ring, J.D.; Huber, G.E.; et al. Full mtGenome reference data: Development and characterization of 588 forensic-quality haplotypes representing three U.S. populations. Forensic Sci. Int. Genet. 2015, 14, 141–155. [Google Scholar] [CrossRef] [Green Version]
- Ye, K.; Lu, J.; Ma, F.; Keinan, A.; Gu, Z. Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proc. Natl. Acad. Sci. USA 2014, 111, 10654–10659. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Schroder, R.; Ni, S.; Madea, B.; Stoneking, M. Extensive tissue-related and allele-related mtDNA heteroplasmy suggests positive selection for somatic mutations. Proc. Natl. Acad. Sci. USA 2015, 112, 2491–2496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Wu, J.; Dressman, D.C.; Iacobuzio-Donahue, C.; Markowitz, S.D.; Velculescu, V.E.; Diaz, L.A., Jr.; Kinzler, K.W.; Vogelstein, B.; Papadopoulos, N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature 2010, 464, 610–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Tuna, S.; Keogh, M.J.; Smith, K.R.; Aitman, T.J.; Beales, P.L.; Bennett, D.L.; Gale, D.P.; Bitner-Glindzicz, M.A.K.; Black, G.C.; et al. NIHR BioResource-Rare Diseases; 100,000 Genomes Project-Rare Diseases Pilot, Germline Selection Shapes Human Mitochondrial DNA Diversity. Science 2019, 364, eaau6520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.G.; Kong, Q.P.; Salas, A.; Bandelt, H.J. Pseudomitochondrial genome haunts disease studies. J. Med. Genet. 2008, 45, 769–772. [Google Scholar] [CrossRef]
- Just, R.S.; Irwin, J.A.; Parson, W. Questioning the prevalence and reliability of human mitochondrial DNA heteroplasmy from massively parallel sequencing data. Proc. Natl. Acad. Sci. USA 2014, 111, E4546–E4547. [Google Scholar] [CrossRef] [Green Version]
- Just, R.S.; Irwin, J.A.; Parson, W. Mitochondrial DNA heteroplasmy in the emerging field of massively parallel sequencing. Forensic Sci. Int. Genet. 2015, 18, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Valencia, C.A.; Zhang, J.; Lee, N.C.; Slone, J.; Gui, B.; Wang, X.; Li, Z.; Dell, S.; Brown, J.; et al. Biparental Inheritance of Mitochondrial DNA in Humans. Proc. Natl. Acad. Sci. USA 2018, 115, 13039–13044. [Google Scholar] [CrossRef] [Green Version]
- Lutz-Bonengel, S.; Parson, W. No further evidence for paternal leakage of mitochondrial DNA in humans yet. Proc. Natl. Acad. Sci. USA 2019, 116, 1821–1822. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Pagnamenta, A.T.; Gleadall, N.; Sanchis-Juan, A.; Stephens, J.; Broxholme, J.; Tuna, S.; Odhams, C.A.; Genomics England Research Consortium; NIHR BioResource; et al. Nuclear-mitochondrial DNA segments resemble paternally inherited mitochondrial DNA in humans. Nat. Commun. 2020, 11, 1–11. [Google Scholar]
- Calvignac, S.; Konecny, L.; Malard, F.; Douady, C.J. Preventing the pollution of mitochondrial datasets with nuclear mitochondrial paralogs (numts). Mitochondrion 2011, 11, 246–254. [Google Scholar] [CrossRef]
- Porter, T.M.; Hajibabaei, M. Profile hidden Markov model sequence analysis can help remove putative pseudogenes from DNA barcoding and metabarcoding datasets. BMC Bioinform. 2021, 22, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Graham, N.R.; Gillespie, R.G.; Krehenwinkel, H. Towards eradicating the nuisance of numts and noise in molecular biodiversity assessment. Mol. Ecol. Resour. 2021, 21, 1755–1758. [Google Scholar] [CrossRef] [PubMed]
- Ring, J.D.; Sturk-Andreaggi, K.; Peck, M.A.; Marshall, C. Bioinformatic removal of NUMT-associated variants in mitotiling next-generation sequencing data from whole blood samples. Electrophoresis 2018, 39, 2785–2797. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.N. Targeted and robust amplification of mitochondrial DNA in the presence of nuclear-encoded mitochondrial pseudogenes using Phi29 DNA polymerases. Methods Mol. Biol. 2014, 1167, 255–263. [Google Scholar]
