
Absent in Melanoma 2 (AIM2) Regulates the Stability of Regulatory T Cells

 , , , ,
, , , ,  , and
, and 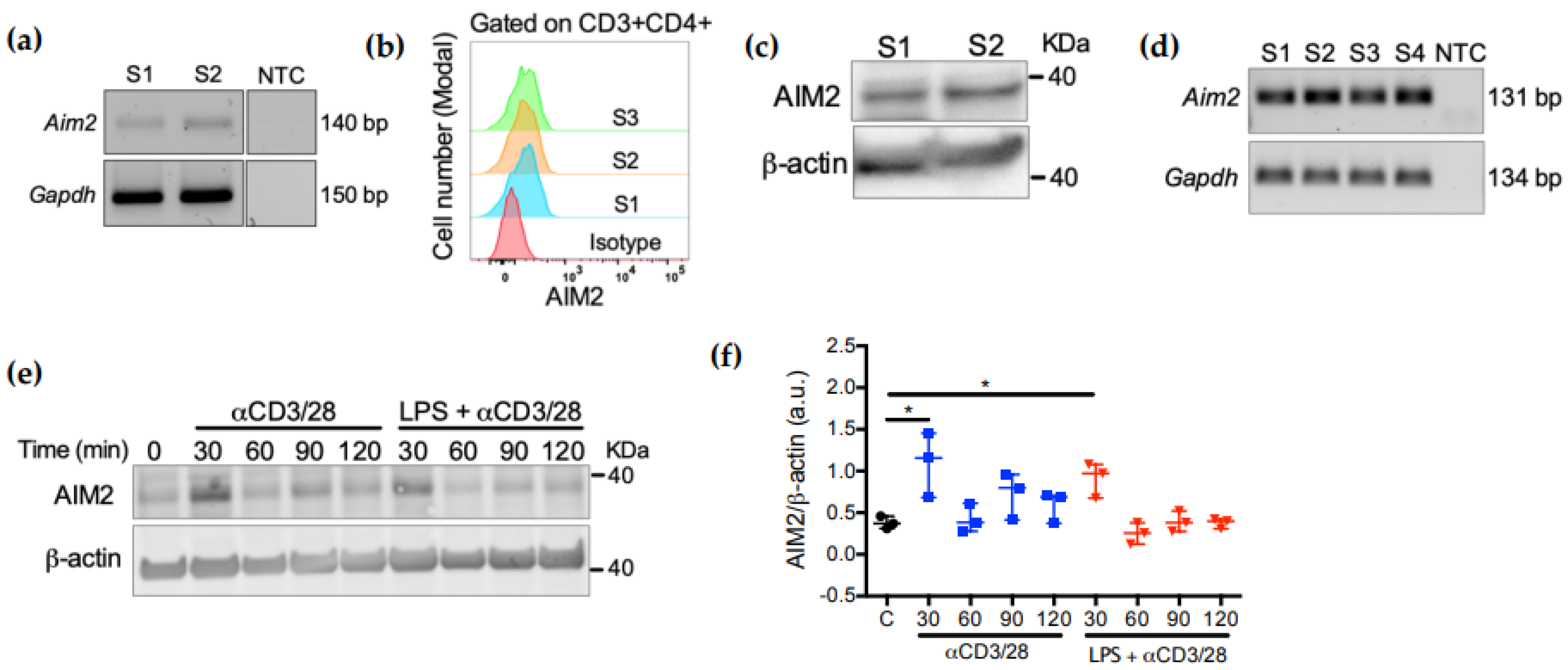{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
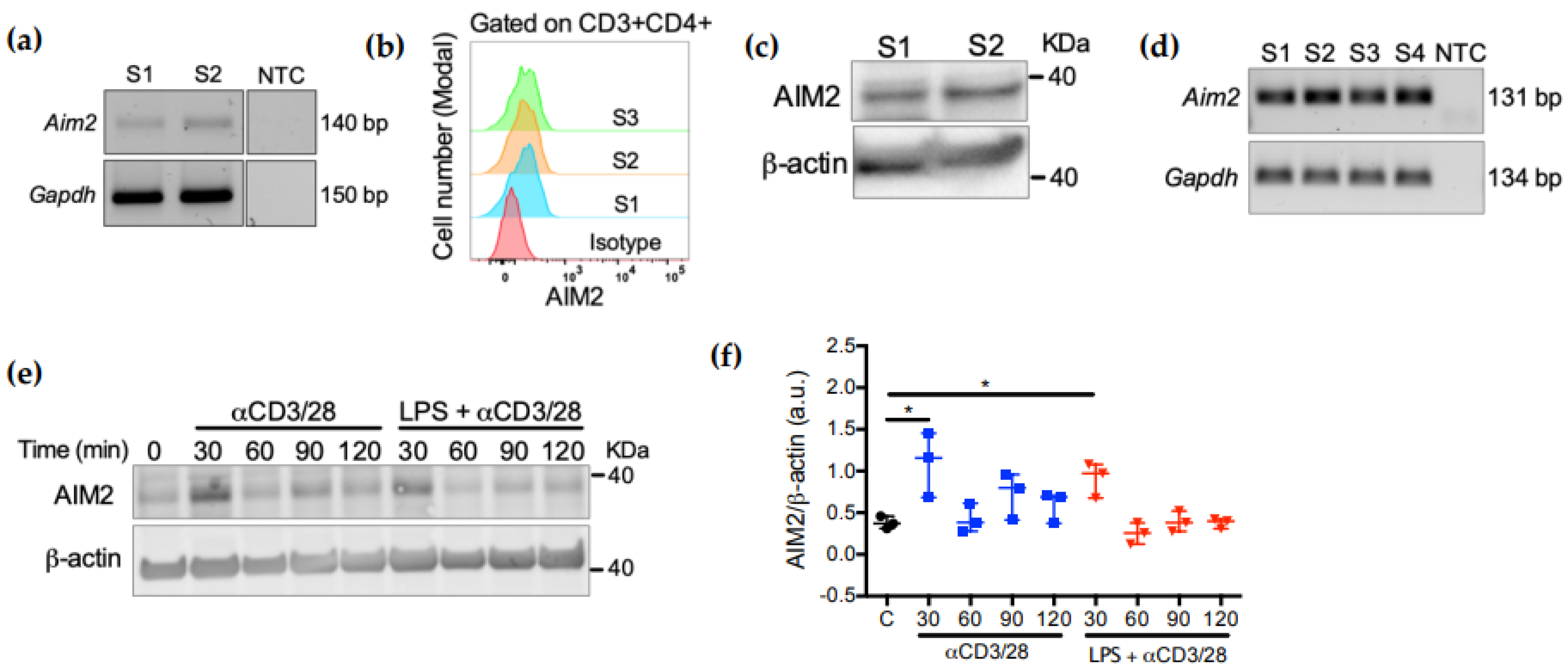
2.1. AIM2 Expression in CD4+ T Cells Is Controlled by TCR Activation
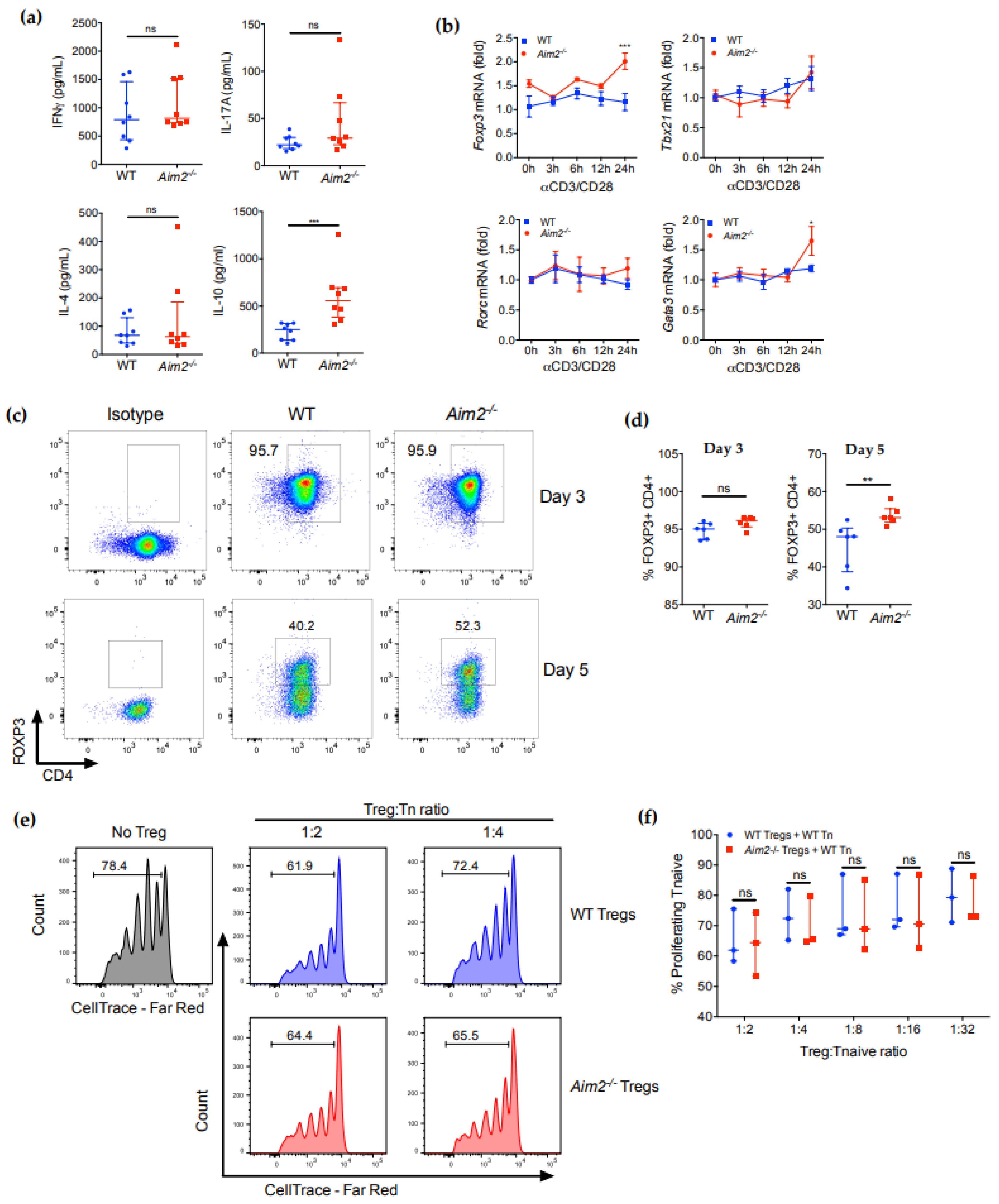
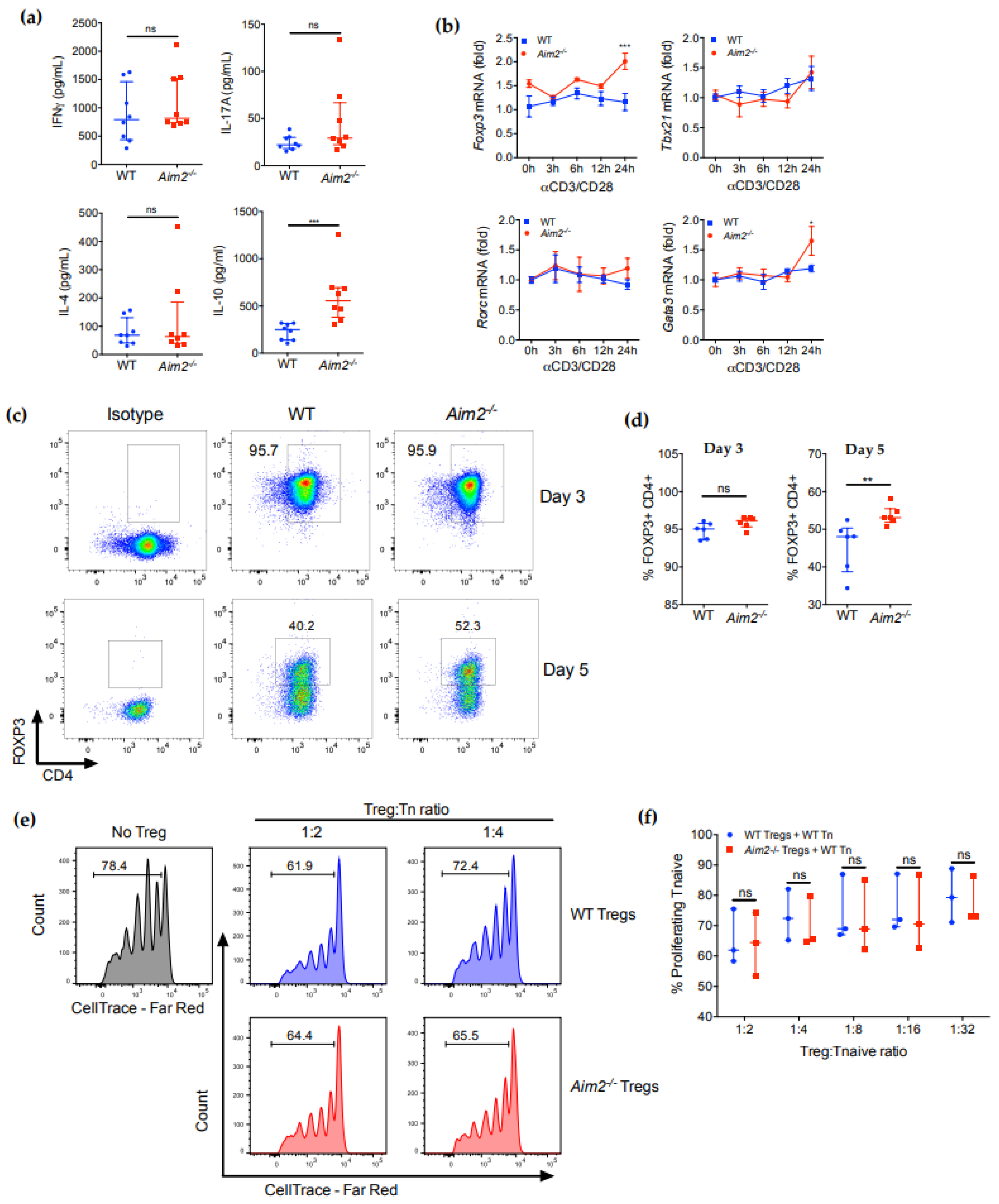
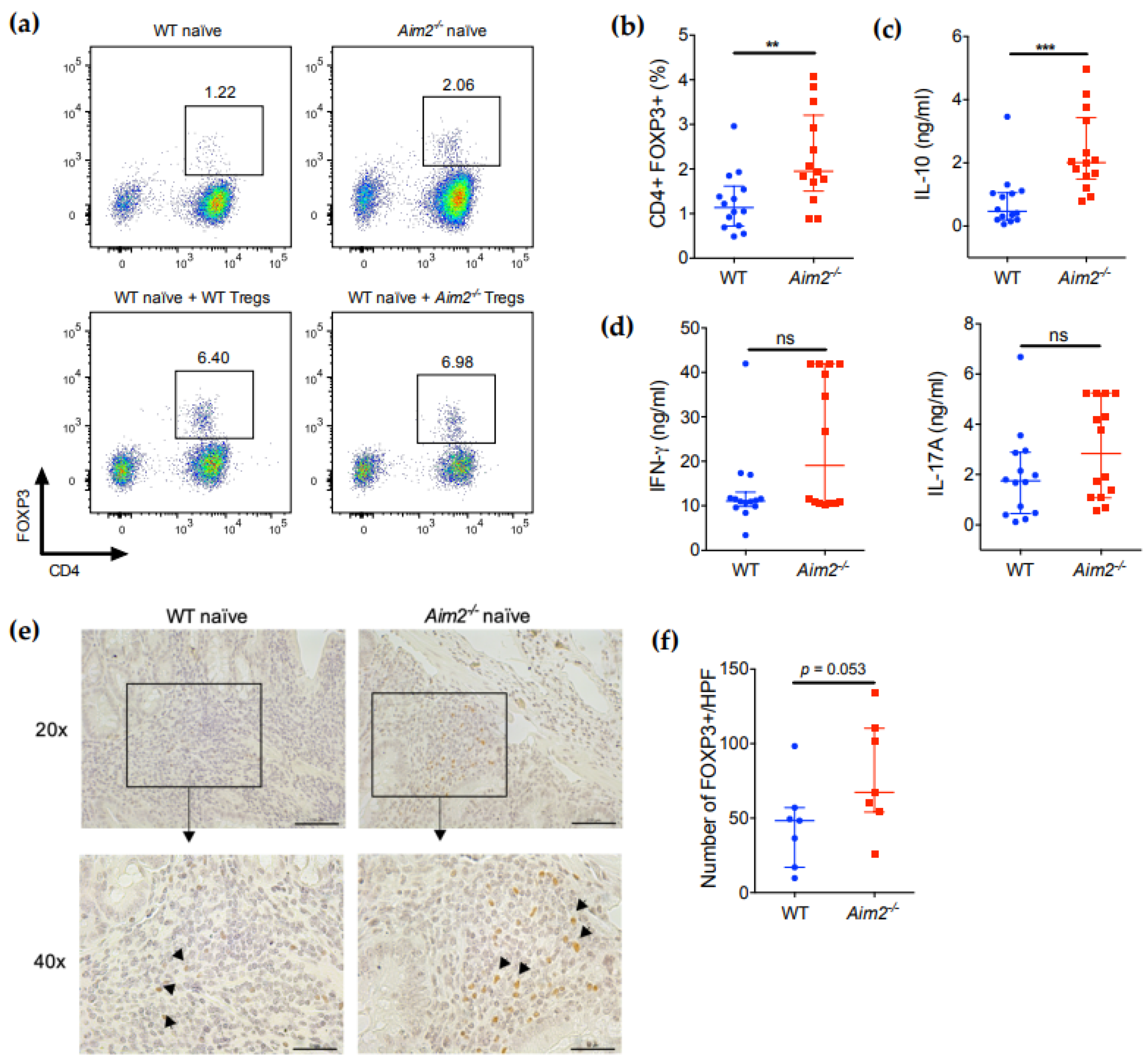
2.2. AIM2 Deficiency Facilitates the Stability of iTreg Cells
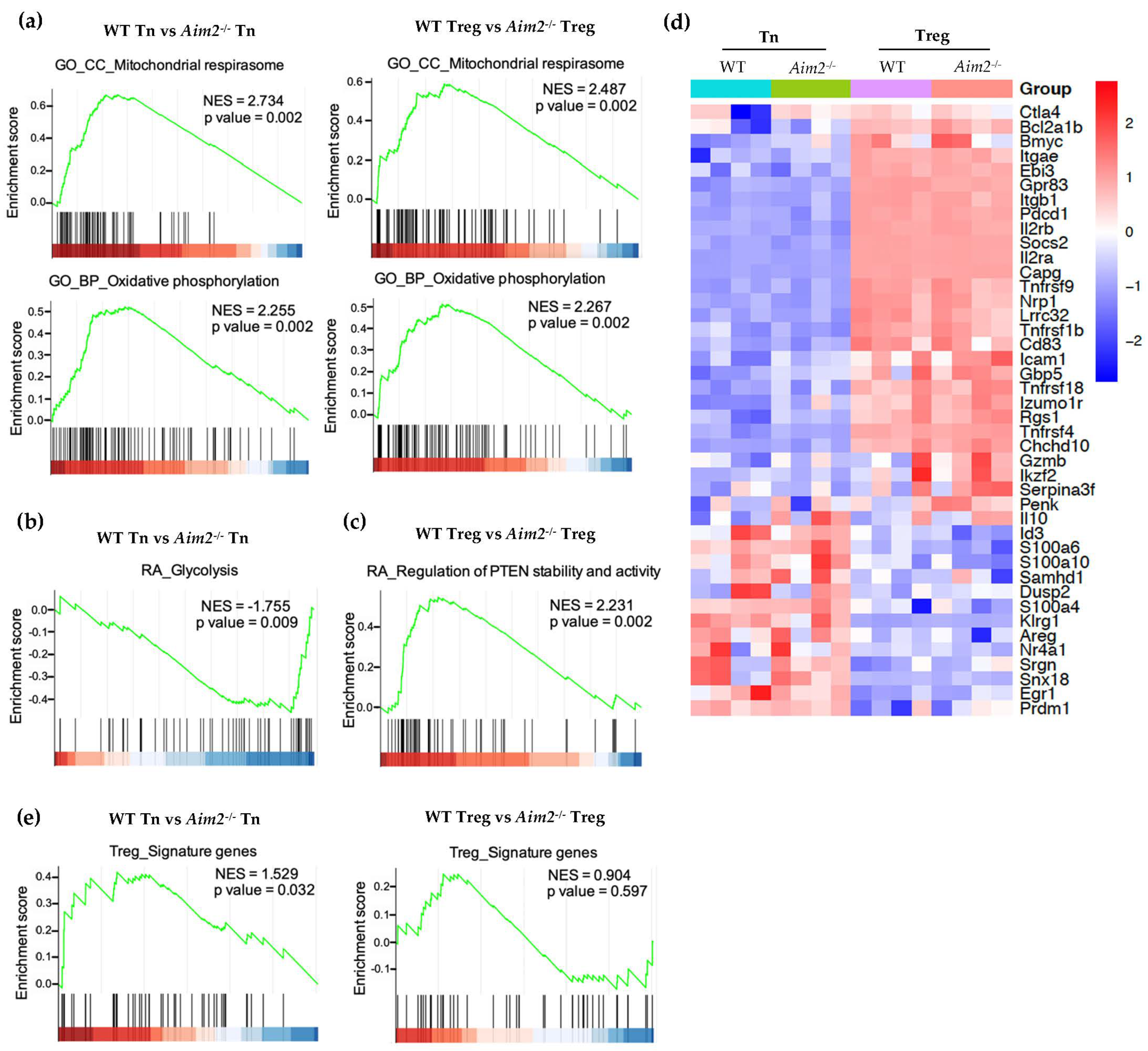
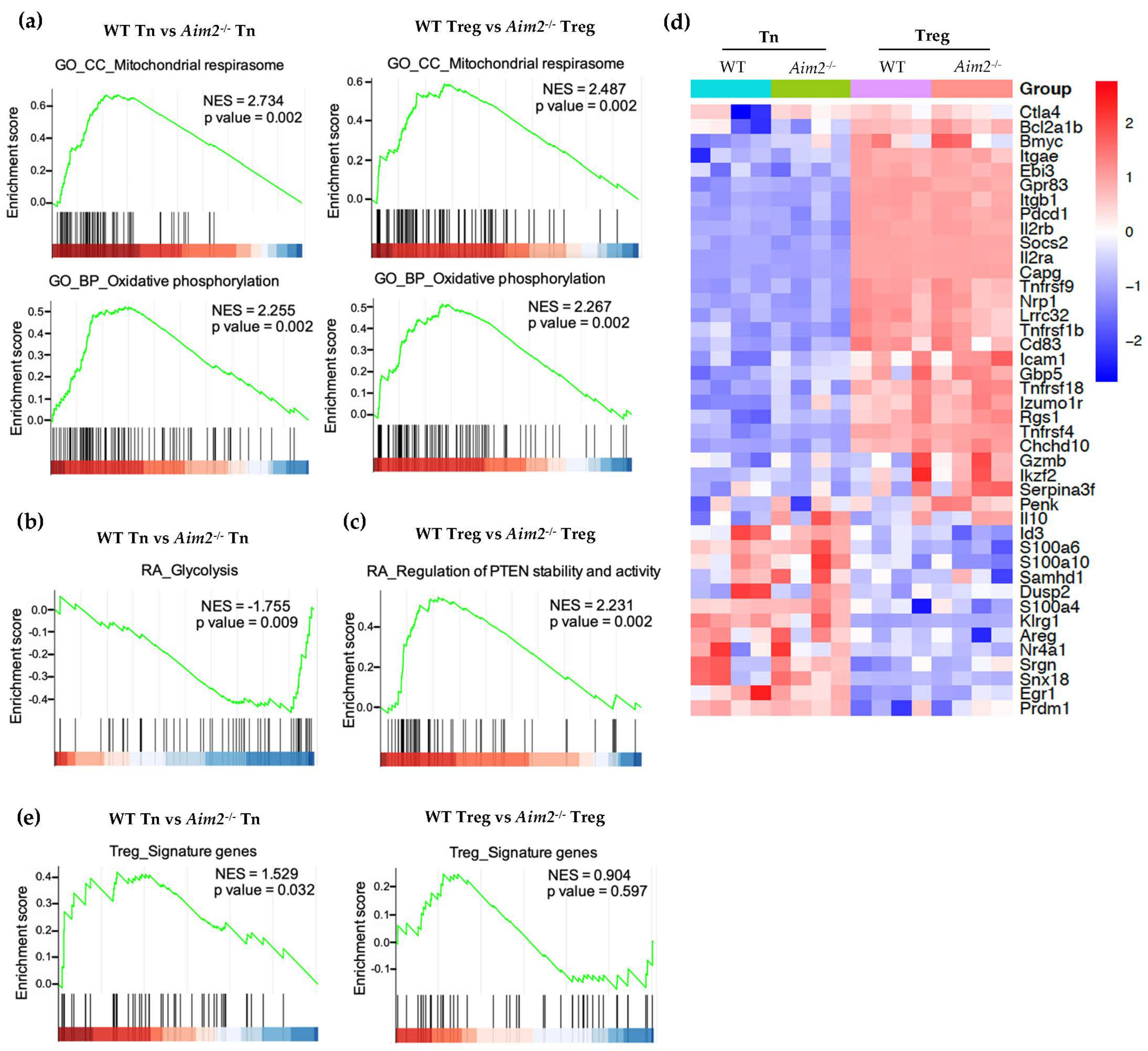
2.3. RNAseq and GSEA Analysis Reveals Different Metabolic Gene Signatures in Aim2−/− T Cells
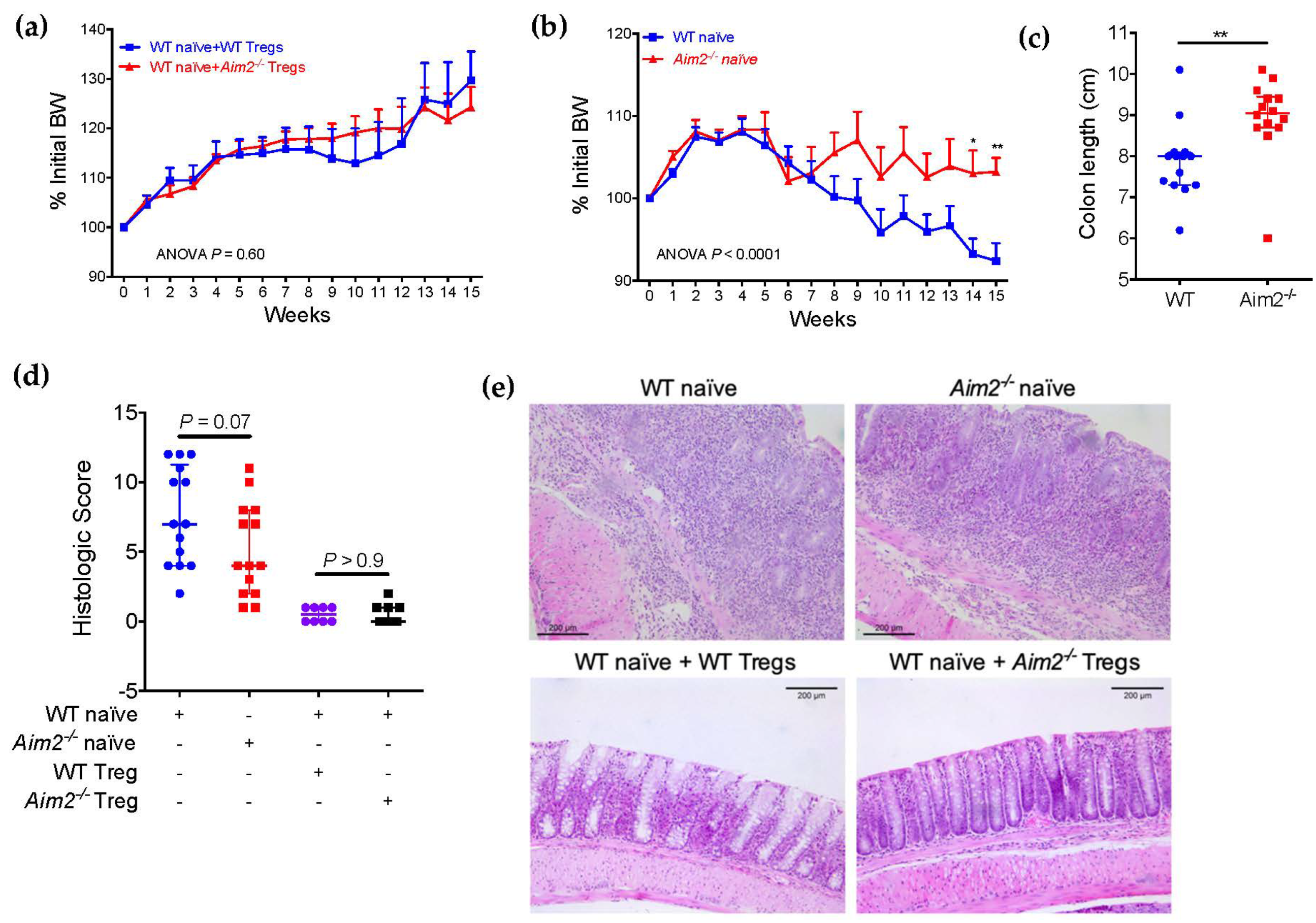
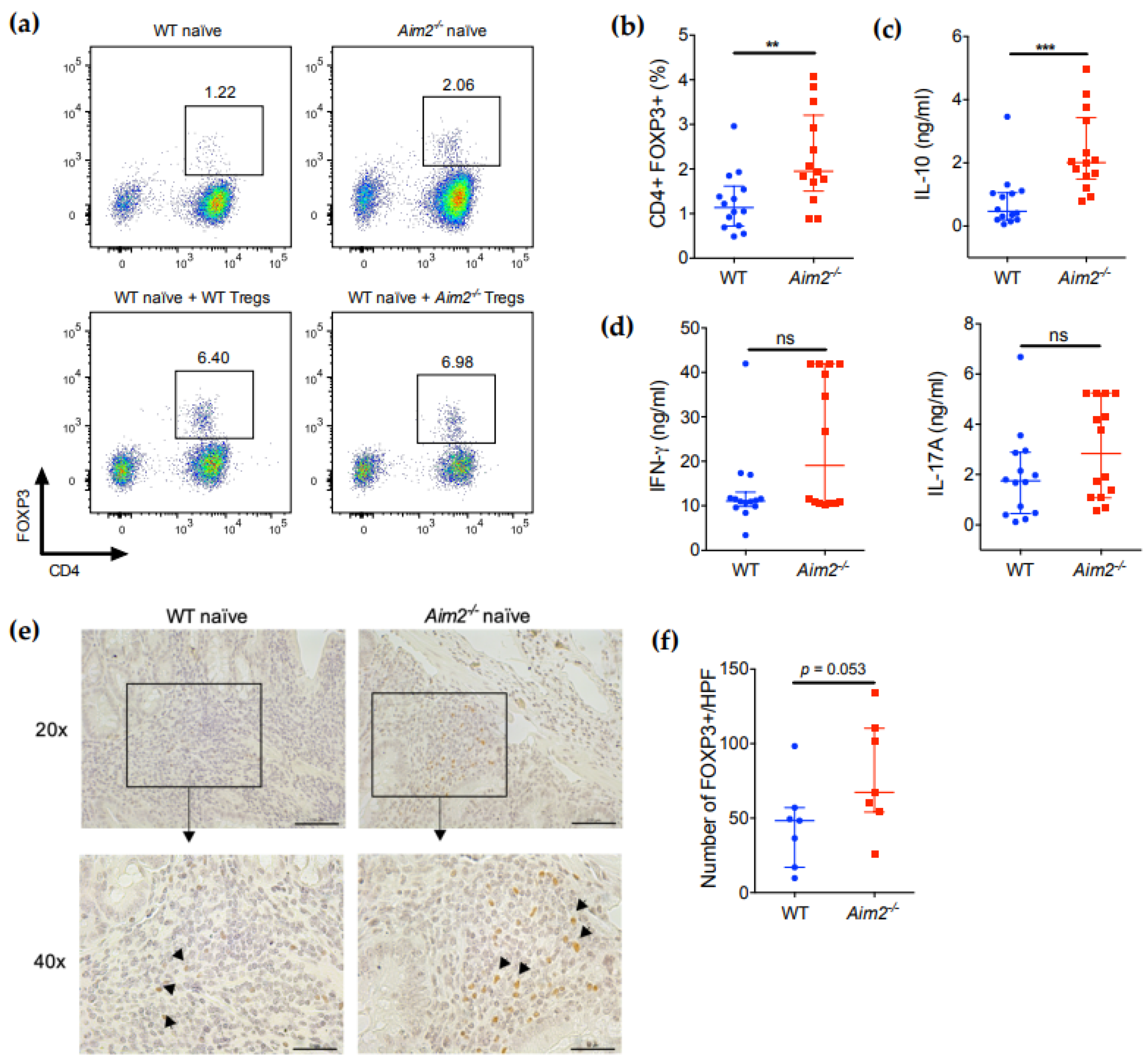
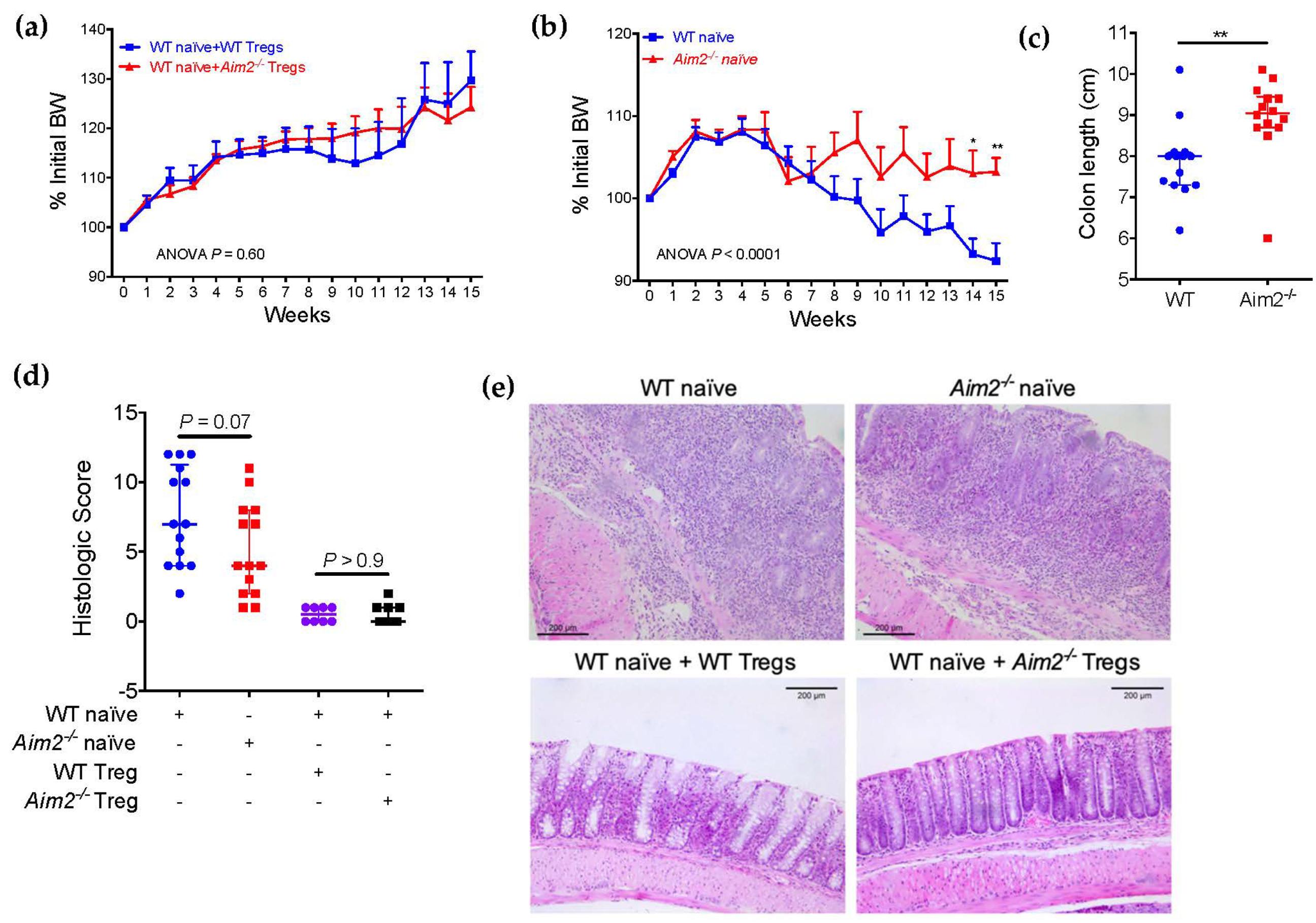
2.4. Aim2−/− Naïve T Cells Show Enhanced Differentiation into FOXP3+ Cells after Adoptive Transfer
3. Discussion
4. Materials and Methods
4.1. Antibodies, Primers and Common Reagents
4.2. Mice
4.3. Human Samples
4.4. CD4+ T Cell Isolation, Culture, Stimulation and Differentiation
4.5. Treg Suppression Assay
4.6. Flow Cytometry Analysis
4.7. Isolation of RNA and Quantitative Real-Time PCR
4.8. Immunoblot
4.9. Quantification of Cytokine Levels
4.10. RNAseq Analysis
4.11. Induction of Colitis by Adoptive Transfer of Naïve T Cells
4.12. Histology, Immunohistochemistry and Evaluation of Colitis
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xiao, T.S. The Nucleic Acid-Sensing Inflammasomes. Immunol. Rev. 2015, 265, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes-Alnemri, T.; Yu, J.-W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 Activates the Inflammasome and Cell Death in Response to Cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathinam, V.A.K.