
Kidney Injury in COVID-19: Epidemiology, Molecular Mechanisms and Potential Therapeutic Targets



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Epidemiology and Clinical Impact of Kidney Injury in COVID-19
2.1. Epidemiology of AKI in COVID-19
2.2. Mortality of COVID-19-Associated AKI
2.3. Relationship between COVID-19, Pre-Existing CKD, and Risk of Future CKD
3. Clinical and Histopathological Features of Kidney Injury in COVID-19
3.1. Tubular Injury and Tubular Dysfunction as Primary Features of COVID-19-Associated AKI
3.2. Hemoproteinuria in COVID-19-Associated AKI
3.3. Time Course of COVID-19-Associated AKI
4. Pathogenesis of Kidney Injury in COVID-19: Indirect vs. Direct Kidney Injury
4.1. Indirect Contributors to COVID-19-Associated AKI
4.2. COVID-19 as a Direct Cause of AKI: Tissue Studies
4.3. COVID-19 as a Direct Cause of AKI: Detection of Virus and Viral Particles in Urine
4.4. COVID-19 as a Direct Cause of AKI: Experiments Using Kidney Spheroids and Organoids
5. Pathogenesis of Kidney Injury in COVID-19: Molecular Mechanisms and Potential Molecular Targets
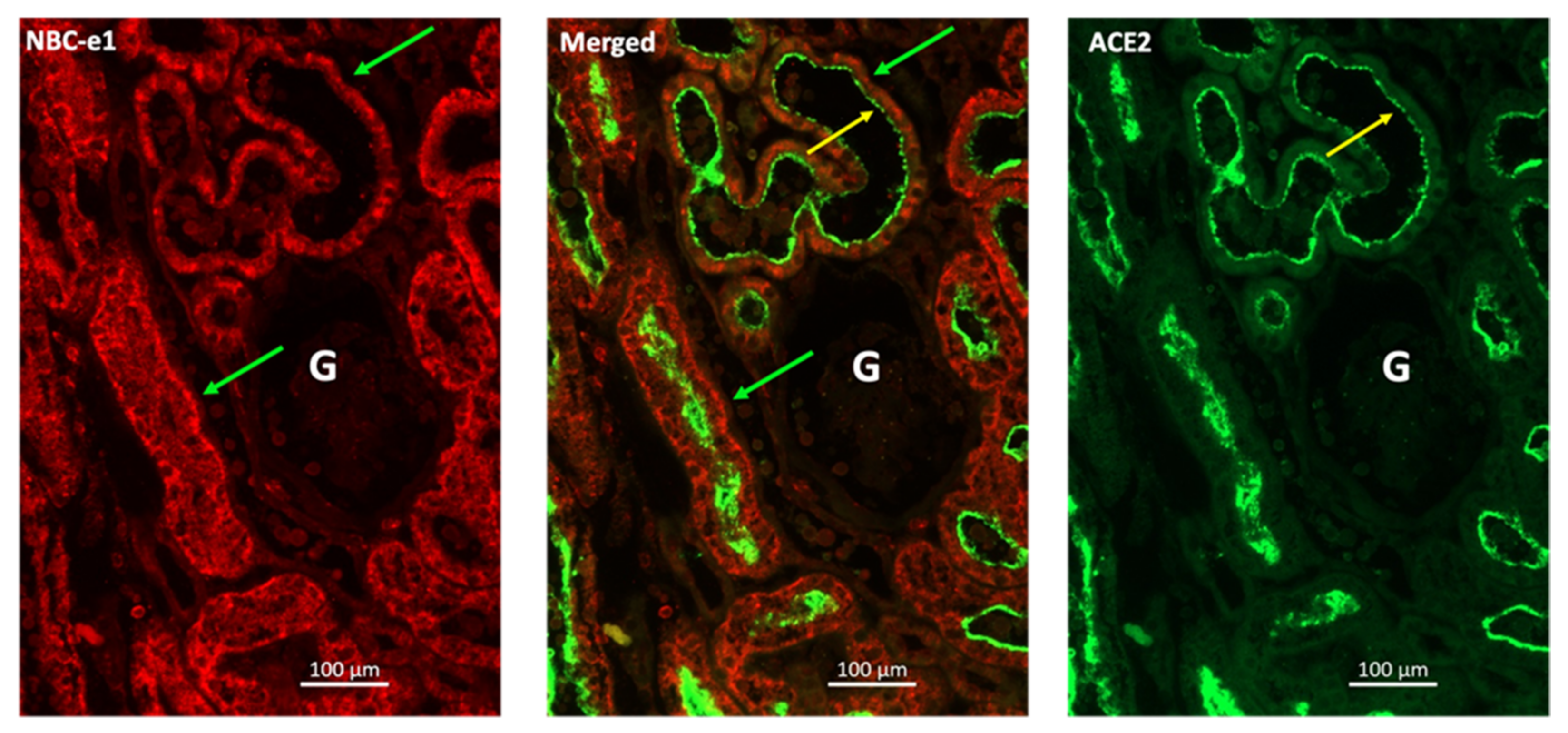
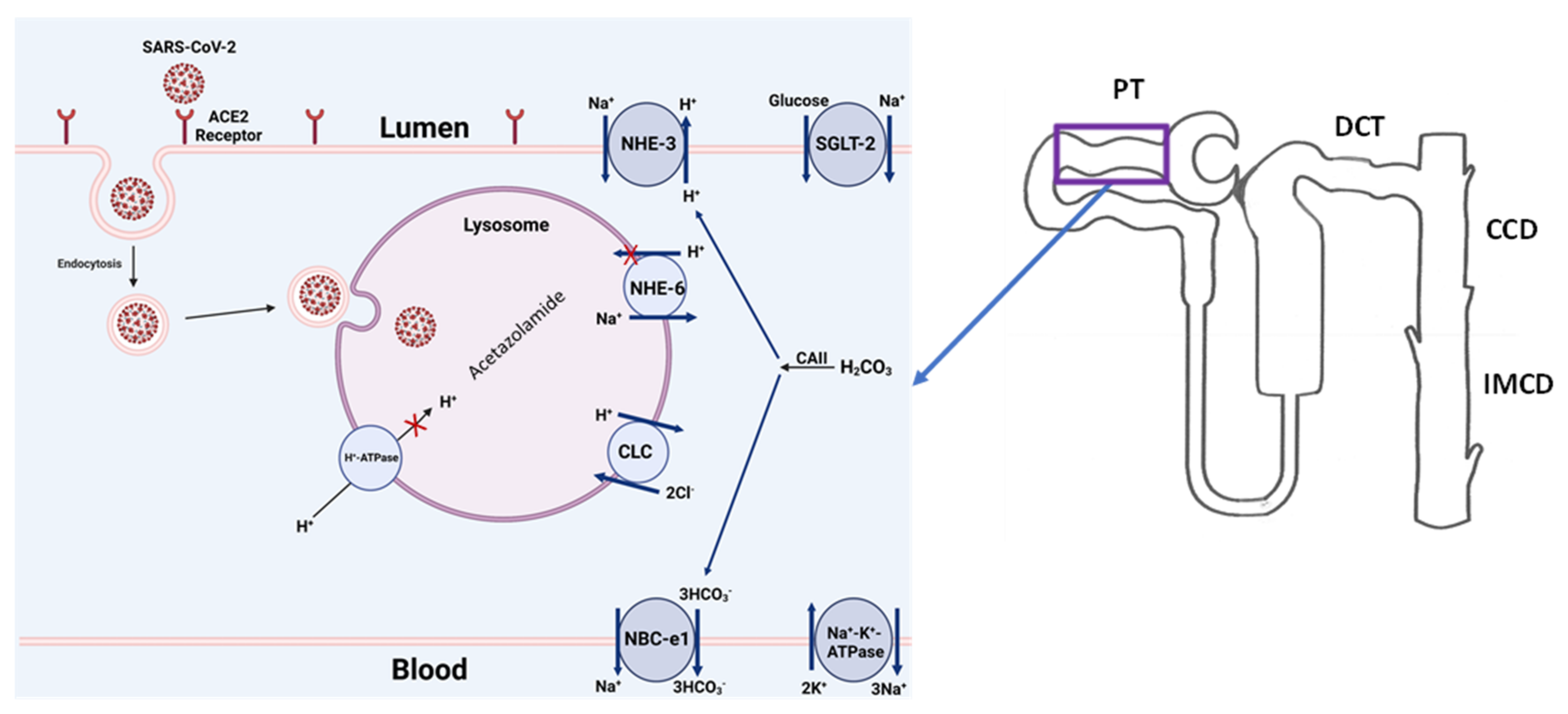
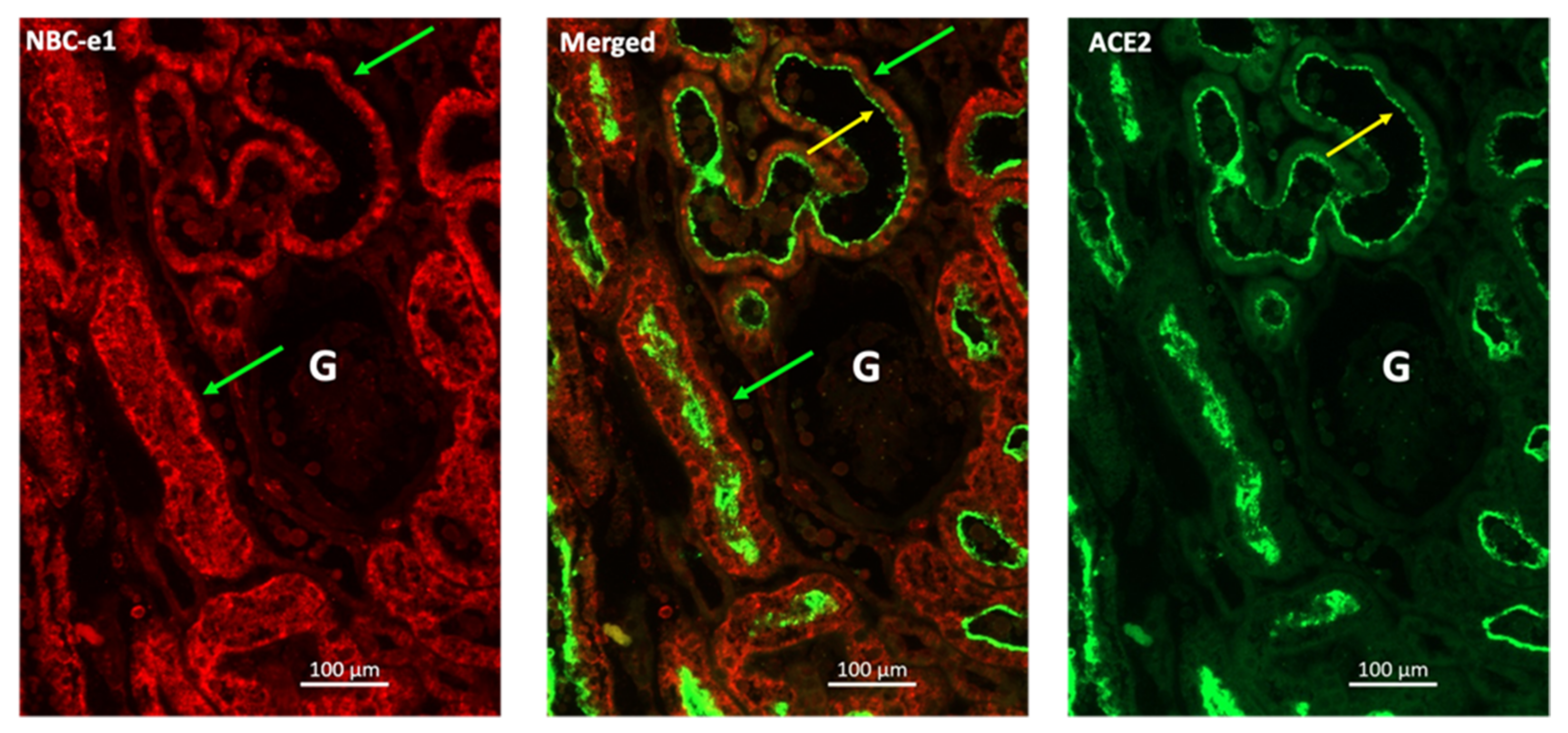
5.1. Interaction of SARS-CoV-2 with the Cell Membrane
5.2. Potential Therapeutic Targets: An Overview
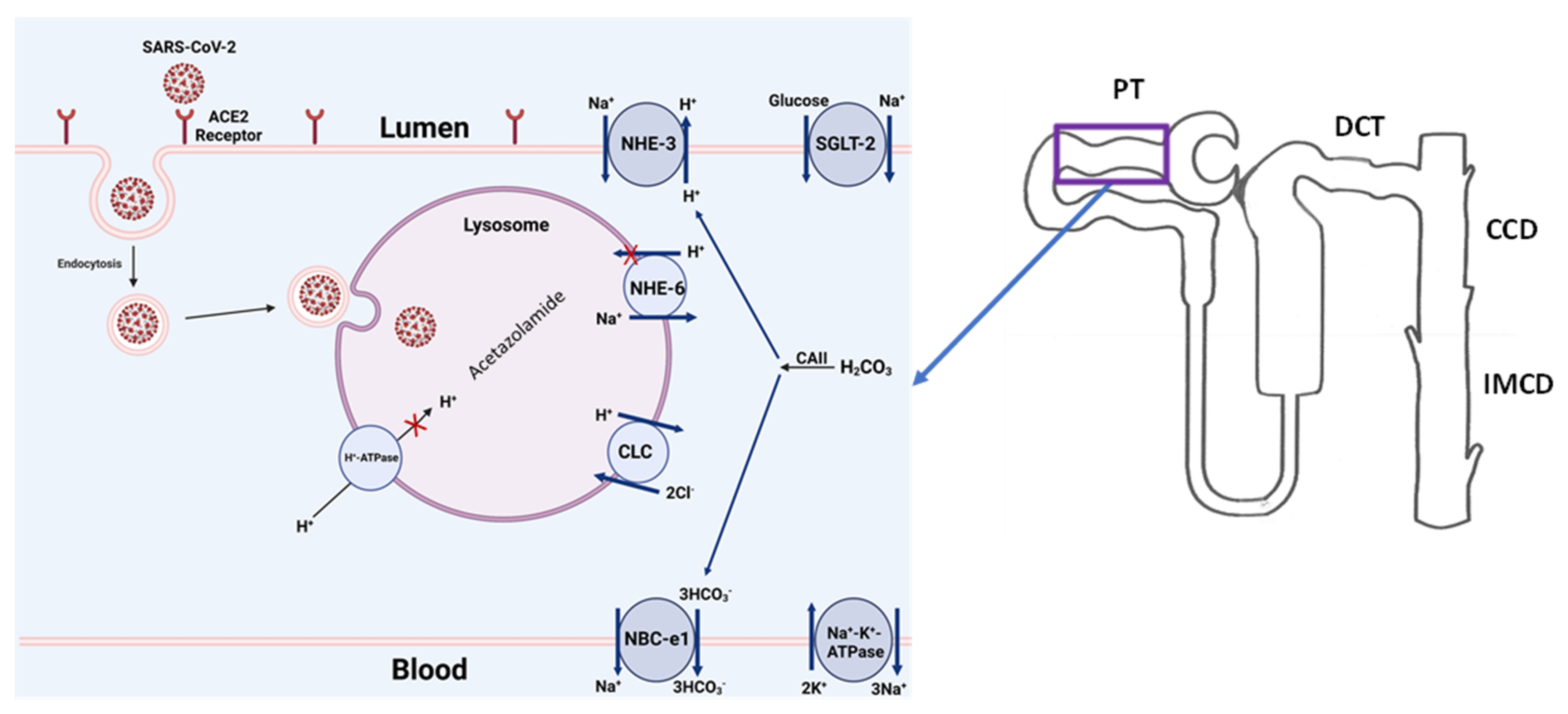
5.3. Potential Therapeutic Targets: Lysosomal Acidification
5.4. Potential Therapeutic Targets: Modulation of the Renin–Angiotensin–Aldosterone System
5.5. Potential Therapeutic Targets: Complement
6. Discussion and Conclusions
6.1. Discussion
6.2. Conclusions
7. Material and Methods
7.1. Materials
7.2. Immunofluorescence Microscopy
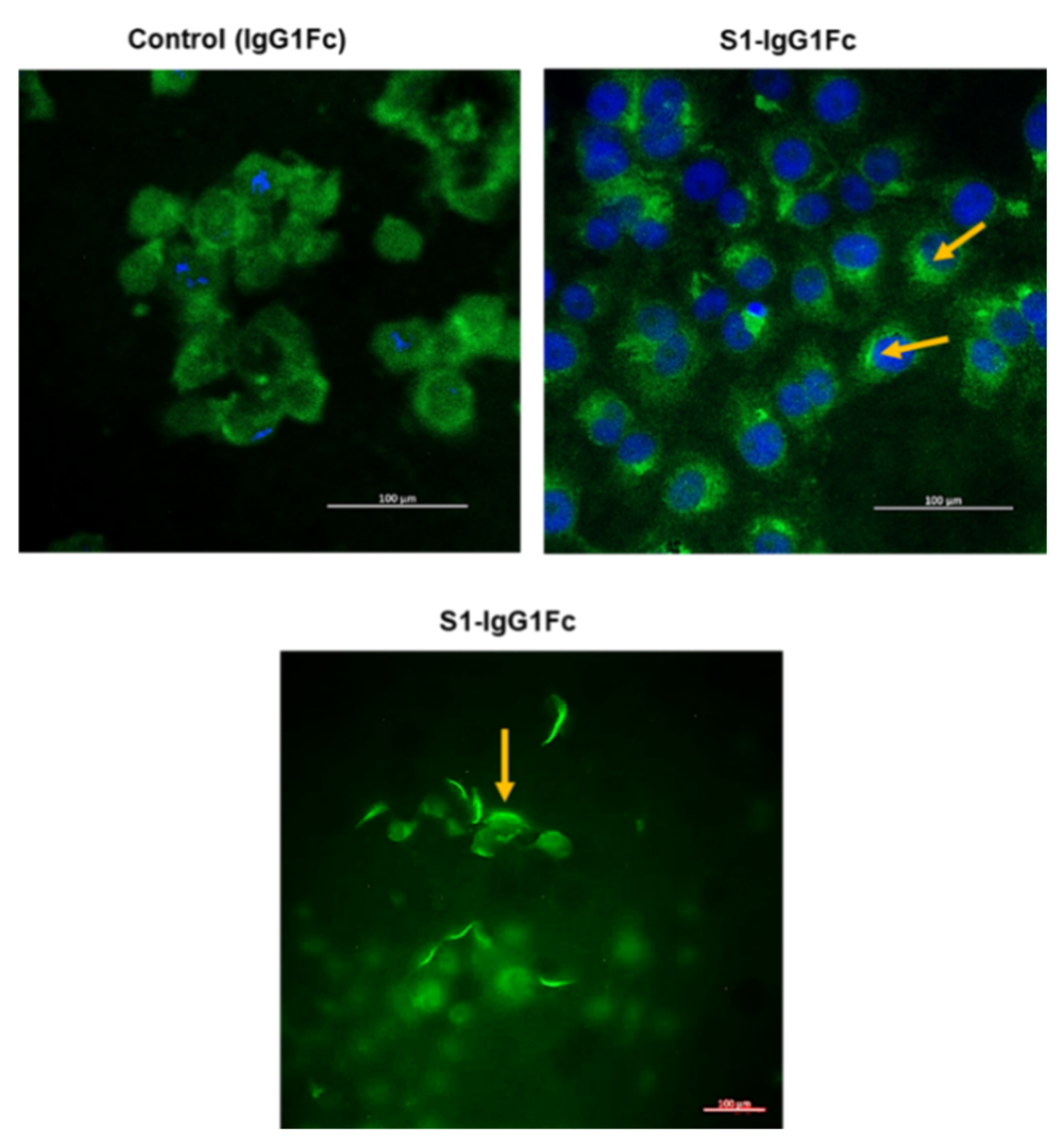
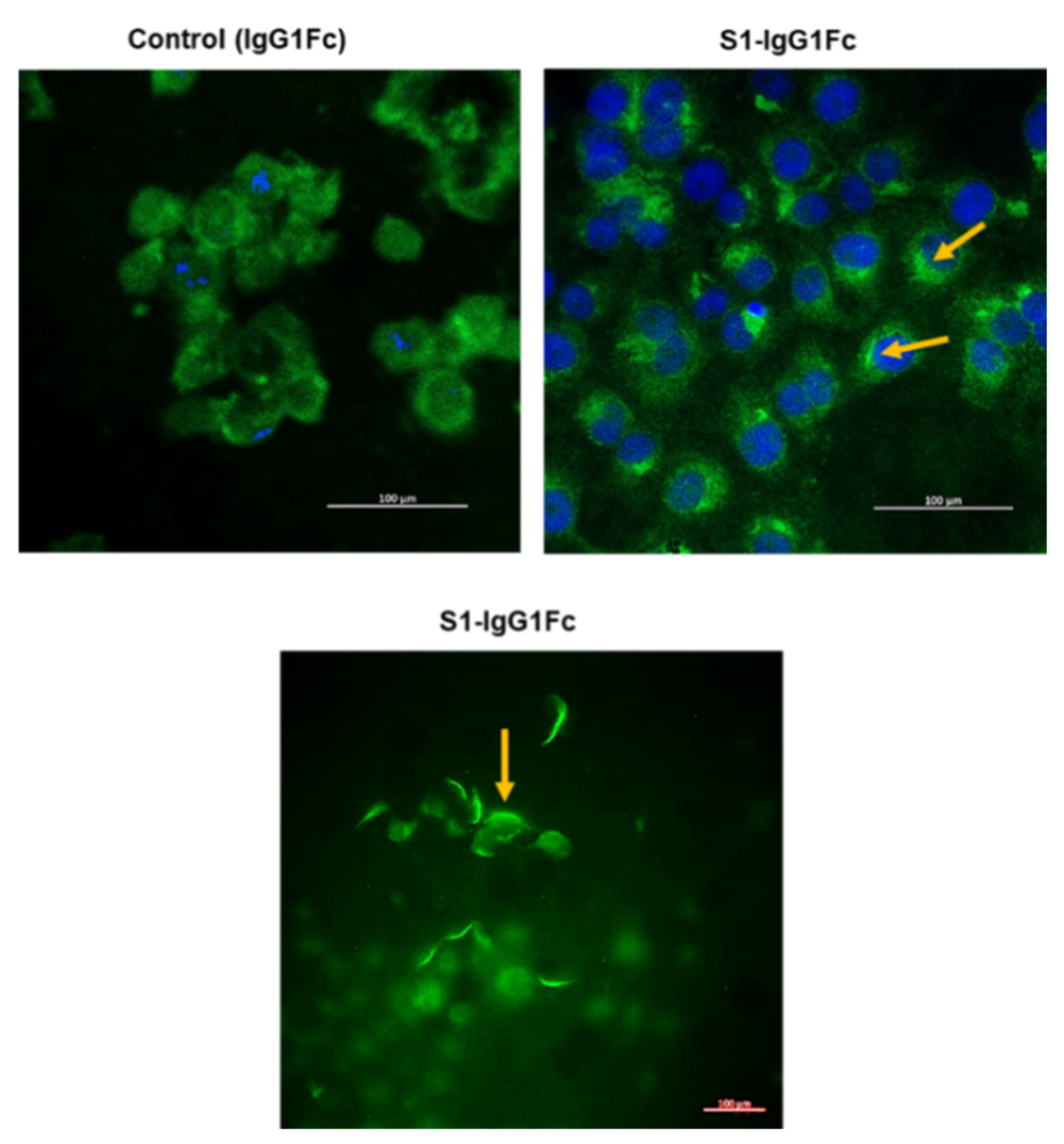
7.3. S1 Fragment Binding to Vero 6 Cells
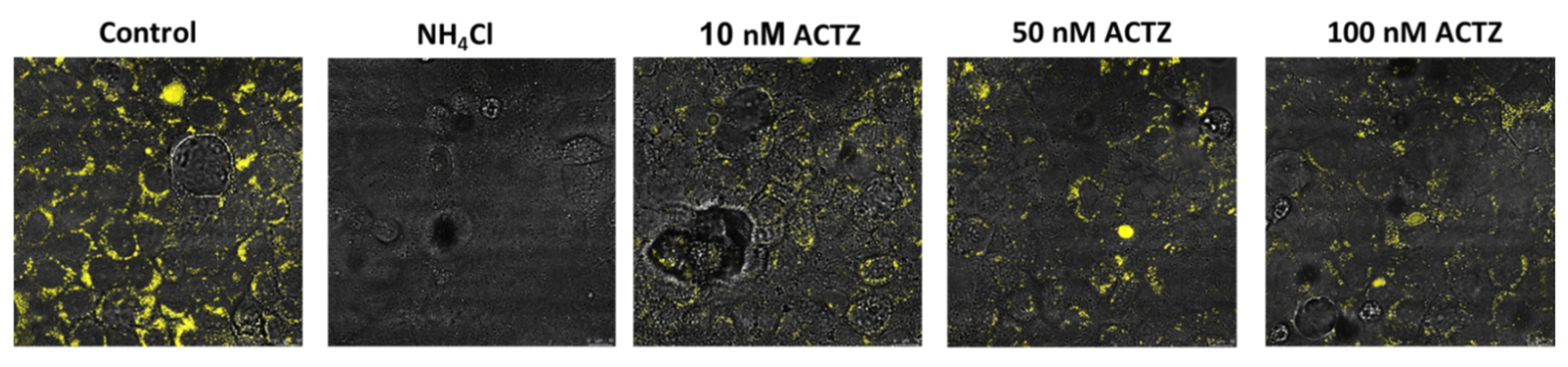
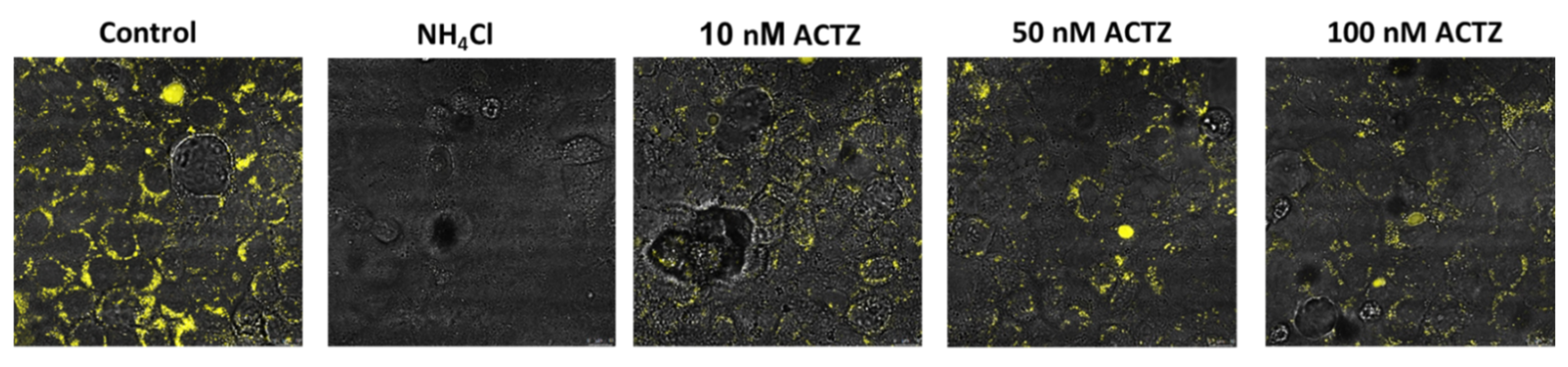
7.4. Effect of Acetazolamide on Lysosomal Acidification
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johns Hopkins University of Medicine Coronavirus Resource Center. Available online: https://coronavirus.jhu.edu/ (accessed on 22 December 2021).
- Nadim, M.K.; Forni, L.G.; Mehta, R.L.; Connor, M.J., Jr.; Liu, K.D.; Ostermann, M.; Rimmele, T.; Zarbock, A.; Bell, S.; Bihorac, A.; et al. COVID-19-associated acute kidney injury: Consensus report of the 25th Acute Disease Quality Initiative (ADQI) Workgroup. Nat. Rev. Nephrol. 2020, 16, 747–764. [Google Scholar] [CrossRef] [PubMed]
- Pfister, F.; Vonbrunn, E.; Ries, T.; Jack, H.M.; Uberla, K.; Lochnit, G.; Sheriff, A.; Herrmann, M.; Buttner-Herold, M.; Amann, K.; et al. Complement Activation in Kidneys of Patients with COVID-19. Front. Immunol. 2020, 11, 594849. [Google Scholar] [CrossRef] [PubMed]
- Hoste, E.A.; Bagshaw, S.M.; Bellomo, R.; Cely, C.M.; Colman, R.; Cruz, D.N.; Edipidis, K.; Forni, L.G.; Gomersall, C.D.; Govil, D.; et al. Epidemiology of acute kidney injury in critically ill patients: The multinational AKI-EPI study. Intensive Care Med. 2015, 41, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Hoste, E.A.J.; Kellum, J.A.; Selby, N.M.; Zarbock, A.; Palevsky, P.M.; Bagshaw, S.M.; Goldstein, S.L.; Cerda, J.; Chawla, L.S. Global epidemiology and outcomes of acute kidney injury. Nat. Rev. Nephrol. 2018, 14, 607–625. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Luo, R.; Wang, K.; Zhang, M.; Wang, Z.; Dong, L.; Li, J.; Yao, Y.; Ge, S.; Xu, G. Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int. 2020, 97, 829–838. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Wang, W.; Xu, Y.; Gao, R.; Lu, R.; Han, K.; Wu, G.; Tan, W. Detection of SARS-CoV-2 in Different Types of Clinical Specimens. JAMA 2020, 323, 1843–1844. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Li, X.; Chen, H.; Yan, S.; Li, D.; Li, Y.; Gong, Z. Coronavirus Disease 19 Infection Does Not Result in Acute Kidney Injury: An Analysis of 116 Hospitalized Patients from Wuhan, China. Am. J. Nephrol. 2020, 51, 343–348. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Wu, D.; Chen, H.; Yan, W.; Yang, D.; Chen, G.; Ma, K.; Xu, D.; Yu, H.; Wang, H.; et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: Retrospective study. BMJ 2020, 368, m1091. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Wang, X.; Ren, J.; Sun, Y.; Yu, R.; Li, K.; Zheng, L.; Yang, J. Risk factors and prognosis for COVID-19-induced acute kidney injury: A meta-analysis. BMJ Open 2020, 10, e042573. [Google Scholar] [CrossRef]
- Hirsch, J.S.; Ng, J.H.; Ross, D.W.; Sharma, P.; Shah, H.H.; Barnett, R.L.; Hazzan, A.D.; Fishbane, S.; Jhaveri, K.D.; Northwell COVID-19 Research Consortium; et al. Acute kidney injury in patients hospitalized with COVID-19. Kidney Int. 2020, 98, 209–218. [Google Scholar] [CrossRef]
- Chan, L.; Chaudhary, K.; Saha, A.; Chauhan, K.; Vaid, A.; Zhao, S.; Paranjpe, I.; Somani, S.; Richter, F.; Miotto, R.; et al. AKI in Hospitalized Patients with COVID-19. J. Am. Soc. Nephrol. 2021, 32, 151–160. [Google Scholar] [CrossRef]
- Mohamed, M.M.B.; Lukitsch, I.; Torres-Ortiz, A.E.; Walker, J.B.; Varghese, V.; Hernandez-Arroyo, C.F.; Alqudsi, M.; LeDoux, J.R.; Velez, J.C.Q. Acute Kidney Injury Associated with Coronavirus Disease 2019 in Urban New Orleans. Kidney360 2020, 1, 614. [Google Scholar] [CrossRef]
- Gupta, S.; Coca, S.G.; Chan, L.; Melamed, M.L.; Brenner, S.K.; Hayek, S.S.; Sutherland, A.; Puri, S.; Srivastava, A.; Leonberg-Yoo, A.; et al. AKI Treated with Renal Replacement Therapy in Critically Ill Patients with COVID-19. J. Am. Soc. Nephrol. 2021, 32, 161–176. [Google Scholar] [CrossRef]
- Fisher, M.; Neugarten, J.; Bellin, E.; Yunes, M.; Stahl, L.; Johns, T.S.; Abramowitz, M.K.; Levy, R.; Kumar, N.; Mokrzycki, M.H.; et al. AKI in Hospitalized Patients with and without COVID-19: A Comparison Study. J. Am. Soc. Nephrol. 2020, 31, 2145–2157. [Google Scholar] [CrossRef]
- Bowe, B.; Cai, M.; Xie, Y.; Gibson, A.K.; Maddukuri, G.; Al-Aly, Z. Acute Kidney Injury in a National Cohort of Hospitalized US Veterans with COVID-19. Clin. J. Am. Soc. Nephrol. 2020, 16, 14–25. [Google Scholar] [CrossRef]
- Kolhe, N.V.; Fluck, R.J.; Selby, N.M.; Taal, M.W. Acute kidney injury associated with COVID-19: A retrospective cohort study. PLoS Med. 2020, 17, e1003406. [Google Scholar] [CrossRef]
- Rubin, S.; Orieux, A.; Prevel, R.; Garric, A.; Bats, M.L.; Dabernat, S.; Camou, F.; Guisset, O.; Issa, N.; Mourissoux, G.; et al. Characterization of acute kidney injury in critically ill patients with severe coronavirus disease 2019. Clin. Kidney J. 2020, 13, 354–361. [Google Scholar] [CrossRef]
- Fominskiy, E.V.; Scandroglio, A.M.; Monti, G.; Calabro, M.G.; Landoni, G.; Dell’Acqua, A.; Beretta, L.; Moizo, E.; Ravizza, A.; Monaco, F.; et al. Prevalence, Characteristics, Risk Factors, and Outcomes of Invasively Ventilated COVID-19 Patients with Acute Kidney Injury and Renal Replacement Therapy. Blood Purif. 2021, 50, 102–109. [Google Scholar] [CrossRef]