- Li, M.; Schroeder, R.; Ko, A.; Stoneking, M. Fidelity of capture-enrichment for mtDNA genome sequencing: Influence of NUMTs. Nucleic Acids. Res. 2012, 40, e137. [Google Scholar] [CrossRef]
- Cihlar, J.C.; Strobl, C.; Lagace, R.; Muenzler, M.; Parson, W.; Budowle, B. Distinguishing mitochondrial DNA and NUMT sequences amplified with the precision ID mtDNA whole genome panel. Mitochondrion 2020, 55, 122–133. [Google Scholar] [CrossRef]
- Holt, C.L.; Stephens, K.M.; Walichiewicz, P.; Fleming, K.D.; Forouzmand, E.; Wu, S.F. Human Mitochondrial Control Region and mtGenome: Design and Forensic Validation of NGS Multiplexes, Sequencing and Analytical Software. Genes 2021, 12, 599. [Google Scholar] [CrossRef]
- Cihlar, J.C.; Amory, C.; Lagace, R.; Roth, C.; Parson, W.; Budowle, B. Developmental Validation of a MPS Workflow with a PCR-Based Short Amplicon Whole Mitochondrial Genome Panel. Genes 2020, 11, 1345. [Google Scholar] [CrossRef]
- Albayrak, L.; Khanipov, K.; Pimenova, M.; Golovko, G.; Rojas, M.; Pavlidis, I.; Chumakov, S.; Aguilar, G.; Chavez, A.; Widger, W.R.; et al. The ability of human nuclear DNA to cause false positive low-abundance heteroplasmy calls varies across the mitochondrial genome. BMC Genom. 2016, 17, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ameur, A.; Dahlberg, J.; Olason, P.; Vezzi, F.; Karlsson, R.; Martin, M.; Viklund, J.; Kahari, A.K.; Lundin, P.; Che, H.; et al. SweGen: A whole-genome data resource of genetic variability in a cross-section of the Swedish population. Eur. J. Hum. Genet. 2017, 25, 1253–1260. [Google Scholar] [CrossRef] [Green Version]
- Magnusson, P.K.; Almqvist, C.; Rahman, I.; Ganna, A.; Viktorin, A.; Walum, H.; Halldner, L.; Lundstrom, S.; Ullen, F.; Langstrom, N.; et al. The Swedish Twin Registry: Establishment of a biobank and other recent developments. Twin Res. Hum. Genet. 2013, 16, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- van Oven, M. PhyloTree Build 17: Growing the human mitochondrial DNA tree. Forensic Sci. Int. Genet. 2015, 5, e392–e394. [Google Scholar] [CrossRef] [Green Version]
- Weissensteiner, H.; Forer, L.; Fendt, L.; Kheirkhah, A.; Salas, A.; Kronenberg, F.; Schoenherr, S. Contamination detection in sequencing studies using the mitochondrial phylogeny. Genome Res. 2021, 31, 309–316. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Lahermo, P.; Richards, M.; Macaulay, V. Detecting errors in mtDNA data by phylogenetic analysis. Int. J. Legal Med. 2001, 115, 64–69. [Google Scholar] [CrossRef]
- Smart, U.; Cihlar, J.C.; Mandape, S.N.; Muenzler, M.; King, J.L.; Budowle, B.; Woerner, A.E. A Continuous Statistical Phasing Framework for the Analysis of Forensic Mitochondrial DNA Mixtures. Genes 2021, 12, 128. [Google Scholar] [CrossRef]
- Bodner, M.; Irwin, J.A.; Coble, M.D.; Parson, W. Inspecting close maternal relatedness: Towards better mtDNA population samples in forensic databases. Forensic Sci. Int. Genet. 2011, 5, 138–141. [Google Scholar] [CrossRef] [Green Version]
- Irwin, J.A.; Saunier, J.L.; Niederstatter, H.; Strouss, K.M.; Sturk, K.A.; Diegoli, T.M.; Brandstatter, A.; Parson, W.; Parsons, T.J. Investigation of heteroplasmy in the human mitochondrial DNA control region: A synthesis of observations from more than 5000 global population samples. J. Mol. Evol. 2009, 68, 516–527. [Google Scholar] [CrossRef]
- Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; 2004. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 3 December 2021).