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 Inflammasome Is Essential for Host Defense against Cytosolic Bacteria and DNA Viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeYoung, K.L.; Ray, M.E.; Su, Y.A.; Anzick, S.L.; Johnstone, R.W.; Trapani, J.A.; Meltzer, P.S.; Trent, J.M. Cloning a Novel Member of the Human Interferon-Inducible Gene Family Associated with Control of Tumorigenicity in a Model of Human Melanoma. Oncogene 1997, 15, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugrin, J.; Martinon, F. The AIM2 Inflammasome: Sensor of Pathogens and Cellular Perturbations. Immunol. Rev. 2018, 281, 99–114. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of ProIL-Beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Mathur, A.; Hayward, J.A.; Man, S.M. Molecular Mechanisms of Inflammasome Signaling. J. Leukoc. Biol. 2018, 103, 233–257. [Google Scholar] [CrossRef]
- de Zoete, M.R.; Palm, N.W.; Zhu, S.; Flavell, R.A. Inflammasomes. Cold Spring Harb. Perspect. Biol. 2014, 6, a016287. [Google Scholar] [CrossRef]
- Place, D.E.; Kanneganti, T.-D. Recent Advances in Inflammasome Biology. Curr. Opin. Immunol. 2018, 50, 32–38. [Google Scholar] [CrossRef]
- Narayan, S.; Kolly, L.; So, A.; Busso, N. Increased Interleukin-10 Production by ASC-Deficient CD4+ T Cells Impairs Bystander T-Cell Proliferation. Immunology 2011, 134, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.N.; Wang, C.; Zhang, C.; Kang, Z.; Gulen, M.F.; Zepp, J.A.; Zhao, J.; Bian, G.; Do, J.; Min, B.; et al. T Cell–Intrinsic ASC Critically Promotes TH17-Mediated Experimental Autoimmune Encephalomyelitis. Nat. Immunol. 2016, 17, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Bruchard, M.; Rebé, C.; Derangère, V.; Togbé, D.; Ryffel, B.; Boidot, R.; Humblin, E.; Hamman, A.; Chalmin, F.; Berger, H.; et al. The Receptor NLRP3 Is a Transcriptional Regulator of TH2 Differentiation. Nat. Immunol. 