
CD44 Expression Intensity Marks Colorectal Cancer Cell Subpopulations with Different Extracellular Vesicle Release Capacity

 , , ,
, , , 
Abstract
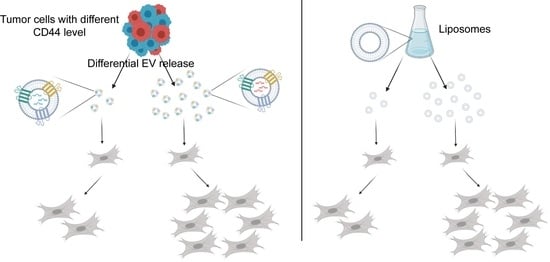
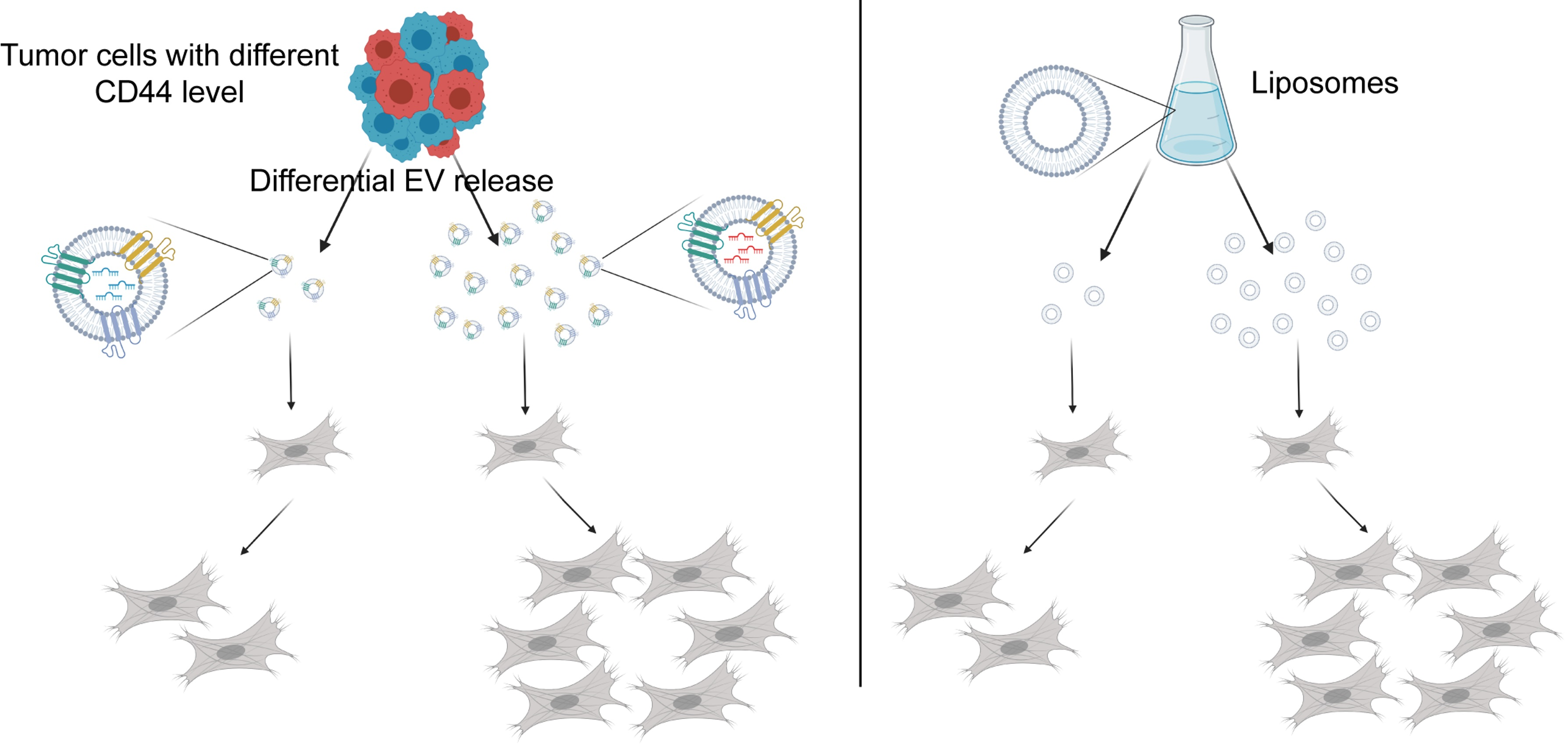{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
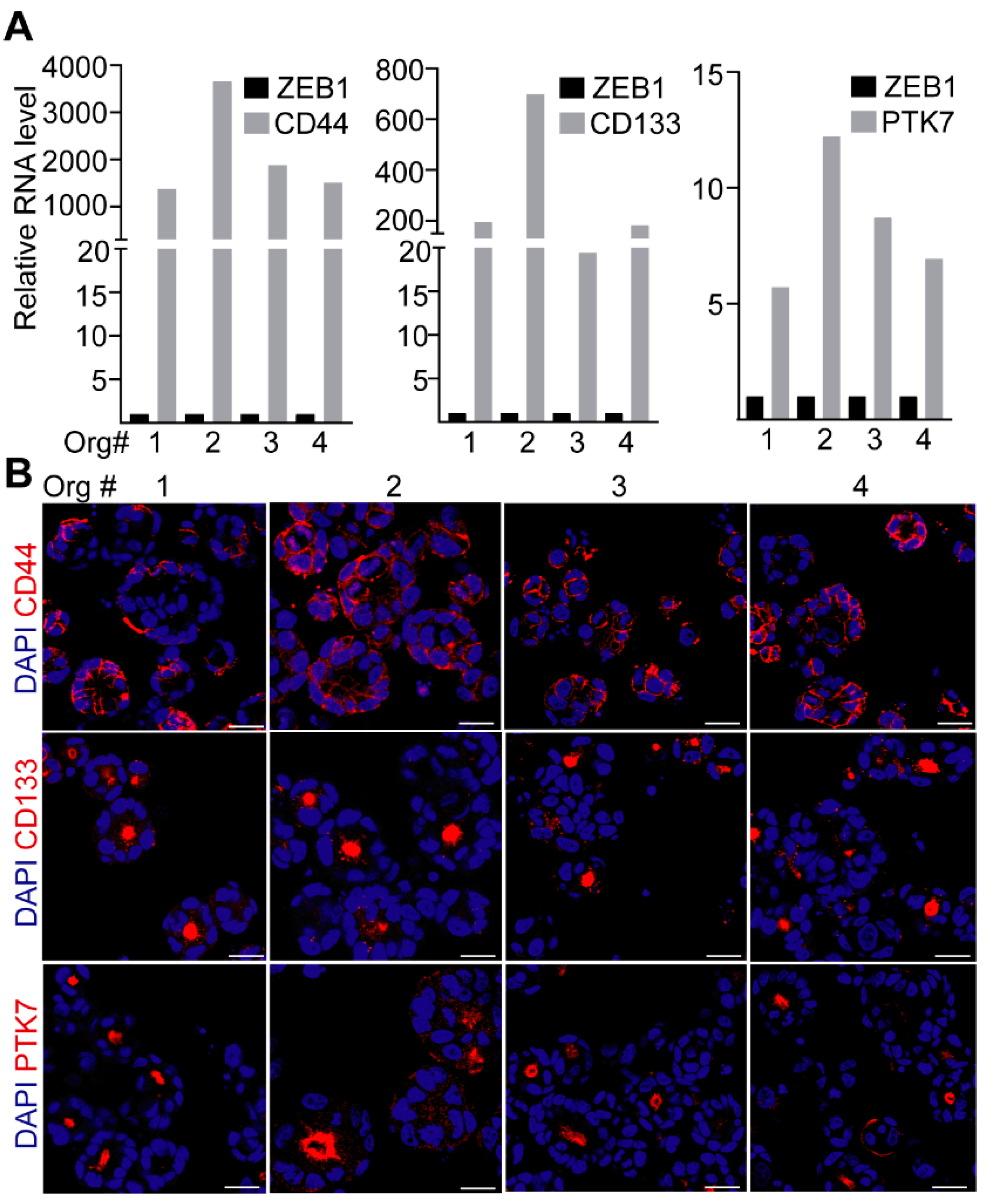
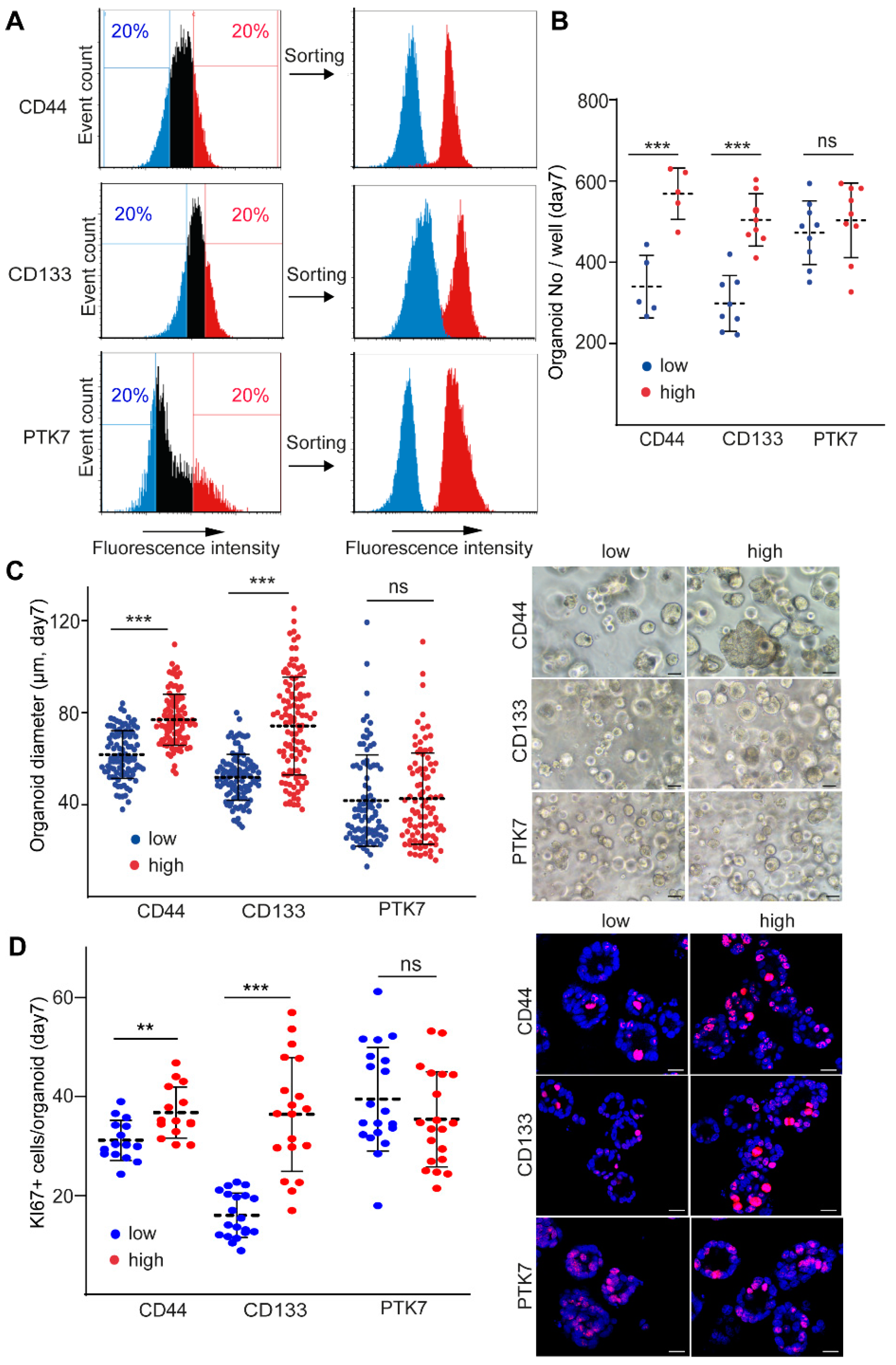
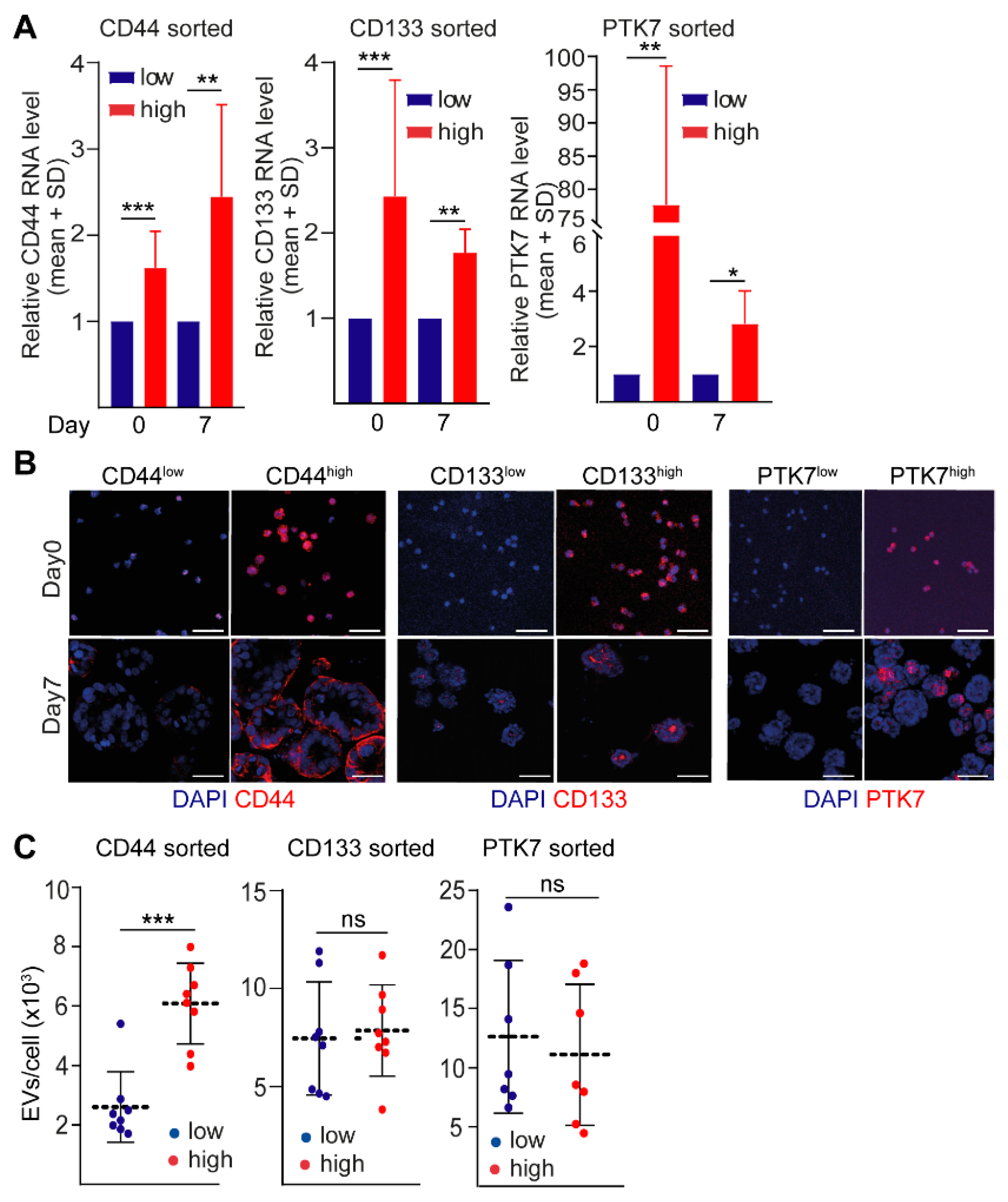
2.1. CD44 and CD133, but Not PTK7 Mark CRC Organoid Cell Populations with a High Proliferative Potential
2.2. CD44high CRC Cell-Derived Organoids Release More EVs Than CD44low Organoids
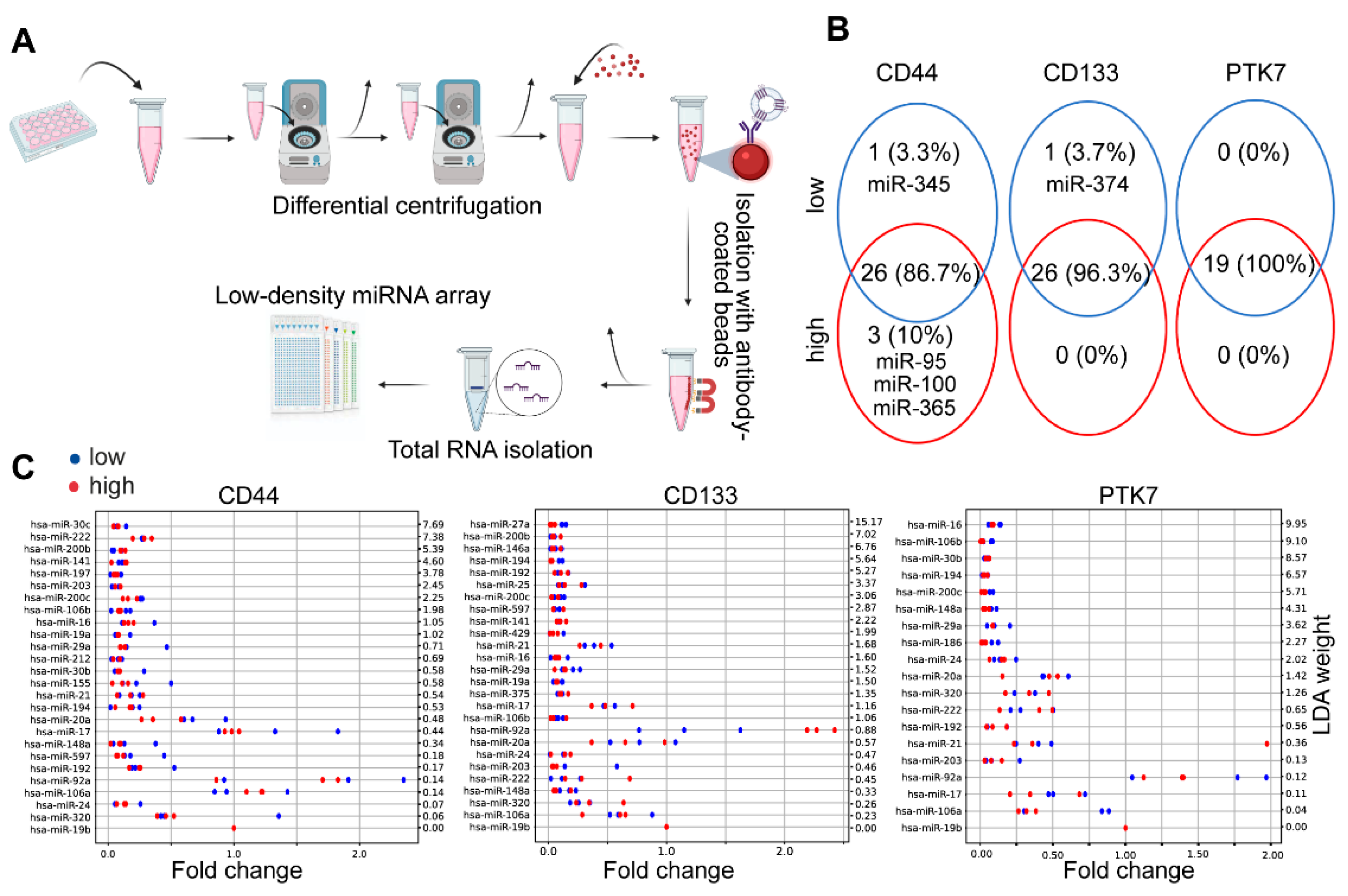
2.3. CD44high and CD44low CRC Cell-Derived EV Cargos Differ Only in a Few miRNAs
2.4. Dose-Dependent Effect of CD44high and CD44low CRC Organoid-Derived EVs on Fibroblasts
3. Discussion
4. Material and Methods
4.1. Cell Cultures
4.2. CRC Organoid Cultures
4.3. Nanoparticle Tracking Analysis (NTA)
4.4. Functional Experiments with EVs
4.5. Liposome Production
4.6. Flow Cytometry and Cell Sorting
4.7. Detecting EVs by Anti-CD63 and Anti-CD81-Coated Beads
4.8. Immunocytochemistry
4.9. Whole-Mount Immunostaining
4.10. Protein Concentration Measurement and Simple Western (WES) Analysis
4.11. Transmission Electron Microscopy
4.12. RNA Isolation and mRNA Measurements from Cells
4.13. TaqMan Low Density Array (TLDA)
4.14. Bioinformatical Analysis