- Husain-Syed, F.; Wilhelm, J.; Kassoumeh, S.; Birk, H.W.; Herold, S.; Vadasz, I.; Walmrath, H.D.; Kellum, J.A.; Ronco, C.; Seeger, W. Acute kidney injury and urinary biomarkers in hospitalized patients with coronavirus disease-2019. Nephrol. Dial. Transplant. 2020, 35, 1271–1274. [Google Scholar] [CrossRef]
- Chaibi, K.; Dao, M.; Pham, T.; Gumucio-Sanguino, V.D.; Di Paolo, F.A.; Pavot, A.; Cohen, Y.; Dreyfuss, D.; Perez-Fernandez, X.; Gaudry, S. Severe Acute Kidney Injury in Patients with COVID-19 and Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2020, 202, 1299–1301. [Google Scholar] [CrossRef]
- Charytan, D.M.; Parnia, S.; Khatri, M.; Petrilli, C.M.; Jones, S.; Benstein, J.; Horwitz, L.I. Decreasing Incidence of Acute Kidney Injury in Patients with COVID-19 Critical Illness in New York City. Kidney Int. Rep. 2021, 6, 916–927. [Google Scholar] [CrossRef]
- Husain-Syed, F.; Birk, H.W.; Ronco, C. Coronavirus Disease 2019 and Acute Kidney Injury: What Have We Learned? Kidney Int. Rep. 2021, 6, 872–874. [Google Scholar] [CrossRef]
- Legrand, M.; Bell, S.; Forni, L.; Joannidis, M.; Koyner, J.L.; Liu, K.; Cantaluppi, V. Pathophysiology of COVID-19-associated acute kidney injury. Nat. Rev. Nephrol. 2021, 17, 751–764. [Google Scholar] [CrossRef]
- Silver, S.A.; Beaubien-Souligny, W.; Shah, P.S.; Harel, S.; Blum, D.; Kishibe, T.; Meraz-Munoz, A.; Wald, R.; Harel, Z. The Prevalence of Acute Kidney Injury in Patients Hospitalized with COVID-19 Infection: A Systematic Review and Meta-analysis. Kidney Med. 2021, 3, 83–98.e1. [Google Scholar] [CrossRef]
- Pelayo, J.; Lo, K.B.; Bhargav, R.; Gul, F.; Peterson, E.; DeJoy Iii, R.; Salacup, G.F.; Albano, J.; Gopalakrishnan, A.; Azmaiparashvili, Z.; et al. Clinical Characteristics and Outcomes of Community- and Hospital-Acquired Acute Kidney Injury with COVID-19 in a US Inner City Hospital System. Cardiorenal Med. 2020, 10, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Pei, G.; Zhang, Z.; Peng, J.; Liu, L.; Zhang, C.; Yu, C.; Ma, Z.; Huang, Y.; Liu, W.; Yao, Y.; et al. Renal Involvement and Early Prognosis in Patients with COVID-19 Pneumonia. J. Am. Soc. Nephrol. 2020, 31, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Chaudhri, I.; Moffitt, R.; Taub, E.; Annadi, R.R.; Hoai, M.; Bolotova, O.; Yoo, J.; Dhaliwal, S.; Sahib, H.; Daccueil, F.; et al. Association of Proteinuria and Hematuria with Acute Kidney Injury and Mortality in Hospitalized Patients with COVID-19. Kidney Blood Press. Res. 2020, 45, 1018–1032. [Google Scholar] [CrossRef] [PubMed]
- Paek, J.H.; Kim, Y.; Park, W.Y.; Jin, K.; Hyun, M.; Lee, J.Y.; Kim, H.A.; Kwon, Y.S.; Park, J.S.; Han, S. Severe acute kidney injury in COVID-19 patients is associated with in-hospital mortality. PLoS ONE 2020, 15, e0243528. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Luo, R.; Wang, X.; Wang, K.; Zhang, N.; Zhang, M.; Wang, Z.; Dong, L.; Li, J.; Zeng, R.; et al. The Incidence, Risk Factors, and Prognosis of Acute Kidney Injury in Adult Patients with Coronavirus Disease 2019. Clin. J. Am. Soc. Nephrol. 2020, 15, 1394–1402. [Google Scholar] [CrossRef]
- Ng, J.H.; Hirsch, J.S.; Hazzan, A.; Wanchoo, R.; Shah, H.H.; Malieckal, D.A.; Ross, D.W.; Sharma, P.; Sakhiya, V.; Fishbane, S.; et al. Outcomes among Patients Hospitalized with COVID-19 and Acute Kidney Injury. Am. J. Kidney Dis. 2021, 77, 204–215.e1. [Google Scholar] [CrossRef]
- Perez Ingles, D.; Illescas, A.; Perryman Collins, N.; Jordyn, A.N.; Marinaro, J.L.; Argyropoulos, C.; Teixeira, J.P. Impact of COVID-19 Pandemic on Crude Mortality Rates Associated with Acute Kidney Injury Requiring Continuous Renal Replacement Therapy: A Single-Center Study [abstract]. Am. J. Respir. Crit. Care Med. 2021, 203, A2582. [Google Scholar]
- Russo, E.; Esposito, P.; Taramasso, L.; Magnasco, L.; Saio, M.; Briano, F.; Russo, C.; Dettori, S.; Vena, A.; Di Biagio, A.; et al. Kidney disease and all-cause mortality in patients with COVID-19 hospitalized in Genoa, Northern Italy. J. Nephrol. 2021, 34, 173–183. [Google Scholar] [CrossRef]
- Domecq, J.P.; Lal, A.; Sheldrick, C.R.; Kumar, V.K.; Boman, K.; Bolesta, S.; Bansal, V.; Harhay, M.O.; Garcia, M.A.; Kaufman, M.; et al. Outcomes of Patients with Coronavirus Disease 2019 Receiving Organ Support Therapies: The International Viral Infection and Respiratory Illness Universal Study Registry. Crit. Care Med. 2021, 49, 437–448. [Google Scholar] [CrossRef]
- Liu, Y.F.; Zhang, Z.; Pan, X.L.; Xing, G.L.; Zhang, Y.; Liu, Z.S.; Tu, S.H. The chronic kidney disease and acute kidney injury involvement in COVID-19 pandemic: A systematic review and meta-analysis. PLoS ONE 2021, 16, e0244779. [Google Scholar] [CrossRef]
- Coca, A.; Burballa, C.; Centellas-Perez, F.J.; Perez-Saez, M.J.; Bustamante-Munguira, E.; Ortega, A.; Duenas, C.; Arenas, M.D.; Perez-Martinez, J.; Ruiz, G.; et al. Outcomes of COVID-19 among Hospitalized Patients with Non-dialysis CKD. Front. Med. 2020, 7, 615312. [Google Scholar] [CrossRef]
- Henry, B.M.; Lippi, G. Chronic kidney disease is associated with severe coronavirus disease 2019 (COVID-19) infection. Int. Urol. Nephrol. 2020, 52, 1193–1194. [Google Scholar] [CrossRef] [Green Version]
- Williamson, E.J.; Walker, A.J.; Bhaskaran, K.; Bacon, S.; Bates, C.; Morton, C.E.; Curtis, H.J.; Mehrkar, A.; Evans, D.; Inglesby, P.; et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020, 584, 430–436. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Locke, E.; Green, P.; Berry, K.; O’Hare, A.M.; Shah, J.A.; Crothers, K.; Eastment, M.C.; Dominitz, J.A.; Fan, V.S. Risk Factors for Hospitalization, Mechanical Ventilation, or Death among 10131 US Veterans with SARS-CoV-2 Infection. JAMA Netw. Open 2020, 3, e2022310. [Google Scholar] [CrossRef] [PubMed]
- Portoles, J.; Marques, M.; Lopez-Sanchez, P.; de Valdenebro, M.; Munez, E.; Serrano, M.L.; Malo, R.; Garcia, E.; Cuervas, V. Chronic kidney disease and acute kidney injury in the COVID-19 Spanish outbreak. Nephrol. Dial. Transplant. 2020, 35, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Brogan, M.; Ross, M.J. The Impact of Chronic Kidney Disease on Outcomes of Patients with COVID-19 Admitted to the Intensive Care Unit. Nephron 2022, 146, 67–71. [Google Scholar] [CrossRef]
- Pakhchanian, H.; Raiker, R.; Mukherjee, A.; Khan, A.; Singh, S.; Chatterjee, A. Outcomes of COVID-19 in CKD Patients: A Multicenter Electronic Medical Record Cohort Study. Clin. J. Am. Soc. Nephrol. 2021, 16, 785–786. [Google Scholar] [CrossRef]
- Gibertoni, D.; Reno, C.; Rucci, P.; Fantini, M.P.; Buscaroli, A.; Mosconi, G.; Rigotti, A.; Giudicissi, A.; Mambelli, E.; Righini, M.; et al. COVID-19 incidence and mortality in non-dialysis chronic kidney disease patients. PLoS ONE 2021, 16, e0254525. [Google Scholar] [CrossRef]