2015, 16, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Arbore, G.; West, E.E.; Spolski, R.; Robertson, A.A.B.; Klos, A.; Rheinheimer, C.; Dutow, P.; Woodruff, T.M.; Yu, Z.X.; ONeill, L.A.; et al. T Helper 1 Immunity Requires Complement-Driven NLRP3 Inflammasome Activity in CD4+ T Cells. Science 2016, 352, aad1210. [Google Scholar] [CrossRef] [Green Version]
- Iizuka-Koga, M.; Nakatsukasa, H.; Ito, M.; Akanuma, T.; Lu, Q.; Yoshimura, A. Induction and Maintenance of Regulatory T Cells by Transcription Factors and Epigenetic Modifications. J. Autoimmun. 2017, 83, 113–121. [Google Scholar] [CrossRef]
- Dutta, A.; Venkataganesh, H.; Love, P.E. New Insights into Epigenetic Regulation of T Cell Differentiation. Cells 2021, 10, 3459. [Google Scholar] [CrossRef]
- Potaczek, D.P.; Harb, H.; Michel, S.; Alhamwe, B.A.; Renz, H.; Tost, J. Epigenetics and Allergy: From Basic Mechanisms to Clinical Applications. Epigenomics 2017, 9, 539–571. [Google Scholar] [CrossRef]
- Suarez-Alvarez, B.; Rodriguez, R.M.; Fraga, M.F.; López-Larrea, C. DNA Methylation: A Promising Landscape for Immune System-Related Diseases. Trends Genet. 2012, 28, 506–514. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Sakaguchi, S. Molecular Control of Regulatory T Cell Development and Function. Curr. Opin. Immunol. 2017, 49, 64–70. [Google Scholar] [CrossRef]
- Kanamori, M.; Nakatsukasa, H.; Okada, M.; Lu, Q.; Yoshimura, A. Induced Regulatory T Cells: Their Development, Stability, and Applications. Trends Immunol. 2016, 37, 803–811. [Google Scholar] [CrossRef]
