
Phosphodiesterases and Compartmentation of cAMP and cGMP Signaling in Regulation of Cardiac Contractility in Normal and Failing Hearts



Abstract
:1. Introduction
2. Cardiac Contractility
3. Cyclic Nucleotide Signaling and Contractility
4. cAMP-Selective PDEs and Compartmentation of cAMP Signaling Regulating Contractility
4.1. PDE4
4.2. PDE8
5. cGMP-Selective PDEs and Compartmentation of cGMP Signaling Regulating Contractility
5.1. PDE5
5.2. PDE9
6. Dual-Substrate PDEs and Compartmentation of Cyclic Nucleotide Signaling Regulating Contractility
6.1. PDE1
6.2. PDE2
6.2.1. PDE2 Regulation of cAMP Relevant for Contractility
6.2.2. PDE2 Regulation of cGMP Relevant for Contractility
6.2.3. PDE2 and cGMP/cAMP Cross-Talk Relevant for Contractility
6.3. PDE3
6.3.1. PDE3 Regulation of cAMP Relevant for Contractility
6.3.2. PDE3 Regulation of cGMP Relevant for Contractility
6.3.3. PDE3 and cGMP/cAMP Cross-Talk Relevant for Contractility
6.4. PDE10
7. Modulation of PDEs in Heart Failure
7.1. PDE2
7.2. PDE3
7.3. PDE4
7.4. PDE5
7.5. PDE9
8. Orchestration of Cyclic Nucleotide Signaling in Nanodomains
9. Summary
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bender, A.T.; Beavo, J.A. Cyclic Nucleotide Phosphodiesterases: Molecular Regulation to Clinical Use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef] [PubMed]
- Kokkonen, K.; Kass, D.A. Nanodomain Regulation of Cardiac Cyclic Nucleotide Signaling by Phosphodiesterases. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 455–479. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, Y.; Lighthouse, J.K.; Mickelsen, D.M.; Wu, J.; Yao, P.; Small, E.M.; Yan, C. A Novel Role of Cyclic Nucleotide Phosphodiesterase 10A in Pathological Cardiac Remodeling and Dysfunction. Circulation 2020, 141, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Katrukha, I.A. Human cardiac troponin complex. Structure and functions. Biochemistry 2013, 78, 1447–1465. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- Taskén, K.; Aandahl, E.M. Localized Effects of cAMP Mediated by Distinct Routes of Protein Kinase A. Physiol. Rev. 2004, 84, 137–167. [Google Scholar] [CrossRef] [Green Version]
- Dodge-Kafka, K.L.; Langeberg, L.; Scott, J.D. Compartmentation of cyclic nucleotide signaling in the heart: The role of A-kinase anchoring proteins. Circ. Res. 2006, 98, 993–1001. [Google Scholar] [CrossRef] [Green Version]
- Biel, M.; Michalakis, S. Cyclic nucleotide-gated channels. Handb. Exp. Pharmacol. 2009, 191, 111–136. [Google Scholar]
- Schindler, R.F.; Brand, T. The Popeye domain containing protein family—A novel class of cAMP effectors with important functions in multiple tissues. Prog. Biophys. Mol. Biol. 2016, 120, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Laudette, M.; Zuo, H.; Lezoualc’H, F.; Schmidt, M. Epac Function and cAMP Scaffolds in the Heart and Lung. J. Cardiovasc. Dev. Dis. 2018, 5, 9. [Google Scholar] [CrossRef] [Green Version]
- Ercu, M.; Klussmann, E. Roles of A-Kinase Anchoring Proteins and Phosphodiesterases in the Cardiovascular System. J. Cardiovasc. Dev. Dis. 2018, 5, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Chen, J.; Fontes, S.K.; Bautista, E.N.; Cheng, Z. Physiological and pathological roles of protein kinase A in the heart. Cardiovasc. Res. 2021, 118, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Potter, L.R. Guanylyl cyclase structure, function and regulation. Cell. Signal. 2011, 23, 1921–1926. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, C.; Leblais, V.; Kobzik, L.; Trochu, J.N.; Khandoudi, N.; Bril, A.; Balligand, J.L.; Le Marec, H. The negative inotropic effect of β3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle. J. Clin. Investig. 1998, 102, 1377–1384. [Google Scholar] [CrossRef]