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Thery, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, K.G.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Tkach, M.; Kowal, J.; Théry, C. Why the need and how to approach the functional diversity of extracellular vesicles. Philos. Trans. R. Soc. B Biol. Sci. 2017, 373, 20160479. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Catalano, V.; Di Franco, S.; Iovino, F.; Dieli, F.; Stassi, G.; Todaro, M. CD133 as a target for colon cancer. Expert Opin. Ther. Targets 2012, 16, 259–267. [Google Scholar] [CrossRef]
- Jung, P.; Sommer, C.; Barriga, F.M.; Buczacki, S.J.; Hernando-Momblona, X.; Sevillano, M.; Duran-Frigola, M.; Aloy, P.; Selbach, M.; Winton, D.J.; et al. Isolation of Human Colon Stem Cells Using Surface Expression of PTK7. Stem Cell Rep. 2015, 5, 979–987. [Google Scholar] [CrossRef]
- Golubkov, V.S.; Prigozhina, N.L.; Zhang, Y.; Stoletov, K.; Lewis, J.D.; Schwartz, P.E.; Hoffman, R.M.; Strongin, A.Y. Protein-tyrosine pseudokinase 7 (PTK7) directs cancer cell motility and metastasis. J. Biol. Chem. 2014, 289, 24238–24249. [Google Scholar] [CrossRef]
- Buczacki, S.J.A.; Popova, S.; Biggs, E.; Koukorava, C.; Buzzelli, J.; Vermeulen, L.; Hazelwood, L.; Francies, H.; Garnett, M.J.; Winton, D.J. Itraconazole targets cell cycle heterogeneity in colorectal cancer. J. Exp. Med. 2018, 215, 1891–1912. [Google Scholar] [CrossRef]