- Lang, M.; Sazzini, M.; Calabrese, F.M.; Simone, D.; Boattini, A.; Romeo, G.; Luiselli, D.; Attimonelli, M.; Gasparre, G. Polymorphic NumtS trace human population relationships. Hum. Genet. 2012, 131, 757–771. [Google Scholar] [CrossRef]
- Delfin, F.; Min-Shan Ko, A.; Li, M.; Gunnarsdottir, E.D.; Tabbada, K.A.; Salvador, J.M.; Calacal, G.C.; Sagum, M.S.; Datar, F.A.; Padilla, S.G.; et al. Complete mtDNA genomes of Filipino ethnolinguistic groups: A melting pot of recent and ancient lineages in the Asia-Pacific region. Eur. J. Hum. Genet. 2014, 22, 228–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, N.; Parson, W.; Dur, A. Next generation database search algorithm for forensic mitogenome analyses. Forensic Sci. Int. Genet. 2018, 37, 204–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaag, K.J.V.; Desmyter, S.; Smit, S.; Prieto, L.; Sijen, T. Reducing the Number of Mismatches between Hairs and Buccal References When Analysing mtDNA Heteroplasmic Variation by Massively Parallel Sequencing. Genes 2020, 11, 1355. [Google Scholar] [CrossRef] [PubMed]
- Lyons, E.A.; Scheible, M.K.; Sturk-Andreaggi, K.; Irwin, J.A.; Just, R.S. A high-throughput Sanger strategy for human mitochondrial genome sequencing. BMC Genom. 2013, 14, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.; Li, J.; Wang, L.; Huang, Q.; Zhu, J.; Wen, S.; Lyu, J.; Wu, W. Comparison of paired cerebrospinal fluid and serum cell-free mitochondrial and nuclear DNA with copy number and fragment length. J. Clin. Lab. Anal. 2020, 34, e23238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Cui, H.; Wong, L.J. Comprehensive one-step molecular analyses of mitochondrial genome by massively parallel sequencing. Clin. Chem. 2012, 58, 1322–1331. [Google Scholar] [CrossRef]
- Santibanez-Koref, M.; Griffin, H.; Turnbull, D.M.; Chinnery, P.F.; Herbert, M.; Hudson, G. Assessing mitochondrial heteroplasmy using next generation sequencing: A note of caution. Mitochondrion 2019, 46, 302–306. [Google Scholar] [CrossRef]
- Singh, L.N.; Ennis, B.; Loneragan, B.; Tsao, N.L.; Lopez Sanchez, M.I.G.; Li, J.; Acheampong, P.; Tran, O.; Trounce, I.A.; Zhu, Y.; et al. MitoScape: A big-data, machine-learning platform for obtaining mitochondrial DNA from next-generation sequencing data. PLoS Comput. Biol. 2021, 17, e1009594. [Google Scholar] [CrossRef]
- Filograna, R.; Mennuni, M.; Alsina, D.; Larsson, N.G. Mitochondrial DNA copy number in human disease: The more the better? FEBS Lett. 2021, 595, 976–1002. [Google Scholar] [CrossRef]
- Chu, H.T.; Hsiao, W.W.; Tsao, T.T.; Chang, C.M.; Liu, Y.W.; Fan, C.C.; Lin, H.; Chang, H.H.; Yeh, T.J.; Chen, J.C.; et al. Quantitative assessment of mitochondrial DNA copies from whole genome sequencing. BMC Genom. 2012, 13, 1–5. [Google Scholar] [CrossRef]
- Longchamps, R.J.; Castellani, C.A.; Yang, S.Y.; Newcomb, C.E.; Sumpter, J.A.; Lane, J.; Grove, M.L.; Guallar, E.; Pankratz, N.; Taylor, K.D.; et al. Evaluation of mitochondrial DNA copy number estimation techniques. PLoS ONE 2020, 15, e0228166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parson, W.; Gusmao, L.; Hares, D.R.; Irwin, J.A.; Mayr, W.R.; Morling, N.; Pokorak, E.; Prinz, M.; Salas, A.; Schneider, P.M.; et al. DNA Commission of the International Society for Forensic Genetics DNA Commission of the International Society for Forensic Genetics: Revised and extended guidelines for mitochondrial DNA typing. Forensic Sci Int Genet. 2014, 13, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Craven, L.; Alston, C.L.; Taylor, R.W.; Turnbull, D.M. Recent Advances in Mitochondrial Disease. Annu. Rev. Genom. Hum. Genet. 2017, 18, 257–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scientific Working Group on DNA Analysis Methods (SWGDAM). Interpretation Guidelines for Mitochondrial DNA Analysis by Forensic DNA Testing Laboratories. 2019. Available online: https://www.swgdam.org/_files/ugd/4344b0_f61de6abf3b94c52b28139bff600ae98.pdf (accessed on 3 December 2021).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar]