- Flythe, J.E.; Assimon, M.M.; Tugman, M.J.; Chang, E.H.; Gupta, S.; Shah, J.; Sosa, M.A.; Renaghan, A.D.; Melamed, M.L.; Wilson, F.P.; et al. Characteristics and Outcomes of Individuals with Pre-existing Kidney Disease and COVID-19 Admitted to Intensive Care Units in the United States. Am. J. Kidney Dis. 2021, 77, 190–203.e1. [Google Scholar] [CrossRef]
- Ozturk, S.; Turgutalp, K.; Arici, M.; Gok, M.; Islam, M.; Altiparmak, M.R.; Aydin, Z.; Doner, B.; Eren, N.; Sengul, E.; et al. Characteristics and outcomes of hospitalised older patients with chronic kidney disease and COVID-19: A multicenter nationwide controlled study. Int. J. Clin. Pract. 2021, 75, e14428. [Google Scholar] [CrossRef]
- Ozturk, S.; Turgutalp, K.; Arici, M.; Odabas, A.R.; Altiparmak, M.R.; Aydin, Z.; Cebeci, E.; Basturk, T.; Soypacaci, Z.; Sahin, G.; et al. Mortality analysis of COVID-19 infection in chronic kidney disease, haemodialysis and renal transplant patients compared with patients without kidney disease: A nationwide analysis from Turkey. Nephrol. Dial. Transplant. 2020, 35, 2083–2095. [Google Scholar] [CrossRef]
- Yang, D.; Xiao, Y.; Chen, J.; Chen, Y.; Luo, P.; Liu, Q.; Yang, C.; Xiong, M.; Zhang, Y.; Liu, X.; et al. COVID-19 and chronic renal disease: Clinical characteristics and prognosis. QJM 2020, 113, 799–805. [Google Scholar] [CrossRef]
- Iaccarino, G.; Grassi, G.; Borghi, C.; Ferri, C.; Salvetti, M.; Volpe, M. SARS-RAS Investigators, Age and Multimorbidity Predict Death among COVID-19 Patients: Results of the SARS-RAS Study of the Italian Society of Hypertension. Hypertension 2020, 76, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Oetjens, M.T.; Luo, J.Z.; Chang, A.; Leader, J.B.; Hartzel, D.N.; Moore, B.S.; Strande, N.T.; Kirchner, H.L.; Ledbetter, D.H.; Justice, A.E.; et al. Electronic health record analysis identifies kidney disease as the leading risk factor for hospitalization in confirmed COVID-19 patients. PLoS ONE 2020, 15, e0242182. [Google Scholar] [CrossRef] [PubMed]
- Carlson, N.; Nelveg-Kristensen, K.E.; Freese Ballegaard, E.; Feldt-Rasmussen, B.; Hornum, M.; Kamper, A.L.; Gislason, G.; Torp-Pedersen, C. Increased vulnerability to COVID-19 in chronic kidney disease. J. Intern. Med. 2021, 290, 166–178. [Google Scholar] [CrossRef]
- Bowe, B.; Xie, Y.; Xu, E.; Al-Aly, Z. Kidney Outcomes in Long COVID. J. Am. Soc. Nephrol. 2021, 32, 2851–2862. [Google Scholar] [CrossRef]
- Al-Aly, Z.; Xie, Y.; Bowe, B. High-dimensional characterization of post-acute sequelae of COVID-19. Nature 2021, 594, 259–264. [Google Scholar] [CrossRef]
- Huang, C.; Huang, L.; Wang, Y.; Li, X.; Ren, L.; Gu, X.; Kang, L.; Guo, L.; Liu, M.; Zhou, X.; et al. 6-month consequences of COVID-19 in patients discharged from hospital: A cohort study. Lancet 2021, 397, 220–232. [Google Scholar] [CrossRef]
- Daugherty, S.E.; Guo, Y.; Heath, K.; Dasmarinas, M.C.; Jubilo, K.G.; Samranvedhya, J.; Lipsitch, M.; Cohen, K. Risk of clinical sequelae after the acute phase of SARS-CoV-2 infection: Retrospective cohort study. BMJ 2021, 373, n1098. [Google Scholar] [CrossRef]
- Nugent, J.; Aklilu, A.; Yamamoto, Y.; Simonov, M.; Li, F.; Biswas, A.; Ghazi, L.; Greenberg, H.; Mansour, G.; Moledina, G.; et al. Assessment of Acute Kidney Injury and Longitudinal Kidney Function after Hospital Discharge among Patients with and without COVID-19. JAMA Netw. Open 2021, 4, e211095. [Google Scholar] [CrossRef]
- Su, H.; Yang, M.; Wan, C.; Yi, L.X.; Tang, F.; Zhu, H.Y.; Yi, F.; Yang, H.C.; Fogo, A.B.; Nie, X.; et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020, 98, 219–227. [Google Scholar] [CrossRef]
- Golmai, P.; Larsen, C.P.; DeVita, M.V.; Wahl, S.J.; Weins, A.; Rennke, H.G.; Bijol, V.; Rosenstock, J.L. Histopathologic and Ultrastructural Findings in Postmortem Kidney Biopsy Material in 12 Patients with AKI and COVID-19. J. Am. Soc. Nephrol. 2020, 31, 1944–1947. [Google Scholar] [CrossRef]
- Alexander, M.P.; Mangalaparthi, K.K.; Madugundu, A.K.; Moyer, A.M.; Adam, B.A.; Mengel, M.; Singh, S.; Herrmann, S.M.; Rule, A.D.; Cheek, E.H.; et al. Acute Kidney Injury in Severe COVID-19 Has Similarities to Sepsis-Associated Kidney Injury: A Multi-Omics Study. Mayo Clin. Proc. 2021, 96, 2561–2575. [Google Scholar] [CrossRef]
- Vasquez-Bonilla, W.O.; Orozco, R.; Argueta, V.; Sierra, M.; Zambrano, L.I.; Munoz-Lara, F.; Lopez-Molina, D.S.; Arteaga-Livias, K.; Grimes, Z.; Bryce, C.; et al. A review of the main histopathological findings in coronavirus disease 2019. Hum. Pathol. 2020, 105, 74–83. [Google Scholar] [CrossRef]
- Xia, P.; Wen, Y.; Duan, Y.; Su, H.; Cao, W.; Xiao, M.; Ma, J.; Zhou, Y.; Chen, G.; Jiang, W.; et al. Clinicopathological Features and Outcomes of Acute Kidney Injury in Critically Ill COVID-19 with Prolonged Disease Course: A Retrospective Cohort. J. Am. Soc. Nephrol. 2020, 31, 2205–2221. [Google Scholar] [CrossRef]
- Nasr, S.H.; Alexander, M.P.; Cornell, L.D.; Herrera, L.H.; Fidler, M.E.; Said, S.M.; Zhang, P.; Larsen, C.P.; Sethi, S. Kidney Biopsy Findings in Patients with COVID-19, Kidney Injury, and Proteinuria. Am. J. Kidney Dis. 2021, 77, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Larsen, C.P.; Hernandez-Arroyo, C.F.; Mohamed, M.M.B.; Caza, T.; Sharshir, M.; Chughtai, A.; Xie, L.; Gimenez, J.M.; Sandow, T.A.; et al. AKI and Collapsing Glomerulopathy Associated with COVID-19 and APOL 1 High-Risk Genotype. J. Am. Soc. Nephrol. 2020, 31, 1688–1695. [Google Scholar] [CrossRef] [PubMed]
- Kudose, S.; Batal, I.; Santoriello, D.; Xu, K.; Barasch, J.; Peleg, Y.; Canetta, P.; Ratner, L.E.; Marasa, M.; Gharavi, A.G.; et al. Kidney Biopsy Findings in Patients with COVID-19. J. Am. Soc. Nephrol. 2020, 31, 1959–1968. [Google Scholar] [CrossRef]
- Larsen, C.P.; Bourne, T.D.; Wilson, J.D.; Saqqa, O.; Sharshir, M.A. Collapsing Glomerulopathy in a Patient with COVID-19. Kidney Int. Rep. 2020, 5, 935–939. [Google Scholar] [CrossRef]
- Ferlicot, S.; Jamme, M.; Gaillard, F.; Oniszczuk, J.; Couturier, A.; May, O.; Grunenwald, A.; Sannier, A.; Moktefi, A.; Le Monnier, O.; et al. The spectrum of kidney biopsies in hospitalized patients with COVID-19, acute kidney injury, and/or proteinuria. Nephrol. Dial. Transplant. 2021, 12, gfab042. [Google Scholar] [CrossRef]
- Sharma, P.; Uppal, N.N.; Wanchoo, R.; Shah, H.H.; Yang, Y.; Parikh, R.; Khanin, Y.; Madireddy, V.; Larsen, C.P.; Jhaveri, K.D.; et al. COVID-19-Associated Kidney Injury: A Case Series of Kidney Biopsy Findings. J. Am. Soc. Nephrol. 2020, 31, 1948–1958. [Google Scholar] [CrossRef]
- May, R.M.; Cassol, C.; Hannoudi, A.; Larsen, C.P.; Lerma, E.V.; Haun, R.S.; Braga, J.R.; Hassen, S.I.; Wilson, J.; VanBeek, C.; et al. A multi-center retrospective cohort study defines the spectrum of kidney pathology in Coronavirus 2019 Disease (COVID-19). Kidney Int. 2021, 100, 1303–1315. [Google Scholar] [CrossRef]