- Todaro, M.; Francipane, M.G.; Medema, J.P.; Stassi, G. Colon cancer stem cells: Promise of targeted therapy. Gastroenterology 2010, 138, 2151–2162. [Google Scholar] [CrossRef]
- Ghatak, S.; Hascall, V.C.; Markwald, R.R.; Misra, S. FOLFOX Therapy Induces Feedback Upregulation of CD44v6 through YB-1 to Maintain Stemness in Colon Initiating Cells. Int. J. Mol. Sci. 2021, 22, 753. [Google Scholar] [CrossRef]
- Zoller, M. CD44: Can a cancer-initiating cell profit from an abundantly expressed molecule? Nat. Rev. Cancer 2011, 11, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Dalerba, P.; Dylla, S.J.; Park, I.K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef] [PubMed]
- Snippert, H.J.; van Es, J.H.; van den Born, M.; Begthel, H.; Stange, D.E.; Barker, N.; Clevers, H. Prominin-1/CD133 marks stem cells and early progenitors in mouse small intestine. Gastroenterology 2009, 136, 2187–2194 e1. [Google Scholar] [CrossRef]
- Ping, Q.; Yan, R.; Cheng, X.; Wang, W.; Zhong, Y.; Hou, Z.; Shi, Y.; Wang, C.; Li, R. Cancer-associated fibroblasts: Overview, progress, challenges, and directions. Cancer Gene Ther. 2021, 28, 984–999. [Google Scholar] [CrossRef] [PubMed]
- Berdiel-Acera, M.; Sanz-Pamplona, R.; Calon, A.; Cuadras, D.; Berenguer, A.; Sanjuan, X.; Paules, M.J.; Salazar, R.; Moreno, V.; Batlle, E.; et al. Differences between CAFs and their paired NCF from adjacent colonic mucosa reveal functional heterogeneity of CAFs, providing prognostic information. Mol. Oncol. 