- Miragaia, R.J.; Gomes, T.; Chomka, A.; Jardine, L.; Riedel, A.; Hegazy, A.N.; Whibley, N.; Tucci, A.; Chen, X.; Lindeman, I.; et al. Single-Cell Transcriptomics of Regulatory T Cells Reveals Trajectories of Tissue Adaptation. Immunity 2019, 50, 493–504.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, W.-C.; Guo, Z.; Guo, H.; Chen, L.; Zhang, G.; Liang, K.; Xie, L.; Tan, X.; Gibson, S.A.; Rampanelli, E.; et al. AIM2 in Regulatory T Cells Restrains Autoimmune Diseases. Nature 2021, 591, 300–305. [Google Scholar] [CrossRef] [PubMed]
- González-Navajas, J.M.; Fine, S.; Law, J.; Datta, S.K.; Nguyen, K.P.; Yu, M.; Corr, M.; Katakura, K.; Eckman, L.; Lee, J.; et al. TLR4 Signaling in Effector CD4+ T Cells Regulates TCR Activation and Experimental Colitis in Mice. J. Clin. Investig. 2010, 120, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Su, M.A.; Wan, Y.Y. An Essential Role of the Transcription Factor GATA-3 for the Function of Regulatory T Cells. Immunity 2011, 35, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Wohlfert, E.A.; Grainger, J.R.; Bouladoux, N.; Konkel, J.E.; Oldenhove, G.; Ribeiro, C.H.; Hall, J.A.; Yagi, R.; Naik, S.; Bhairavabhotla, R.; et al. GATA3 Controls Foxp3+ Regulatory T Cell Fate during Inflammation in Mice. J. Clin. Investig. 2011, 121, 4503–4515. [Google Scholar] [CrossRef]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4+ T Cell Subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [Green Version]
- Beier, U.H.; Angelin, A.; Akimova, T.; Wang, L.; Liu, Y.; Xiao, H.; Koike, M.A.; Hancock, S.A.; Bhatti, T.R.; Han, R.; et al. Essential Role of Mitochondrial Energy Metabolism in Foxp3+ T-Regulatory Cell Function and Allograft Survival. FASEB J. 2015, 29, 2315–2326. [Google Scholar] [CrossRef] [Green Version]
- Howie, D.; Cobbold, S.P.; Adams, E.; Ten Bokum, A.; Necula, A.S.; Zhang, W.; Huang, H.; Roberts, D.J.; Thomas, B.; Hester, S.S.; et al. Foxp3 Drives Oxidative Phosphorylation and Protection from Lipotoxicity. JCI Insight 2017, 2, e89160. [Google Scholar] [CrossRef] [Green Version]