- Akilesh, S.; Nast, C.C.; Yamashita, M.; Henriksen, K.; Charu, V.; Troxell, M.L.; Kambham, N.; Bracamonte, E.; Houghton, D.; Ahmed, N.I.; et al. Multicenter Clinicopathologic Correlation of Kidney Biopsies Performed in COVID-19 Patients Presenting with Acute Kidney Injury or Proteinuria. Am. J. Kidney Dis. 2021, 77, 82–93.e1. [Google Scholar] [CrossRef]
- Hanley, B.; Naresh, K.N.; Roufosse, C.; Nicholson, A.G.; Weir, J.; Cooke, G.S.; Thursz, M.; Manousou, P.; Corbett, R.; Goldin, R.; et al. Histopathological findings and viral tropism in UK patients with severe fatal COVID-19: A post-mortem study. Lancet Microbe 2020, 1, e245–e253. [Google Scholar] [CrossRef]
- Schurink, B.; Roos, E.; Radonic, T.; Barbe, E.; Bouman, C.S.C.; de Boer, H.H.; de Bree, G.J.; Bulle, E.B.; Aronica, E.M.; Florquin, S.; et al. Viral presence and immunopathology in patients with lethal COVID-19: A prospective autopsy cohort study. Lancet Microbe 2020, 1, e290–e299. [Google Scholar] [CrossRef]
- Jhaveri, K.D.; Meir, L.R.; Flores Chang, B.S.; Parikh, R.; Wanchoo, R.; Barilla-LaBarca, M.L.; Bijol, V.; Hajizadeh, N. Thrombotic microangiopathy in a patient with COVID-19. Kidney Int. 2020, 98, 509–512. [Google Scholar] [CrossRef]
- El Shamy, O.; Munoz-Casablanca, N.; Coca, S.; Sharma, S.; Lookstein, R.; Uribarri, J. Bilateral Renal Artery Thrombosis in a Patient with COVID-19. Kidney Med. 2021, 3, 116–119. [Google Scholar] [CrossRef]
- Hernandez-Arroyo, C.F.; Varghese, V.; Mohamed, M.M.B.; Velez, J.C.Q. Urinary Sediment Microscopy in Acute Kidney Injury Associated with COVID-19. Kidney360 2020, 1, 819. [Google Scholar] [CrossRef]
- Johnson, A.C.M.; Zager, R.A. Mechanisms Underlying Increased TIMP2 and IGFBP7 Urinary Excretion in Experimental AKI. J. Am. Soc. Nephrol. 2018, 29, 2157–2167. [Google Scholar] [CrossRef]
- Werion, A.; Belkhir, L.; Perrot, M.; Schmit, G.; Aydin, S.; Chen, Z.; Penaloza, A.; De Greef, J.; Yildiz, H.; Pothen, L.; et al. SARS-CoV-2 causes a specific dysfunction of the kidney proximal tubule. Kidney Int. 2020, 98, 1296–1307. [Google Scholar] [CrossRef]
- Kormann, R.; Jacquot, A.; Alla, A.; Corbel, A.; Koszutski, M.; Voirin, P.; Garcia Parrilla, M.; Bevilacqua, S.; Schvoerer, E.; Gueant, J.L.; et al. Coronavirus disease 2019: Acute Fanconi syndrome precedes acute kidney injury. Clin. Kidney J. 2020, 13, 362–370. [Google Scholar] [CrossRef]
- Morell-Garcia, D.; Ramos-Chavarino, D.; Bauca, J.M.; Argente Del Castillo, P.; Ballesteros-Vizoso, M.A.; Garcia de Guadiana-Romualdo, L.; Gomez-Cobo, C.; Pou, J.A.; Amezaga-Menendez, R.; Alonso-Fernandez, A.; et al. Urine biomarkers for the prediction of mortality in COVID-19 hospitalized patients. Sci. Rep. 2021, 11, 11134. [Google Scholar] [CrossRef]
- Sundaram, S.; Soni, M.; Annigeri, R. Urine abnormalities predict acute kidney injury in COVID-19 patients: An analysis of 110 cases in Chennai, South India. Diabetes Metab. Syndr. 2021, 15, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Caceres, P.S.; Savickas, G.; Murray, S.L.; Umanath, K.; Uduman, J.; Yee, J.; Liao, T.D.; Bolin, S.; Levin, A.M.; Khan, M.N.; et al. High SARS-CoV-2 Viral Load in Urine Sediment Correlates with Acute Kidney Injury and Poor COVID-19 Outcome. J. Am. Soc. Nephrol. 2021, 32, 2517–2528. [Google Scholar] [CrossRef] [PubMed]
- Omer, D.; Pleniceanu, O.; Gnatek, Y.; Namestnikov, M.; Cohen-Zontag, O.; Goldberg, S.; Friedman, Y.E.; Friedman, N.; Mandelboim, M.; Vitner, E.B.; et al. Human Kidney Spheroids and Monolayers Provide Insights into SARS-CoV-2 Renal Interactions. J. Am. Soc. Nephrol. 2021, 32, 2242–2254. [Google Scholar] [CrossRef]
- Peng, S.; Wang, H.Y.; Sun, X.; Li, P.; Ye, Z.; Li, Q.; Wang, J.; Shi, X.; Liu, L.; Yao, Y.; et al. Early versus late acute kidney injury among patients with COVID-19-a multicenter study from Wuhan, China. Nephrol. Dial. Transplant. 2020, 35, 2095–2102. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Chen, L.; Yang, C.R.; Raghuram, V.; Khundmiri, S.J.; Knepper, M.A. Does SARS-CoV-2 Infect the Kidney? J. Am. Soc. Nephrol. 2020, 31, 2746–2748. [Google Scholar] [CrossRef] [PubMed]
- Sang, L.; Chen, S.; Zheng, X.; Guan, W.; Zhang, Z.; Liang, W.; Zhong, M.; Jiang, L.; Pan, C.; Zhang, W.; et al. The incidence, risk factors and prognosis of acute kidney injury in severe and critically ill patients with COVID-19 in mainland China: A retrospective study. BMC Pulm. Med. 2020, 20, 290. [Google Scholar] [CrossRef] [PubMed]
- Van den Akker, J.P.; Egal, M.; Groeneveld, A.B. Invasive mechanical ventilation as a risk factor for acute kidney injury in the critically ill: A systematic review and meta-analysis. Crit. Care 2013, 17, R98. [Google Scholar] [CrossRef] [Green Version]
- Kuiper, J.W.; Groeneveld, A.B.; Slutsky, A.S.; Plotz, F.B. Mechanical ventilation and acute renal failure. Crit. Care Med. 2005, 33, 1408–1415. [Google Scholar] [CrossRef]
- Strom, T.; Johansen, R.R.; Prahl, J.O.; Toft, P. Sedation and renal impairment in critically ill patients: A post hoc analysis of a randomized trial. Crit. Care 2011, 15, R119. [Google Scholar] [CrossRef] [Green Version]
- Joannidis, M.; Druml, W.; Forni, L.G.; Groeneveld, A.B.J.; Honore, P.M.; Hoste, E.; Ostermann, M.; Oudemans-van Straaten, H.M.; Schetz, M. Prevention of acute kidney injury and protection of renal function in the intensive care unit: Update 2017: Expert opinion of the Working Group on Prevention, AKI section, European Society of Intensive Care Medicine. Intensive Care Med. 2017, 43, 730–749. [Google Scholar] [CrossRef]
- Fogagnolo, A.; Grasso, S.; Dres, M.; Gesualdo, L.; Murgolo, F.; Morelli, E.; Ottaviani, I.; Marangoni, E.; Volta, C.A.; Spadaro, S. Focus on renal blood flow in mechanically ventilated patients with SARS-CoV-2: A prospective pilot study. J. Clin. Monit. Comput. 2021, 1–7. [Google Scholar] [CrossRef]
- Bhasin, B.; Veitla, V.; Dawson, A.Z.; Garacci, Z.; Sturgill, D.; Ozieh, M.N.; Regner, K.R. AKI in Hospitalized Patients with COVID-19 and Seasonal Influenza: A Comparative Analysis. Kidney360 2021, 2, 619. [Google Scholar] [CrossRef]
- Taha, M.; Sano, D.; Hanoudi, S.; Esber, Z.; Elahi, M.; Gabali, A.; Chopra, T.; Draghici, S.; Samavati, L. Platelets and renal failure in the SARS-CoV-2 syndrome. Platelets 2021, 32, 130–137. [Google Scholar] [CrossRef]
- Macor, P.; Durigutto, P.; Mangogna, A.; Bussani, R.; De Maso, L.; D’Errico, S.; Zanon, M.; Pozzi, N.; Meroni, P.L.; Tedesco, F. Multiple-Organ Complement Deposition on Vascular Endothelium in COVID-19 Patients. Biomedicines 2021, 9, 1003. [Google Scholar] [CrossRef]
- Vijayan, A.; Humphreys, B.D. SARS-CoV-2 in the kidney: Bystander or culprit? Nat. Rev. Nephrol. 2020, 16, 703–704. [Google Scholar] [CrossRef]
- Batlle, D.; Soler, M.J.; Sparks, M.A.; Hiremath, S.; South, A.M.; Welling, P.A.; Swaminathan, S. COVID-19 and ACE2 in Cardiovascular, Lung, and Kidney Working Group, Acute Kidney Injury in COVID-19: Emerging Evidence of a Distinct Pathophysiology. J. Am. Soc. Nephrol. 2020, 31, 1380–1383. [Google Scholar] [CrossRef]