- Tsai, E.J.; Kass, D.A. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol. Ther. 2009, 122, 216–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, J.; Kuret, A.; Längst, N.; Lukowski, R. Targets of cGMP/cGKI in Cardiac Myocytes. J. Cardiovasc. Pharmacol. 2020, 75, 494–507. [Google Scholar] [CrossRef]
- Brusq, J.-M.; Mayoux, E.; Guigui, L.; Kirilovsky, J. Effects of C-type natriuretic peptide on rat cardiac contractility. J. Cereb. Blood Flow Metab. 1999, 128, 206–212. [Google Scholar] [CrossRef] [Green Version]
- Pierkes, M.; Gambaryan, S.; Bokník, P.; Lohmann, S.M.; Schmitz, W.; Potthast, R.; Holtwick, R.; Kuhn, M. Increased effects of C-type natriuretic peptide on cardiac ventricular contractility and relaxation in guanylyl cyclase A-deficient mice. Cardiovasc. Res. 2002, 53, 852–861. [Google Scholar] [CrossRef] [Green Version]
- Wollert, K.C.; Yurukova, S.; Kilic, A.; Begrow, F.; Fiedler, B.; Gambaryan, S.; Walter, U.; Lohmann, S.M.; Kuhn, M. Increased effects of C-type natriuretic peptide on contractility and calcium regulation in murine hearts overexpressing cyclic GMP-dependent protein kinase I. Br. J. Pharmacol. 2003, 140, 1227–1236. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Moalem, J.; Tse, J.; Scholz, P.M.; Weiss, H.R. Effects of natriuretic peptides on ventricular myocyte contraction and role of cyclic GMP signaling. Eur. J. Pharmacol. 2005, 510, 209–215. [Google Scholar] [CrossRef]
- Su, J.; Scholz, P.M.; Weiss, H.R. Differential Effects of cGMP Produced by Soluble and Particulate Guanylyl Cyclase on Mouse Ventricular Myocytes. Exp. Biol. Med. 2005, 230, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Scholz, P.M.; Pilzak, A.; Su, J.; Weiss, H.R. Role of Phospholamban in Cyclic GMP Mediated Signaling in Cardiac Myocytes. Cell. Physiol. Biochem. 2006, 20, 157–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moltzau, L.R.; Aronsen, J.M.; Meier, S.; Nguyen, C.H.T.; Hougen, K.; Ørstavik, Ø.; Sjaastad, I.; Christensen, G.; Skomedal, T.; Osnes, J.-B.; et al. SERCA2 activity is involved in the CNP-mediated functional responses in failing rat myocardium. J. Cereb. Blood Flow Metab. 2013, 170, 366–379. [Google Scholar] [CrossRef] [Green Version]
- Manfra, O.; Calamera, G.; Froese, A.; Arunthavarajah, D.; Surdo, N.C.; Meier, S.; Melleby, A.O.; Aasrum, M.; Aronsen, J.M.; Nikolaev, V.O.; et al. CNP regulates cardiac contractility and increases cGMP near both SERCA and TnI: Difference from BNP visualized by targeted cGMP biosensors. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Gisbert, M.P.; Fischmeister, R. Atrial natriuretic factor regulates the calcium current in frog isolated cardiac cells. Circ. Res. 1988, 62, 660–667. [Google Scholar] [CrossRef] [Green Version]
- Méry, P.-F.; Lohmann, S.M.; Walter, U.; Fischmeister, R. Ca2+ current is regulated by cyclic GMP-dependent protein kinase in mammalian cardiac myocytes. Proc. Natl. Acad. Sci. USA 1991, 88, 1197–1201. [Google Scholar] [CrossRef] [Green Version]
- Tohse, N.; Nakaya, H.; Takeda, Y.; Kanno, M. Cyclic GMP-mediated inhibition of L-type Ca2+ channel activity by human natriuretic peptide in rabbit heart cells. Br. J. Pharmacol. 1995, 114, 1076–1082. [Google Scholar] [CrossRef] [Green Version]