- Sturk-Andreaggi, K.; Peck, M.A.; Boysen, C.; Dekker, P.; McMahon, T.P.; Marshall, C.K. AQME: A forensic mitochondrial DNA analysis tool for next-generation sequencing data. Forensic Sci. Int. Genet. 2017, 31, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Sturk-Andreaggi, K.; Parson, W.; Allen, M.; Marshall, C. Impact of the sequencing method on the detection and interpretation of mitochondrial DNA length heteroplasmy. Forensic Sci. Int. Genet. 2020, 44, 102205. [Google Scholar] [CrossRef] [Green Version]
- Stoler, N.; Nekrutenko, A. Sequencing error profiles of Illumina sequencing instruments. NAR Genom. Bioinform. 2021, 3, lqab019. [Google Scholar] [CrossRef]
- van Oven, M.; Kayser, M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum. Mutat. 2009, 30, E386–E394. [Google Scholar] [CrossRef]
- Parson, W.; Dur, A. EMPOP—A forensic mtDNA database. Forensic Sci. Int. Genet. 2007, 1, 88–92. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. circlize Implements and Enhances Circular Visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Subclassification | Count | Total Reads | % Mapped Reads | % mtDNA Reads | Average Read Depth |
|---|---|---|---|---|---|---|
| Passing | Complete | 858 | 865,518,368.5 | 99.16% | 0.036% | 2365.9 |
| Nearly Complete | 59 | 833,866,169.5 | 99.20% | 0.010% | 652.5 | |
| Excluded | Incomplete | 17 | 784,241,829.7 | 99.32% | 0.010% | 592.0 |
| Mixed | 7 | 762,731,157.9 | 98.72% | 0.019% | 1191.1 | |
| Related | 1 | 869,537,450 | 99.52% | 0.029% | 1870.7 |
| NUMT Variants | Count | Total Reads | % Reads Mapped | % Mapped mtDNA | Average Read Depth |
|---|---|---|---|---|---|
| None | 561 | 869,724,465.0 | 99.17% | 0.044% | 2919.1 |
| 2–5% | 265 | 854,695,905.9 | 99.16% | 0.021% | 1390.1 |
| ≥5% | 91 | 855,009,718.4 | 99.16% | 0.011% | 686.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sturk-Andreaggi, K.; Ring, J.D.; Ameur, A.; Gyllensten, U.; Bodner, M.; Parson, W.; Marshall, C.; Allen, M. The Value of Whole-Genome Sequencing for Mitochondrial DNA Population Studies: Strategies and Criteria for Extracting High-Quality Mitogenome Haplotypes. Int. J. Mol. Sci. 2022, 23, 2244. https://doi.org/10.3390/ijms23042244
Sturk-Andreaggi K, Ring JD, Ameur A, Gyllensten U, Bodner M, Parson W, Marshall C, Allen M. The Value of Whole-Genome Sequencing for Mitochondrial DNA Population Studies: Strategies and Criteria for Extracting High-Quality Mitogenome Haplotypes. International Journal of Molecular Sciences. 2022; 23(4):2244. https://doi.org/10.3390/ijms23042244
Chicago/Turabian StyleSturk-Andreaggi, Kimberly, Joseph D. Ring, Adam Ameur, Ulf Gyllensten, Martin Bodner, Walther Parson, Charla Marshall, and Marie Allen. 2022. "The Value of Whole-Genome Sequencing for Mitochondrial DNA Population Studies: Strategies and Criteria for Extracting High-Quality Mitogenome Haplotypes" International Journal of Molecular Sciences 23, no. 4: 2244. https://doi.org/10.3390/ijms23042244
APA StyleSturk-Andreaggi, K., Ring, J. D., Ameur, A., Gyllensten, U., Bodner, M., Parson, W., Marshall, C., & Allen, M. (2022). The Value of Whole-Genome Sequencing for Mitochondrial DNA Population Studies: Strategies and Criteria for Extracting High-Quality Mitogenome Haplotypes. International Journal of Molecular Sciences, 23(4), 2244. https://doi.org/10.3390/ijms23042244