- Farkash, E.A.; Wilson, A.M.; Jentzen, J.M. Ultrastructural Evidence for Direct Renal Infection with SARS-CoV-2. J. Am. Soc. Nephrol. 2020, 31, 1683–1687. [Google Scholar] [CrossRef]
- Couturier, A.; Ferlicot, S.; Chevalier, K.; Guillet, M.; Essig, M.; Jaureguiberry, S.; Collarino, R.; Dargelos, M.; Michaut, A.; Geri, G.; et al. Indirect effects of severe acute respiratory syndrome coronavirus 2 on the kidney in coronavirus disease patients. Clin. Kidney J. 2020, 13, 347–353. [Google Scholar] [CrossRef]
- Rossi, G.M.; Delsante, M.; Pilato, F.P.; Gnetti, L.; Gabrielli, L.; Rossini, G.; Re, M.C.; Cenacchi, G.; Affanni, P.; Colucci, M.E.; et al. Kidney Biopsy Findings in a Critically Ill COVID-19 Patient with Dialysis-Dependent Acute Kidney Injury: A Case Against “SARS-CoV-2 Nephropathy”. Kidney Int. Rep. 2020, 5, 1100–1105. [Google Scholar] [CrossRef]
- Smith, K.D.; Akilesh, S.; Alpers, C.E.; Nicosia, R.F. Am I a coronavirus? Kidney Int. 2020, 98, 506–507. [Google Scholar] [CrossRef]
- Roufosse, C.; Curtis, E.; Moran, L.; Hollinshead, M.; Cook, T.; Hanley, B.; Horsfield, C.; Neil, D. Electron microscopic investigations in COVID-19: Not all crowns are coronas. Kidney Int. 2020, 98, 505–506. [Google Scholar] [PubMed]
- Calomeni, E.; Satoskar, A.; Ayoub, I.; Brodsky, S.; Rovin, B.H.; Nadasdy, T. Multivesicular bodies mimicking SARS-CoV-2 in patients without COVID-19. Kidney Int. 2020, 98, 233–234. [Google Scholar] [PubMed]
- Miller, S.E.; Brealey, J.K. Visualization of putative coronavirus in kidney. Kidney Int. 2020, 98, 231–232. [Google Scholar]
- Cassol, C.A.; Gokden, N.; Larsen, C.P.; Bourne, T.D. Appearances Can Be Deceiving—Viral-like Inclusions in COVID-19 Negative Renal Biopsies by Electron Microscopy. Kidney360 2020, 1, 824. [Google Scholar] [CrossRef]
- Diao, B.; Wang, C.; Wang, R.; Feng, Z.; Zhang, J.; Yang, H.; Tan, Y.; Wang, H.; Wang, C.; Liu, L.; et al. Human kidney is a target for novel severe acute respiratory syndrome coronavirus 2 infection. Nat. Commun. 2021, 12, 2506. [Google Scholar] [CrossRef] [PubMed]
- Frithiof, R.; Bergqvist, A.; Jarhult, J.D.; Lipcsey, M.; Hultstrom, M. Presence of SARS-CoV-2 in urine is rare and not associated with acute kidney injury in critically ill COVID-19 patients. Crit. Care 2020, 24, 587. [Google Scholar] [CrossRef] [PubMed]
- Hassler, L.; Reyes, F.; Sparks, M.A.; Welling, P.; Batlle, D. Evidence for and Against Direct Kidney Infection by SARS-CoV-2 in Patients with COVID-19. Clin. J. Am. Soc. Nephrol. 2021, 16, 1755–1765. [Google Scholar] [PubMed]
- Zhang, Y.M.; Zhang, H. Genetic Roadmap for Kidney Involvement of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection. Clin. J. Am. Soc. Nephrol. 2020, 15, 1044–1046. [Google Scholar]
- Pan, X.W.; Xu, D.; Zhang, H.; Zhou, W.; Wang, L.H.; Cui, X.G. Identification of a potential mechanism of acute kidney injury during the COVID-19 outbreak: A study based on single-cell transcriptome analysis. Intensive Care Med. 2020, 46, 1114–1116. [Google Scholar] [CrossRef] [Green Version]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- Lin, W.; Fan, J.; Hu, L.F.; Zhang, Y.; Ooi, J.D.; Meng, T.; Jin, P.; Ding, X.; Peng, L.K.; Song, L.; et al. Single-cell analysis of angiotensin-converting enzyme II expression in human kidneys and bladders reveals a potential route of 2019 novel coronavirus infection. Chin. Med. J. 2021, 134, 935–943. [Google Scholar] [CrossRef]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Kissling, S.; Rotman, S.; Gerber, C.; Halfon, M.; Lamoth, F.; Comte, D.; Lhopitallier, L.; Sadallah, S.; Fakhouri, F. Collapsing glomerulopathy in a COVID-19 patient. Kidney Int. 2020, 98, 228–231. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Bradley, B.T.; Maioli, H.; Johnston, R.; Chaudhry, I.; Fink, S.L.; Xu, H.; Najafian, B.; Deutsch, G.; Lacy, J.M.; Williams, T.; et al. Histopathology and ultrastructural findings of fatal COVID-19 infections in Washington State: A case series. Lancet 2020, 396, 320–332. [Google Scholar] [CrossRef]
- Pesaresi, M.; Pirani, F.; Tagliabracci, A.; Valsecchi, M.; Procopio, A.D.; Busardo, F.P.; Graciotti, L. SARS-CoV-2 identification in lungs, heart and kidney specimens by transmission and scanning electron microscopy. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 5186–5188. [Google Scholar]
- Puelles, V.G.; Lutgehetmann, M.; Lindenmeyer, M.T.; Sperhake, J.P.; Wong, M.N.; Allweiss, L.; Chilla, S.; Heinemann, A.; Wanner, N.; Liu, S.; et al. Multiorgan and Renal Tropism of SARS-CoV-2. N. Engl. J. Med. 2020, 383, 590–592. [Google Scholar] [CrossRef] [PubMed]
- Braun, F.; Lutgehetmann, M.; Pfefferle, S.; Wong, M.N.; Carsten, A.; Lindenmeyer, M.T.; Norz, D.; Heinrich, F.; Meissner, K.; Wichmann, D.; et al. SARS-CoV-2 renal tropism associates with acute kidney injury. Lancet 2020, 396, 597–598. [Google Scholar] [CrossRef]
- Santoriello, D.; Khairallah, P.; Bomback, A.S.; Xu, K.; Kudose, S.; Batal, I.; Barasch, J.; Radhakrishnan, J.; D’Agati, V.; Markowitz, G. Postmortem Kidney Pathology Findings in Patients with COVID-19. J. Am. Soc. Nephrol. 2020, 31, 2158–2167. [Google Scholar] [CrossRef]
- Dargelos, M.; Couturier, A.; Ferlicot, S.; Goujon, J.M.; Roque-Afonso, A.M.; Gault, E.; Touchard, G.; Ory, C.; Kaaki, S.; Vilaine, E.; et al. Severe acute respiratory syndrome coronavirus 2 indirectly damages kidney structures. Clin. Kidney J. 2020, 13, 1101–1104. [Google Scholar] [CrossRef] [PubMed]
- Best Rocha, A.; Stroberg, E.; Barton, L.M.; Duval, E.J.; Mukhopadhyay, S.; Yarid, N.; Caza, T.; Wilson, J.D.; Kenan, D.J.; Kuperman, M.; et al. Detection of SARS-CoV-2 in formalin-fixed paraffin-embedded tissue sections using commercially available reagents. Lab. Investig. 2020, 100, 1485–1489. [Google Scholar] [CrossRef] [PubMed]
- Delorey, T.M.; Ziegler, C.G.K.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, A.; Abbondanza, D.; Fleming, S.J.; Subramanian, A.; Montoro, D.T.; et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 2021, 595, 107–113. [Google Scholar] [CrossRef]
- Dorward, D.A.; Russell, C.D.; Um, I.H.; Elshani, M.; Armstrong, S.D.; Penrice-Randal, R.; Millar, T.; Lerpiniere, C.E.B.; Tagliavini, G.; Hartley, C.S.; et al. Tissue-Specific Immunopathology in Fatal COVID-19. Am. J. Respir. Crit. Care Med. 2021, 203, 192–201. [Google Scholar] [CrossRef]
- Peng, L.; Liu, J.; Xu, W.; Luo, Q.; Chen, D.; Lei, Z.; Huang, Z.; Li, X.; Deng, K.; Lin, B.; et al. SARS-CoV-2 can be detected in urine, blood, anal swabs, and oropharyngeal swabs specimens. J. Med. Virol. 2020, 92, 1676–1680. [Google Scholar] [CrossRef]
- Sun, J.; Zhu, A.; Li, H.; Zheng, K.; Zhuang, Z.; Chen, Z.; Shi, Y.; Zhang, Z.; Chen, S.B.; Liu, X.; et al. Isolation of infectious SARS-CoV-2 from urine of a COVID-19 patient. Emerg. Microbes Infect. 2020, 9, 991–993. [Google Scholar] [CrossRef]