- Sodi, R.; Dubuis, E.; Shenkin, A.; Hart, G. B-type natriuretic peptide (BNP) attenuates the L-type calcium current and regulates ventricular myocyte function. Regul. Pept. 2008, 151, 95–105. [Google Scholar] [CrossRef]
- Moltzau, L.R.; Aronsen, J.M.; Meier, S.; Skogestad, J.; Ørstavik, Ø.; Lothe, G.B.; Sjaastad, I.; Skomedal, T.; Osnes, J.-B.; Levy, F.O.; et al. Different Compartmentation of Responses to Brain Natriuretic Peptide and C-Type Natriuretic Peptide in Failing Rat Ventricle. J. Pharmacol. Exp. Ther. 2014, 350, 681–690. [Google Scholar] [CrossRef] [Green Version]
- Suko, J.; Maurer-Fogy, I.; Plank, B.; Bertel, O.; Wyskovsky, W.; Hohenegger, M.; Hellmann, G. Phosphorylation of serine 2843 in ryanodine receptor-calcium release channel of skeletal muscle by cAMP-, cGMP- and CaM-dependent protein kinase. Biochim. Biophys. Acta 1993, 1175, 193–206. [Google Scholar] [CrossRef]
- Xiao, B.; Zhong, G.; Obayashi, M.; Yang, D.; Chen, K.; Walsh, M.P.; Shimoni, Y.; Cheng, H.; Ter Keurs, H.; Chen, S.R.W. Ser-2030, but not Ser-2808, is the major phosphorylation site in cardiac ryanodine receptors responding to protein kinase A activation upon β-adrenergic stimulation in normal and failing hearts. Biochem. J. 2006, 396, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Kaumann, A.J.; Birnbaumer, L. Prostaglandin E1 action on sinus pacemaker and adenylyl cyclase in kitten myocardium. Nature 1974, 251, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.S.; Brunton, L.L.; Mayer, S.E. Selective activation of particulate cAMP-dependent protein kinase by isoproterenol and prostaglandin E1. J. Biol. Chem. 1980, 255, 5113–5119. [Google Scholar] [CrossRef]
- Hayes, J.; Bowling, N.; King, K.; Boder, G. Evidence for selective regulation of the phosphorylation of myocyte proteins by isoproterenol and prostaglandin E1. Biochim. Biophys. Acta 1982, 714, 136–142. [Google Scholar] [CrossRef]
- Buxton, I.L.; Brunton, L.L. Compartments of cyclic AMP and protein kinase in mammalian cardiomyocytes. J. Biol. Chem. 1983, 258, 10233–10239. [Google Scholar] [CrossRef]
- Aass, H.; Skomedal, T.; Osnes, J. Increase of cyclic AMP in subcellular fractions of rat heart muscle after β-adrenergic stimulation: Prenalterol and isoprenaline caused different distribution of bound cyclic AMP. J. Mol. Cell. Cardiol. 1988, 20, 847–860. [Google Scholar] [CrossRef]
- Defer, N.; Best-Belpomme, M.; Hanoune, J. Tissue specificity and physiological relevance of various isoforms of adenylyl cyclase. Am. J. Physiol. Physiol. 2000, 279, F400–F416. [Google Scholar] [CrossRef]
- Steinberg, S.F. The Molecular Basis for Distinct β-Adrenergic Receptor Subtype Actions in Cardiomyocytes. Circ. Res. 1999, 85, 1101–1111. [Google Scholar] [CrossRef] [Green Version]
- Xiao, R.P. β-adrenergic signaling in the heart: Dual coupling of the β2-adrenergic receptor to Gs and Gi proteins. Sci. STKE 2001, 2001, re15. [Google Scholar] [CrossRef]
- Afzal, F.; Aronsen, J.M.; Moltzau, L.R.; Sjaastad, I.; Levy, F.O.; Skomedal, T.; Osnes, J.B.; Qvigstad, E. Differential regulation of β2 -adrenoceptor-mediated inotropic and lusitropic response by PDE3 and PDE4 in failing and non-failing rat cardiac ventricle. Br. J. Pharmacol. 2011, 162, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Chen-Izu, Y.; Xiao, R.-P.; Izu, L.T.; Cheng, H.; Kuschel, M.; Spurgeon, H.; Lakatta, E.G. Gi-Dependent Localization of β2-Adrenergic Receptor Signaling to L-Type Ca2+ Channels. Biophys. J. 2000, 79, 2547–2556. [Google Scholar] [CrossRef] [Green Version]