2014, 8, 1290–1305. [Google Scholar] [CrossRef]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.F.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.F.; Iglesias, M.; Céspedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.-F.; et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef]
- Vermeulen, L.; Melo, F.d.E.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef]
- Oszvald, A.; Szvicsek, Z.; Pápai, M.; Kelemen, A.; Varga, Z.; Tölgyes, T.; Dede, K.; Bursics, A.; Buzás, E.I.; Wiener, Z. Fibroblast-Derived Extracellular Vesicles Induce Colorectal Cancer Progression by Transmitting Amphiregulin. Front. Cell Dev. Biol. 2020, 8, 558. [Google Scholar] [CrossRef]
- Schutgens, F.; Clevers, H. Human Organoids: Tools for Understanding Biology and Treating Diseases. Annu. Rev. Pathol. 2020, 15, 211–234. [Google Scholar] [CrossRef]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Szvicsek, Z.; Oszvald, Á.; Szabó, L.; Sándor, G.O.; Kelemen, A.; Soós, A.Á.; Pálóczi, K.; Harsányi, L.; Tölgyes, T.; Dede, K.; et al. Extracellular vesicle release from intestinal organoids is modulated by Apc mutation and other colorectal cancer progression factors. Cell Mol. Life Sci. 2019, 76, 2463–2476. [Google Scholar] [CrossRef] [PubMed]