- Jeong, H.W.; Kim, S.M.; Kim, H.S.; Kim, Y.I.; Kim, J.H.; Cho, J.Y.; Kim, S.H.; Kang, H.; Kim, S.G.; Park, S.J.; et al. Viable SARS-CoV-2 in various specimens from COVID-19 patients. Clin. Microbiol. Infect. 2020, 26, 1520–1524. [Google Scholar] [CrossRef]
- Kim, J.M.; Kim, H.M.; Lee, E.J.; Jo, H.J.; Yoon, Y.; Lee, N.J.; Son, J.; Lee, Y.J.; Kim, M.S.; Lee, Y.P.; et al. Detection and Isolation of SARS-CoV-2 in Serum, Urine, and Stool Specimens of COVID-19 Patients from the Republic of Korea. Osong Public Health Res. Perspect. 2020, 11, 112–117. [Google Scholar] [CrossRef]
- Ling, Y.; Xu, S.B.; Lin, Y.X.; Tian, D.; Zhu, Z.Q.; Dai, F.H.; Wu, F.; Song, Z.G.; Huang, W.; Chen, J.; et al. Persistence and clearance of viral RNA in 2019 novel coronavirus disease rehabilitation patients. Chin. Med. J. 2020, 133, 1039–1043. [Google Scholar] [CrossRef]
- Zhang, N.; Gong, Y.; Meng, F.; Shi, Y.; Wang, J.; Mao, P.; Chuai, X.; Bi, Y.; Yang, P.; Wang, F. Comparative study on virus shedding patterns in nasopharyngeal and fecal specimens of COVID-19 patients. Sci. China Life Sci. 2021, 64, 486–488. [Google Scholar] [CrossRef]
- Zheng, S.; Fan, J.; Yu, F.; Feng, B.; Lou, B.; Zou, Q.; Xie, G.; Lin, S.; Wang, R.; Yang, X.; et al. Viral load dynamics and disease severity in patients infected with SARS-CoV-2 in Zhejiang province, China, January-March 2020: Retrospective cohort study. BMJ 2020, 369, m1443. [Google Scholar] [CrossRef] [Green Version]
- Wolfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Muller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020, 581, 465–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lescure, F.X.; Bouadma, L.; Nguyen, D.; Parisey, M.; Wicky, P.H.; Behillil, S.; Gaymard, A.; Bouscambert-Duchamp, M.; Donati, F.; Le Hingrat, Q.; et al. Clinical and virological data of the first cases of COVID-19 in Europe: A case series. Lancet Infect. Dis. 2020, 20, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Mondanizadeh, M.; Hrahimi, E.; Sarmadian, H.; Jamalian, M.; Khansarinejad, B. Evaluation of SARS-CoV-2 Existence in Blood, Urine, and Rectal Swab in Positive Patients with Different Virus Titers. Jundishapur J. Microbiol. 2020, 13, e106534. [Google Scholar] [CrossRef]
- To, K.K.; Tsang, O.T.; Leung, W.S.; Tam, A.R.; Wu, T.C.; Lung, D.C.; Yip, C.C.; Cai, J.P.; Chan, J.M.; Chik, T.S.; et al. Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: An observational cohort study. Lancet Infect. Dis. 2020, 20, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Kujawski, S.A.; Wong, K.K.; Collins, J.P.; Epstein, L.; Killerby, M.; Midgley, C.M.; Abedi, G.R.; Ahmed, N.S.; Almendares, O.; Alvarez, F.N.; et al. COVID-19 Investigation Team, Clinical and virologic characteristics of the first 12 patients with coronavirus disease 2019 (COVID-19) in the United States. Nat. Med. 2020, 26, 861–868. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.F.; Yip, C.C.; To, K.K.; Tang, T.H.; Wong, S.C.; Leung, K.H.; Fung, A.Y.; Ng, A.C.; Zou, Z.; Tsoi, H.W.; et al. Improved Molecular Diagnosis of COVID-19 by the Novel, Highly Sensitive and Specific COVID-19-RdRp/Hel Real-Time Reverse Transcription-PCR Assay Validated In Vitro and with Clinical Specimens. J. Clin. Microbiol. 2020, 58, e00310-20. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Jiang, L.; Huang, G.; Pu, H.; Gong, B.; Lin, H.; Ma, S.; Chen, X.; Long, B.; Si, G.; et al. Comparison of different samples for 2019 novel coronavirus detection by nucleic acid amplification tests. Int. J. Infect. Dis. 2020, 93, 264–267. [Google Scholar] [CrossRef]
- Trypsteen, W.; Van Cleemput, J.; Snippenberg, W.V.; Gerlo, S.; Vandekerckhove, L. On the whereabouts of SARS-CoV-2 in the human body: A systematic review. PLoS Pathog. 2020, 16, e1009037. [Google Scholar] [CrossRef]
- Bwire, G.M.; Majigo, M.V.; Njiro, B.J.; Mawazo, A. Detection profile of SARS-CoV-2 using RT-PCR in different types of clinical specimens: A systematic review and meta-analysis. J. Med. Virol. 2021, 93, 719–725. [Google Scholar] [CrossRef]
- De Souza, N. Organoids. Nat. Methods 2018, 15, 23. [Google Scholar] [CrossRef]
- Buzhor, E.; Harari-Steinberg, O.; Omer, D.; Metsuyanim, S.; Jacob-Hirsch, J.; Noiman, T.; Dotan, Z.; Goldstein, R.S.; Dekel, B. Kidney spheroids recapitulate tubular organoids leading to enhanced tubulogenic potency of human kidney-derived cells. Tissue Eng. Part A 2011, 17, 2305–2319. [Google Scholar] [CrossRef]
- Garreta, E.; Prado, P.; Tarantino, C.; Oria, R.; Fanlo, L.; Marti, E.; Zalvidea, D.; Trepat, X.; Roca-Cusachs, P.; Gavalda-Navarro, A.; et al. Fine tuning the extracellular environment accelerates the derivation of kidney organoids from human pluripotent stem cells. Nat. Mater. 2019, 18, 397–405. [Google Scholar] [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkruys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado Del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e7. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Ogando, N.S.; Dalebout, T.J.; Zevenhoven-Dobbe, J.C.; Limpens, R.; van der Meer, Y.; Caly, L.; Druce, J.; de Vries, J.J.C.; Kikkert, M.; Barcena, M.; et al. SARS-coronavirus-2 replication in Vero E6 cells: Replication kinetics, rapid adaptation and cytopathology. J. Gen. Virol. 2020, 101, 925–940. [Google Scholar] [CrossRef]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. 2020, 176, 104742. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.; Paray, B.A.; Hussain, A.; Qadir, F.A.; Attar, F.; Aziz, F.M.; Sharifi, M.; Derakhshankhah, H.; Rasti, B.; Mehrabi, M.; et al. A review on the cleavage priming of the spike protein on coronavirus by angiotensin-converting enzyme-2 and furin. J. Biomol. Struct. Dyn. 2021, 39, 3025–3033. [Google Scholar] [CrossRef] [Green Version]
- Yamada, Y.; Liu, D.X. Proteolytic activation of the spike protein at a novel RRRR/S motif is implicated in furin-dependent entry, syncytium formation, and infectivity of coronavirus infectious bronchitis virus in cultured cells. J. Virol. 2009, 83, 8744–8758. [Google Scholar] [CrossRef] [Green Version]
- Tchesnokov, E.P.; Feng, J.Y.; Porter, D.P.; Gotte, M. Mechanism of Inhibition of Ebola Virus RNA-Dependent RNA Polymerase by Remdesivir. Viruses 2019, 11, 326. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of COVID-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Heald-Sargent, T.; Gallagher, T. Ready, set, fuse! The coronavirus spike protein and acquisition of fusion competence. Viruses 2012, 4, 557–580. [Google Scholar] [CrossRef] [Green Version]
- Soleimani, M. Acute Kidney Injury in SARS-CoV-2 Infection: Direct Effect of Virus on Kidney Proximal Tubule Cells. Int. J. Mol. Sci. 2020, 21, 3275. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Yao, X.; Ye, F.; Zhang, M.; Cui, C.; Huang, B.; Niu, P.; Liu, X.; Zhao, L.; Dong, E.; Song, C.; et al. In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). Clin. Infect. Dis. 2020, 71, 732–739. [Google Scholar] [CrossRef] [Green Version]