- Angelin, A.; Gil-de-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J.; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.e7. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, S.E.; Singer, B.D.; Steinert, E.M.; Martinez, C.A.; Mehta, M.M.; Martínez-Reyes, I.; Gao, P.; Helmin, K.A.; Abdala-Valencia, H.; Sena, L.A.; et al. Mitochondrial Complex III Is Essential for Suppressive Function of Regulatory T Cells. Nature 2019, 565, 495–499. [Google Scholar] [CrossRef]
- Gerriets, V.A.; Kishton, R.J.; Johnson, M.O.; Cohen, S.; Siska, P.J.; Nichols, A.G.; Warmoes, M.O.; de Cubas, A.A.; MacIver, N.J.; Locasale, J.W.; et al. Foxp3 and Toll-like Receptor Signaling Balance Treg Cell Anabolic Metabolism for Suppression. Nat. Immunol. 2016, 17, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.Y.; Turka, L.A. Immunometabolism and PI(3)K Signaling As a Link between IL-2, Foxp3 Expression, and Suppressor Function in Regulatory T Cells. Front. Immunol. 2018, 9, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, A.; DuPage, M.; Priyadharshini, B.; Sage, P.T.; Quiros, J.; Borges, C.M.; Townamchai, N.; Gerriets, V.A.; Rathmell, J.C.; Sharpe, A.H.; et al. Control of PI(3) Kinase in Treg Cells Maintains Homeostasis and Lineage Stability. Nat. Immunol. 2015, 16, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Zemmour, D.; Zilionis, R.; Kiner, E.; Klein, A.M.; Mathis, D.; Benoist, C. Single-Cell Gene Expression Reveals a Landscape of Regulatory T Cell Phenotypes Shaped by the TCR. Nat. Immunol. 2018, 19, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Powrie, F.; Leach, M.W.; Mauze, S.; Caddle, L.B.; Coffman, R.L. Phenotypically Distinct Subsets of CD4+ T Cells Induce or Protect from Chronic Intestinal Inflammation in C. B-17 Scid Mice. Int. Immunol. 1993, 5, 1461–1471. [Google Scholar] [CrossRef]