- Zeöld, A.; Sándor, G.O.; Kiss, A.; Soós, A.Á.; Tölgyes, T.; Bursics, A.; Szűcs, Á.; Harsányi, L.; Kittel, Á.; Gézsi, A.; et al. Shared extracellular vesicle miRNA profiles of matched ductal pancreatic adenocarcinoma organoids and blood plasma samples show the power of organoid technology. Cell. Mol. Life Sci. 2021, 78, 3005–3020. [Google Scholar] [CrossRef]
- Kelemen, A.; Carmi, I.; Oszvald, Á.; Lőrincz, P.; Petővári, G.; Tölgyes, T.; Dede, K.; Bursics, A.; Buzás, E.I.; Wiener, Z. l IFITM1 expression determines extracellular vesicle uptake in colorectal cancer. Cell. Mol. Life Sci. 2021, 78, 7009–7024. [Google Scholar] [CrossRef] [PubMed]
- Gallo, A.; Tandon, M.; Alevizos, I.; Illei, G.G. The majority of microRNAs detectable in serum and saliva is concentrated in exosomes. PLoS ONE 2012, 7, e30679. [Google Scholar]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef]
- Carter, J.V.; Galbraith, N.J.; Yang, D.; Burton, J.F.; Walker, S.P.; Galandiuk, S. Blood-based microRNAs as biomarkers for the diagnosis of colorectal cancer: A systematic review and meta-analysis. Br. J. Cancer 2017, 116, 762–774. [Google Scholar] [CrossRef]
- Huang, Z.; Huang, S.; Wang, Q.; Liang, L.; Ni, S.; Wang, L.; Sheng, W.; He, X.; Du, X. MicroRNA-95 promotes cell proliferation and targets sorting Nexin 1 in human colorectal carcinoma. Cancer Res. 2011, 71, 2582–2589. [Google Scholar] [CrossRef]
- Lu, Y.; Zhao, X.; Liu, Q.; Li, C.; Graves-Deal, R.; Cao, Z.; Singh, B.; Franklin, J.L.; Wang, J.; Hu, H.; et al. lncRNA MIR100HG-derived miR-100 and miR-125b mediate cetuximab resistance via Wnt/beta-catenin signaling. Nat. Med. 2017, 23, 1331–1341. [Google Scholar] [CrossRef]