- Shang, C.; Zhuang, X.; Zhang, H.; Li, Y.; Zhu, Y.; Lu, J.; Ge, C.; Cong, J.; Li, T.; Tian, M.; et al. Inhibitors of endosomal acidification suppress SARS-CoV-2 replication and relieve viral pneumonia in hACE2 transgenic mice. Virol. J. 2021, 18, 46. [Google Scholar] [CrossRef]
- RECOVERY Collaborative Group; Horby, P.; Mafham, M.; Linsell, L.; Bell, J.L.; Staplin, N.; Emberson, J.R.; Wiselka, M.; Ustianowski, A.; Elmahi, E.; et al. Effect of Hydroxychloroquine in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2020, 383, 2030–2040. [Google Scholar]
- Tang, W.; Cao, Z.; Han, M.; Wang, Z.; Chen, J.; Sun, W.; Wu, Y.; Xiao, W.; Liu, S.; Chen, E.; et al. Hydroxychloroquine in patients with mainly mild to moderate coronavirus disease 2019: Open label, randomised controlled trial. BMJ 2020, 369, m1849. [Google Scholar] [CrossRef]
- Cavalcanti, A.B.; Zampieri, F.G.; Rosa, R.G.; Azevedo, L.C.P.; Veiga, V.C.; Avezum, A.; Damiani, L.P.; Marcadenti, A.; Kawano-Dourado, L.; Lisboa, T.; et al. Hydroxychloroquine with or without Azithromycin in Mild-to-Moderate COVID-19. N. Engl. J. Med. 2020, 383, 2041–2052. [Google Scholar] [CrossRef] [PubMed]
- Self, W.H.; Semler, M.W.; Leither, L.M.; Casey, J.D.; Angus, D.C.; Brower, R.G.; Chang, S.Y.; Collins, S.P.; Eppensteiner, J.C.; Filbin, M.R.; et al. Effect of Hydroxychloroquine on Clinical Status at 14 Days in Hospitalized Patients with COVID-19: A Randomized Clinical Trial. JAMA 2020, 324, 2165–2176. [Google Scholar] [CrossRef] [PubMed]
- Arabi, Y.M.; Gordon, A.C.; Derde, L.P.G.; Nichol, A.D.; Murthy, S.; Beidh, F.A.; Annane, D.; Swaidan, L.A.; Beane, A.; Beasley, R.; et al. Lopinavir-ritonavir and hydroxychloroquine for critically ill patients with COVID-19: REMAP-CAP randomized controlled trial. Intensive Care Med. 2021, 47, 867–886. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, X.; Xu, Y.; Xu, Z.; Huang, Y.; Chen, S.; Li, S.; Liu, D.; Lin, Z.; Li, Y. Ventilatory Ratio in Hypercapnic Mechanically Ventilated Patients with COVID-19-associated Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2020, 201, 1297–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oppenheimer, B.W.; Bakker, J.; Goldring, R.M.; Teter, K.; Green, D.L.; Berger, K.I. Increased Dead Space Ventilation and Refractory Hypercapnia in Patients with Coronavirus Disease 2019: A Potential Marker of Thrombosis in the Pulmonary Vasculature. Crit. Care Explor. 2020, 2, e0208. [Google Scholar] [CrossRef]
- South, A.M.; Tomlinson, L.; Edmonston, D.; Hiremath, S.; Sparks, M.A. Controversies of renin-angiotensin system inhibition during the COVID-19 pandemic. Nat. Rev. Nephrol. 2020, 16, 305–307. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Eales, J.M.; Scannali, D.; Nazgiewicz, A.; Prestes, P.; Maier, M.; Denniff, M.; Xu, X.; Saluja, S.; Cano-Gamez, E.; et al. Hypertension and renin-angiotensin system blockers are not associated with expression of angiotensin-converting enzyme 2 (ACE2) in the kidney. Eur. Heart J. 2020, 41, 4580–4588. [Google Scholar] [CrossRef]
- Milne, S.; Yang, C.X.; Timens, W.; Bosse, Y.; Sin, D.D. SARS-CoV-2 receptor ACE2 gene expression and RAAS inhibitors. Lancet Respir. Med. 2020, 8, e50–e51. [Google Scholar] [CrossRef]
- Wysocki, J.; Lores, E.; Ye, M.; Soler, M.J.; Batlle, D. Kidney and Lung ACE2 Expression after an ACE Inhibitor or an Ang II Receptor Blocker: Implications for COVID-19. J. Am. Soc. Nephrol. 2020, 31, 1941–1943. [Google Scholar] [CrossRef]
- Mancia, G.; Rea, F.; Ludergnani, M.; Apolone, G.; Corrao, G. Renin-Angiotensin-Aldosterone System Blockers and the Risk of COVID-19. N. Engl. J. Med. 2020, 382, 2431–2440. [Google Scholar] [CrossRef]
- Morales, D.R.; Conover, M.M.; You, S.C.; Pratt, N.; Kostka, K.; Duarte-Salles, T.; Fernandez-Bertolin, S.; Aragon, M.; DuVall, S.L.; Lynch, K.; et al. Renin-angiotensin system blockers and susceptibility to COVID-19: An international, open science, cohort analysis. Lancet Digit. Health 2021, 3, e98–e114. [Google Scholar] [CrossRef]
- Reynolds, H.R.; Adhikari, S.; Pulgarin, C.; Troxel, A.B.; Iturrate, E.; Johnson, S.B.; Hausvater, A.; Newman, J.D.; Berger, J.S.; Bangalore, S.; et al. Renin-Angiotensin-Aldosterone System Inhibitors and Risk of COVID-19. N. Engl. J. Med. 2020, 382, 2441–2448. [Google Scholar] [CrossRef]
- Zhang, P.; Zhu, L.; Cai, J.; Lei, F.; Qin, J.J.; Xie, J.; Liu, Y.M.; Zhao, Y.C.; Huang, X.; Lin, L.; et al. Association of Inpatient Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers with Mortality among Patients with Hypertension Hospitalized with COVID-19. Circ. Res. 2020, 126, 1671–1681. [Google Scholar] [CrossRef]
- Baral, R.; Tsampasian, V.; Debski, M.; Moran, B.; Garg, P.; Clark, A.; Vassiliou, V.S. Association Between Renin-Angiotensin-Aldosterone System Inhibitors and Clinical Outcomes in Patients with COVID-19: A Systematic Review and Meta-analysis. JAMA Netw. Open 2021, 4, e213594. [Google Scholar] [CrossRef]
- Bavishi, C.; Whelton, P.K.; Mancia, G.; Corrao, G.; Messerli, F.H. Renin-angiotensin-system inhibitors and all-cause mortality in patients with COVID-19: A systematic review and meta-analysis of observational studies. J. Hypertens. 2021, 39, 784–794. [Google Scholar] [CrossRef]
- Pranata, R.; Permana, H.; Huang, I.; Lim, M.A.; Soetedjo, N.N.M.; Supriyadi, R.; Soeroto, A.Y.; Alkatiri, A.A.; Firman, D.; Lukito, A.A. The use of renin angiotensin system inhibitor on mortality in patients with coronavirus disease 2019 (COVID-19): A systematic review and meta-analysis. Diabetes Metab. Syndr. 2020, 14, 983–990. [Google Scholar] [CrossRef]
- Cohen, J.B.; Hanff, T.C.; William, P.; Sweitzer, N.; Rosado-Santander, N.R.; Medina, C.; Rodriguez-Mori, J.E.; Renna, N.; Chang, T.I.; Corrales-Medina, V.; et al. Continuation versus discontinuation of renin-angiotensin system inhibitors in patients admitted to hospital with COVID-19: A prospective, randomised, open-label trial. Lancet Respir. Med. 2021, 9, 275–284. [Google Scholar] [CrossRef]
- Lopes, R.D.; Macedo, A.V.S.; de Barros, E.S.P.G.M.; Moll-Bernardes, R.J.; Dos Santos, T.M.; Mazza, L.; Feldman, A.; D’Andrea Saba Arruda, G.; de Albuquerque, D.C.; Camiletti, A.S.; et al. Effect of Discontinuing vs Continuing Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers on Days Alive and Out of the Hospital in Patients Admitted with COVID-19: A Randomized Clinical Trial. JAMA 2021, 325, 254–264. [Google Scholar] [CrossRef]
- Duarte, M.; Pelorosso, F.; Nicolosi, L.N.; Salgado, M.V.; Vetulli, H.; Aquieri, A.; Azzato, F.; Castro, M.; Coyle, J.; Davolos, I.; et al. Telmisartan for treatment of COVID-19 patients: An open multicenter randomized clinical trial. EClinicalMedicine 2021, 37, 100962. [Google Scholar] [CrossRef]
- Touyz, R.M.; Montezano, A.C. Angiotensin-(1-7) and Vascular Function: The Clinical Context. Hypertension 2018, 71, 68–69. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Wang, T.; Zhou, Y.; Zhao, Y.; Zhang, Y.; Li, J. Potential Role of ACE2 in Coronavirus Disease 2019 (COVID-19) Prevention and Management. J. Transl. Int. Med. 2020, 8, 9–19. [Google Scholar] [CrossRef]