- Ostanin, D.V.; Bao, J.; Koboziev, I.; Gray, L.; Robinson-Jackson, S.A.; Kosloski-Davidson, M.; Price, V.H.; Grisham, M.B. T Cell Transfer Model of Chronic Colitis: Concepts, Considerations, and Tricks of the Trade. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G135–G146. [Google Scholar] [CrossRef] [Green Version]
- Lucca, L.E.; Dominguez-Villar, M. Modulation of Regulatory T Cell Function and Stability by Co-Inhibitory Receptors. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef]
- Oldenhove, G.; Bouladoux, N.; Wohlfert, E.A.; Hall, J.A.; Chou, D.; Dos Santos, L.; O’Brien, S.; Blank, R.; Lamb, E.; Natarajan, S.; et al. Decrease of Foxp3+ Treg Cell Number and Acquisition of Effector Cell Phenotype during Lethal Infection. Immunity 2009, 31, 772–786. [Google Scholar] [CrossRef] [Green Version]
- Koenecke, C.; Lee, C.-W.; Thamm, K.; Föhse, L.; Schafferus, M.; Mittrücker, H.-W.; Floess, S.; Huehn, J.; Ganser, A.; Förster, R.; et al. IFN-γ Production by Allogeneic Foxp3+ Regulatory T Cells Is Essential for Preventing Experimental Graft-versus-Host Disease. J. Immunol. 2012, 189, 2890–2896. [Google Scholar] [CrossRef] [Green Version]
- Kitz, A.; de Marcken, M.; Gautron, A.-S.; Mitrovic, M.; Hafler, D.A.; Dominguez-Villar, M. AKT Isoforms Modulate Th1-like Treg Generation and Function in Human Autoimmune Disease. EMBO Rep. 2016, 17, 1169–1183. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.E.; Petrucelli, A.S.; Chen, L.; Koblansky, A.A.; Truax, A.D.; Oyama, Y.; Rogers, A.B.; Brickey, W.J.; Wang, Y.; Schneider, M.; et al. Inflammasome-Independent Role of AIM2 in Suppressing Colon Tumorigenesis via DNA-PK and Akt. Nat. Med. 2015, 21, 906–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pompura, S.L.; Dominguez-Villar, M. The PI3K/AKT Signaling Pathway in Regulatory T-Cell Development, Stability, and Function. J. Leukoc. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Qi, M.; Dai, D.; Liu, J.; Li, Z.; Liang, P.; Wang, Y.; Cheng, L.; Zhan, Y.; An, Z.; Song, Y.; et al. AIM2 Promotes the Development of Non-Small Cell Lung Cancer by Modulating Mitochondrial Dynamics. Oncogene 2020, 39, 2707–2723. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Zhang, Q.; Liu, V.; Xia, Z.; Pothoven, K.L.; Lee, C. Cutting Edge: TGF-Beta-Induced Expression of Foxp3 in T Cells Is Mediated through Inactivation of ERK. J. Immunol. 2008, 180, 2757–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-F.; D’Souza, W.N.; Ch’en, I.L.; Pages, G.; Pouyssegur, J.; Hedrick, S.M. Polar Opposites: Erk Direction of CD4 T Cell Subsets. J. Immunol. 2012, 189, 721–731. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yao, S.; Dann, S.M.; Qin, H.; Elson, C.O.; Cong, Y. ERK Differentially Regulates Th17- and Treg-Cell Development and Contributes to the Pathogenesis of Colitis. Eur. J. Immunol. 2013, 43, 1716–1726. [Google Scholar] [CrossRef]
- Bertin, S.; Lozano-Ruiz, B.; Bachiller, V.; García-Martínez, I.; Herdman, S.; Zapater, P.; Francés, R.; Such, J.; Lee, J.; Raz, E.; et al. Dual-Specificity Phosphatase 6 Regulates CD4+ T-Cell Functions and Restrains Spontaneous Colitis in IL-10-Deficient Mice. Mucosal Immunol. 2015, 8, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Cossarizza, A.; Chang, H.; Radbruch, A.; Acs, A.; Adam, D.; Adam-Klages, S.; Agace, W.W.; Aghaeepour, N.; Akdis, M.; Allez, M.; et al. Guidelines for the Use of Flow Cytometry and Cell Sorting in Immunological Studies (Second Edition). Eur. J. Immunol. 