- Zhou, M.K.; Liu, X.J.; Zhao, Z.G.; Cheng, Y.M. MicroRNA-100 functions as a tumor suppressor by inhibiting Lgr5 expression in colon cancer cells. Mol. Med. Rep. 2015, 11, 2947–2952. [Google Scholar] [CrossRef]
- Chen, D.; Sun, Y.; Yuan, Y.; Han, Z.; Zhang, P.; Zhang, J.; You, M.J.; Teruya-Feldstein, J.; Wang, M.; Gupta, S.; et al. miR-100 induces epithelial-mesenchymal transition but suppresses tumorigenesis, migration and invasion. PLoS Genet. 2014, 10, e1004177. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Liu, L.; Zheng, W.; Chen, L.; Wu, X.; Xu, Y.; Du, X.; Han, W. microRNA-365, down-regulated in colon cancer, inhibits cell cycle progression and promotes apoptosis of colon cancer cells by probably targeting Cyclin D1 and Bcl-2. Carcinogenesis 2011, 33, 220–225. [Google Scholar] [CrossRef]
- Zhou, M.; Liu, W.; Ma, S.; Cao, H.; Peng, X.; Guo, L.; Zhou, X.; Zheng, L.; Guo, L.; Wan, M.; et al. A novel onco-miR-365 induces cutaneous squamous cell carcinoma. Carcinogenesis 2013, 34, 1653–1659. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, Y.; Ou, C.; Lin, Z.; Wang, J.; Liu, H.; Zhou, M.; Ding, Z. microRNA-365-targeted nuclear factor I/B transcriptionally represses cyclin-dependent kinase 6 and 4 to inhibit the progression of cutaneous squamous cell carcinoma. Int. J. Biochem. Cell Biol. 2015, 65, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, S.; Bao, H.; Mu, S.; Zhang, B.; Ma, H.; Ma, S. MicroRNA-365 promotes lung carcinogenesis by downregulating the USP33/SLIT2/ROBO1 signalling pathway. Cancer Cell Int. 2018, 18, 64. [Google Scholar] [CrossRef] [PubMed]
- Coon, J.; Kingsley, K.; Howard, K.M. miR-365 (microRNA): Potential Biomarker in Oral Squamous Cell Carcinoma Exosomes and Extracellular Vesicles. Int. J. Mol. Sci. 2020, 21, 5317. [Google Scholar] [CrossRef]
- Tang, J.-T.; Wang, J.-L.; Du, W.; Hong, J.; Zhao, S.-L.; Wang, Y.-C.; Xiong, H.; Chen, H.-M.; Fang, J.-Y. MicroRNA 345, a methylation-sensitive microRNA is involved in cell proliferation and invasion in human colorectal cancer. Carcinogenesis 2011, 32, 1207–1215. [Google Scholar] [CrossRef]
- Sun, Y.; Sukumaran, P.; Varma, A.; Derry, S.; Sahmoun, A.E.; Singh, B.B. Cholesterol-induced activation of TRPM7 regulates cell proliferation, migration, and viability of human prostate cells. Biochim. Biophys. Acta 2014, 1843, 1839–1850. [Google Scholar] [CrossRef]
- Huang, B.; Song, B.L.; Xu, C. Cholesterol metabolism in cancer: Mechanisms and therapeutic opportunities. Nat. Metab. 2020, 2, 132–141. [Google Scholar] [CrossRef]
- Zettler, M.E.; Prociuk, M.A.; Austria, J.A.; Massaeli, H.; Zhong, G.; Pierce, G.N. OxLDL stimulates cell proliferation through a general induction of cell cycle proteins. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H644–H653. [Google Scholar] [CrossRef][Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kelemen, A.; Carmi, I.; Seress, I.; Lőrincz, P.; Tölgyes, T.; Dede, K.; Bursics, A.; Buzás, E.I.; Wiener, Z. CD44 Expression Intensity Marks Colorectal Cancer Cell Subpopulations with Different Extracellular Vesicle Release Capacity. Int. J. Mol. Sci. 2022, 23, 2180. https://doi.org/10.3390/ijms23042180
Kelemen A, Carmi I, Seress I, Lőrincz P, Tölgyes T, Dede K, Bursics A, Buzás EI, Wiener Z. CD44 Expression Intensity Marks Colorectal Cancer Cell Subpopulations with Different Extracellular Vesicle Release Capacity. International Journal of Molecular Sciences. 2022; 23(4):2180. https://doi.org/10.3390/ijms23042180
Chicago/Turabian StyleKelemen, Andrea, Idan Carmi, Iván Seress, Péter Lőrincz, Tamás Tölgyes, Kristóf Dede, Attila Bursics, Edit I. Buzás, and Zoltán Wiener. 2022. "CD44 Expression Intensity Marks Colorectal Cancer Cell Subpopulations with Different Extracellular Vesicle Release Capacity" International Journal of Molecular Sciences 23, no. 4: 2180. https://doi.org/10.3390/ijms23042180
APA StyleKelemen, A., Carmi, I., Seress, I., Lőrincz, P., Tölgyes, T., Dede, K., Bursics, A., Buzás, E. I., & Wiener, Z. (2022). CD44 Expression Intensity Marks Colorectal Cancer Cell Subpopulations with Different Extracellular Vesicle Release Capacity. International Journal of Molecular Sciences, 23(4), 2180. https://doi.org/10.3390/ijms23042180