- Wosten-van Asperen, R.M.; Lutter, R.; Specht, P.A.; Moll, G.N.; van Woensel, J.B.; van der Loos, C.M.; van Goor, H.; Kamilic, J.; Florquin, S.; Bos, A.P. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1-7) or an angiotensin II receptor antagonist. J. Pathol. 2011, 225, 618–627. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef]
- Zou, Z.; Yan, Y.; Shu, Y.; Gao, R.; Sun, Y.; Li, X.; Ju, X.; Liang, Z.; Liu, Q.; Zhao, Y.; et al. Angiotensin-converting enzyme 2 protects from lethal avian influenza A H5N1 infections. Nat. Commun. 2014, 5, 3594. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Xie, Z.; Li, T.; Zhang, S.; Lai, C.; Zhu, P.; Wang, K.; Han, L.; Duan, Y.; Zhao, Z.; et al. Angiotensin-converting enzyme 2 inhibits lung injury induced by respiratory syncytial virus. Sci. Rep. 2016, 6, 19840. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Benthin, C.; Zeno, B.; Albertson, T.E.; Boyd, J.; Christie, J.D.; Hall, R.; Poirier, G.; Ronco, J.J.; Tidswell, M.; et al. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Crit. Care 2017, 21, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozkan, S.; Cakmak, F.; Konukoglu, D.; Biberoglu, S.; Ipekci, A.; Akdeniz, Y.S.; Bolayirli, I.M.; Balkan, I.I.; Dumanli, G.Y.; Ikizceli, I. Efficacy of Serum Angiotensin II Levels in Prognosis of Patients with Coronavirus Disease 2019. Crit. Care Med. 2021, 49, e613–e623. [Google Scholar] [CrossRef]
- Wang, K.; Gheblawi, M.; Nikhanj, A.; Munan, M.; MacIntyre, E.; O’Neil, C.; Poglitsch, M.; Colombo, D.; Del Nonno, F.; Kassiri, Z.; et al. Dysregulation of ACE (Angiotensin-Converting Enzyme)-2 and Renin-Angiotensin Peptides in SARS-CoV-2 Mediated Mortality and End-Organ Injuries. Hypertension 2022, 79, 365–378. [Google Scholar] [CrossRef]
- Haga, S.; Yamamoto, N.; Nakai-Murakami, C.; Osawa, Y.; Tokunaga, K.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc. Natl Acad Sci. USA 2008, 105, 7809–7814. [Google Scholar] [CrossRef] [Green Version]
- Palau, V.; Riera, M.; Soler, M.J. ADAM17 inhibition may exert a protective effect on COVID-19. Nephrol. Dial. Transplant. 2020, 35, 1071–1072. [Google Scholar] [CrossRef]
- Dudoignon, E.; Moreno, N.; Deniau, B.; Coutrot, M.; Longer, R.; Amiot, Q.; Mebazaa, A.; Pirracchio, R.; Depret, F.; Legrand, M. Activation of the renin-angiotensin-aldosterone system is associated with Acute Kidney Injury in COVID-19. Anaesth. Crit. Care Pain Med. 2020, 39, 453–455. [Google Scholar] [CrossRef]
- Jayk Bernal, A.; Gomes da Silva, M.M.; Musungaie, D.B.; Kovalchuk, E.; Gonzalez, A.; Delos Reyes, V.; Martin-Quiros, A.; Caraco, Y.; Williams-Diaz, A.; Brown, M.L.; et al. Molnupiravir for Oral Treatment of COVID-19 in Nonhospitalized Patients. N. Engl. J. Med. 2021. [Google Scholar] [CrossRef]
- Ng, S.; Cowling, B.J.; Fang, V.J.; Chan, K.H.; Ip, D.K.; Cheng, C.K.; Uyeki, T.M.; Houck, P.M.; Malik Peiris, J.S.; Leung, G.M. Effects of oseltamivir treatment on duration of clinical illness and viral shedding and household transmission of influenza virus. Clin. Infect. Dis. 2010, 50, 707–714. [Google Scholar] [CrossRef] [Green Version]
- Siddiqi, H.K.; Mehra, M.R. COVID-19 illness in native and immunosuppressed states: A clinical-therapeutic staging proposal. J. Heart Lung Transplant. 2020, 39, 405–407. [Google Scholar] [CrossRef] [Green Version]
- Sinha, P.; Matthay, M.A.; Calfee, C.S. Is a “Cytokine Storm” Relevant to COVID-19? JAMA Intern. Med. 2020, 180, 1152–1154. [Google Scholar] [CrossRef]
- Kox, M.; Waalders, N.J.B.; Kooistra, E.J.; Gerretsen, J.; Pickkers, P. Cytokine Levels in Critically Ill Patients with COVID-19 and Other Conditions. JAMA 2020, 324, 1565–1567. [Google Scholar] [CrossRef]
- Leisman, D.E.; Ronner, L.; Pinotti, R.; Taylor, M.D.; Sinha, P.; Calfee, C.S.; Hirayama, A.V.; Mastroiani, F.; Turtle, C.J.; Harhay, M.O.; et al. Cytokine elevation in severe and critical COVID-19: A rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir. Med. 2020, 8, 1233–1244. [Google Scholar] [CrossRef]
- Mudd, P.A.; Crawford, J.C.; Turner, J.S.; Souquette, A.; Reynolds, D.; Bender, D.; Bosanquet, J.P.; Anand, N.J.; Striker, D.A.; Martin, R.S.; et al. Distinct inflammatory profiles distinguish COVID-19 from influenza with limited contributions from cytokine storm. Sci. Adv. 2020, 6, eabe3024. [Google Scholar] [CrossRef]
- RECOVERY Collaborative Group; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar]
- WHO Rapid Evidence Appraisal for COVID-19 Therapies (REACT) Working Group; Sterne, J.A.C.; Murthy, S.; Diaz, J.V.; Slutsky, A.S.; Villar, J.; Angus, D.C.; Annane, D.; Azevedo, L.C.P.; Berwanger, O.; et al. Association Between Administration of Systemic Corticosteroids and Mortality among Critically Ill Patients with COVID-19: A Meta-analysis. JAMA 2020, 324, 1330–1341. [Google Scholar]
- REMAP-CAP Investigators; Gordon, A.C.; Mouncey, P.R.; Al-Beidh, F.; Rowan, K.M.; Nichol, A.D.; Arabi, Y.M.; Annane, D.; Beane, A.; van Bentum-Puijk, W.; et al. Interleukin-6 Receptor Antagonists in Critically Ill Patients with COVID-19. N. Engl. J. Med. 2021, 384, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- RECOVERY Collaborative Group. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2021, 397, 1637–1645. [Google Scholar] [CrossRef]
- Marconi, V.C.; Ramanan, A.V.; de Bono, S.; Kartman, C.E.; Krishnan, V.; Liao, R.; Piruzeli, M.L.B.; Goldman, J.D.; Alatorre-Alexander, J.; de Cassia Pellegrini, R.; et al. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): A randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir. Med. 2021, 9, 1407–1418. [Google Scholar] [CrossRef]
- Katzourakis, A. COVID-19: Endemic doesn’t mean harmless. Nature 2022, 601, 485. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teixeira, J.P.; Barone, S.; Zahedi, K.; Soleimani, M. Kidney Injury in COVID-19: Epidemiology, Molecular Mechanisms and Potential Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 2242. https://doi.org/10.3390/ijms23042242
Teixeira JP, Barone S, Zahedi K, Soleimani M. Kidney Injury in COVID-19: Epidemiology, Molecular Mechanisms and Potential Therapeutic Targets. International Journal of Molecular Sciences. 2022; 23(4):2242. https://doi.org/10.3390/ijms23042242
Chicago/Turabian StyleTeixeira, J. Pedro, Sharon Barone, Kamyar Zahedi, and Manoocher Soleimani. 2022. "Kidney Injury in COVID-19: Epidemiology, Molecular Mechanisms and Potential Therapeutic Targets" International Journal of Molecular Sciences 23, no. 4: 2242. https://doi.org/10.3390/ijms23042242
APA StyleTeixeira, J. P., Barone, S., Zahedi, K., & Soleimani, M. (2022). Kidney Injury in COVID-19: Epidemiology, Molecular Mechanisms and Potential Therapeutic Targets. International Journal of Molecular Sciences, 23(4), 2242. https://doi.org/10.3390/ijms23042242