2019, 49, 1457–1973. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Frankish, A.; Diekhans, M.; Ferreira, A.-M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE Reference Annotation for the Human and Mouse Genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision Weights Unlock Linear Model Analysis Tools for RNA-Seq Read Counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lozano-Ruiz, B.; Tzoumpa, A.; Martínez-Cardona, C.; Moreno, D.; Aransay, A.M.; Cortazar, A.R.; Picó, J.; Peiró, G.; Lozano, J.; Zapater, P.; et al. Absent in Melanoma 2 (AIM2) Regulates the Stability of Regulatory T Cells. Int. J. Mol. Sci. 2022, 23, 2230. https://doi.org/10.3390/ijms23042230
Lozano-Ruiz B, Tzoumpa A, Martínez-Cardona C, Moreno D, Aransay AM, Cortazar AR, Picó J, Peiró G, Lozano J, Zapater P, et al. Absent in Melanoma 2 (AIM2) Regulates the Stability of Regulatory T Cells. International Journal of Molecular Sciences. 2022; 23(4):2230. https://doi.org/10.3390/ijms23042230
Chicago/Turabian StyleLozano-Ruiz, Beatriz, Amalia Tzoumpa, Claudia Martínez-Cardona, David Moreno, Ana M. Aransay, Ana R. Cortazar, Joanna Picó, Gloria Peiró, Juanjo Lozano, Pedro Zapater, and et al. 2022. "Absent in Melanoma 2 (AIM2) Regulates the Stability of Regulatory T Cells" International Journal of Molecular Sciences 23, no. 4: 2230. https://doi.org/10.3390/ijms23042230
APA StyleLozano-Ruiz, B., Tzoumpa, A., Martínez-Cardona, C., Moreno, D., Aransay, A. M., Cortazar, A. R., Picó, J., Peiró, G., Lozano, J., Zapater, P., Francés, R., & González-Navajas, J. M. (2022). Absent in Melanoma 2 (AIM2) Regulates the Stability of Regulatory T Cells. International Journal of Molecular Sciences, 23(4), 2230. https://doi.org/10.3390/ijms23042230