- Xiao, R.P.; Ji, X.; Lakatta, E.G. Functional coupling of the β2-adrenoceptor to a pertussis toxin-sensitive G protein in cardiac myocytes. Mol. Pharmacol. 1995, 47, 322–329. [Google Scholar] [PubMed]
- Xiao, R.P.; Avdonin, P.; Zhou, Y.Y.; Cheng, H.; Akhter, S.A.; Eschenhagen, T.; Lefkowitz, R.J.; Koch, W.J.; Lakatta, E.G. Coupling of β2-adrenoceptor to Gi proteins and its physiological relevance in murine cardiac myocytes. Circ. Res. 1999, 84, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Kuschel, M.; Zhou, Y.Y.; Cheng, H.; Zhang, S.J.; Chen, Y.; Lakatta, E.G.; Xiao, R.P. Gi protein-mediated functional com-partmentalization of cardiac β2-adrenergic signaling. J. Biol. Chem. 1999, 274, 22048–22052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolaev, V.O.; Bünemann, M.; Schmitteckert, E.; Lohse, M.J.; Engelhardt, S. Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching β1-adrenergic but locally confined β2-adrenergic receptor-mediated signaling. Circ. Res. 2006, 99, 1084–1091. [Google Scholar] [CrossRef] [Green Version]
- Nikolaev, V.O.; Moshkov, A.; Lyon, A.R.; Miragoli, M.; Novak, P.; Paur, H.; Lohse, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. β2-Adrenergic Receptor Redistribution in Heart Failure Changes cAMP Compartmentation. Science 2010, 327, 1653–1657. [Google Scholar] [CrossRef]
- MacDougall, D.A.; Agarwal, S.R.; Stopford, E.A.; Chu, H.; Collins, J.A.; Longster, A.L.; Colyer, J.; Harvey, R.D.; Calaghan, S. Caveolae compartmentalise β2-adrenoceptor signals by curtailing cAMP production and maintaining phosphatase activity in the sarcoplasmic reticulum of the adult ventricular myocyte. J. Mol. Cell. Cardiol. 2012, 52, 388–400. [Google Scholar] [CrossRef] [Green Version]
- Wright, P.T.; Nikolaev, V.O.; O’hara, T.; Diakonov, I.; Bhargava, A.; Tokar, S.; Schobesberger, S.; Shevchuk, A.I.; Sikkel, M.B.; Wilkinson, R.; et al. Caveolin-3 regulates compartmentation of cardiomyocyte β2-adrenergic receptor-mediated cAMP signaling. J. Mol. Cell. Cardiol. 2014, 67, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Brattelid, T.; Qvigstad, E.; Lynham, J.A.; Molenaar, P.; Aass, H.; Geiran, O.; Skomedal, T.; Levy, F.O.; Kaumann, A.J. Functional serotonin 5-HT4 receptors in porcine and human ventricular myocardium with increased 5-HT4 mRNA in heart failure. Naunyn-Schmiedebergs Arch. Exp. Pathol. Pharmakol. 2004, 370, 157–166. [Google Scholar] [CrossRef]
- Qvigstad, E.; Brattelid, T.; Sjaastad, I.; Andressen, K.W.; Krobert, K.A.; Birkeland, J.A.; Sejersted, O.M.; Kaumann, A.J.; Skomedal, T.; Osnes, J.-B.; et al. Appearance of a ventricular 5-HT receptor-mediated inotropic response to serotonin in heart failure. Cardiovasc. Res. 2005, 65, 869–878. [Google Scholar] [CrossRef] [Green Version]
- Kaumann, A.J.; Levy, F.O. 5-Hydroxytryptamine receptors in the human cardiovascular system. Pharmacol. Ther. 2006, 111, 674–706. [Google Scholar] [CrossRef] [PubMed]
- Afzal, F.; Andressen, K.W.; Mørk, H.K.; Aronsen, J.M.; Sjaastad, I.; Dahl, C.P.; Skomedal, T.; Levy, F.O.; Osnes, J.B.; Qvigstad, E. 5-HT4-elicited positive inotropic response is mediated by cAMP and regulated by PDE3 in failing rat and human cardiac ventricles. Br. J. Pharmacol. 2008, 155, 1005–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afzal, F.; Qvigstad, E.; Aronsen, J.M.; Moltzau, L.R.; Sjaastad, I.; Skomedal, T.; Osnes, J.-B.; Levy, F.O. Agents increasing cyclic GMP amplify 5-HT4-elicited positive inotropic response in failing rat cardiac ventricle. Naunyn Schmiedebergs Arch. Exp. Pathol. Pharmakol. 2011, 384, 543–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostic, M.; Erdogan, S.; Rena, G.; Borchert, G.; Hoch, B.; Bartel, S.; Scotland, G.; Huston, E.; Houslay, M.; Krause, E.-G. Altered Expression of PDE1 and PDE4 Cyclic Nucleotide Phosphodiesterase Isoforms in 7-oxo-prostacyclin-preconditioned Rat Heart. J. Mol. Cell. Cardiol. 1997, 29, 3135–3146. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.; Baillie, G.S.; MacKenzie, S.J.; Yarwood, S.J.; Houslay, M.D. The MAP kinase ERK2 inhibits the cyclic AMP-specific phosphodiesterase HSPDE4D3 by phosphorylating it at Ser579. EMBO J. 1999, 18, 893–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sette, C.; Conti, M. Phosphorylation and activation of a cAMP-specific phosphodiesterase by the cAMP-dependent protein kinase. Involvement of serine 54 in the enzyme activation. J. Biol. Chem. 1996, 271, 16526–16534. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, S.J.; Baillie, G.; McPhee, I.; MacKenzie, C.; Seamons, R.; McSorley, T.; Millen, J.; Beard, M.B.; Van Heeke, G.; Houslay, M.D. Long PDE4 cAMP specific phosphodiesterases are activated by protein kinase A-mediated phosphorylation of a single serine residue in Upstream Conserved Region 1 (UCR1). J. Cereb. Blood Flow Metab. 2002, 136, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Houslay, M.D.; Adams, D.R. PDE4 cAMP phosphodiesterases: Modular enzymes that orchestrate signalling cross-talk, desensitization and compartmentalization. Biochem. J. 2003, 370, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Huston, E.; Gall, I.; Houslay, T.M.; Houslay, M.D. Helix-1 of the cAMP-specific phosphodiesterase PDE4A1 regulates its phospholipase-D-dependent redistribution in response to release of Ca2+. J. Cell Sci. 2006, 119, 3799–3810. [Google Scholar] [CrossRef] [Green Version]
- Wills, L.; Ehsan, M.; Whiteley, E.L.; Baillie, G.S. Location, location, location: PDE4D5 function is directed by its unique N-terminal region. Cell. Signal. 2016, 28, 701–705. [Google Scholar] [CrossRef] [Green Version]
- Richter, W.; Xie, M.; Scheitrum, C.; Krall, J.; Movsesian, M.A.; Conti, M. Conserved expression and functions of PDE4 in rodent and human heart. Basic Res. Cardiol. 2010, 106, 249–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qvigstad, E.; Moltzau, L.R.; Aronsen, J.M.; Nguyen, C.H.; Hougen, K.; Sjaastad, I.; Levy, F.O.; Skomedal, T.; Osnes, J.-B. Natriuretic peptides increase β1-adrenoceptor signalling in failing hearts through phosphodiesterase 3 inhibition. Cardiovasc. Res. 2009, 85, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Molina, C.E.; Leroy, J.; Richter, W.; Xie, M.; Scheitrum, C.; Lee, I.O.; Maack, C.; Rucker-Martin, C.; Donzeau-Gouge, P.; Verde, I.; et al. Cyclic adenosine monophosphate phosphodiesterase type 4 protects against atrial arrhythmias. J. Am. Coll. Cardiol. 2012, 59, 2182–2190. [Google Scholar] [CrossRef] [Green Version]
- Moltzau, L.R.; Meier, S.; Aronsen, J.M.; Afzal, F.; Sjaastad, I.; Skomedal, T.; Osnes, J.-B.; Levy, F.O.; Qvigstad, E. Differential regulation of C-type natriuretic peptide-induced cGMP and functional responses by PDE2 and PDE3 in failing myocardium. Naunyn Schmiedebergs Arch. Exp. Pathol. Pharmakol. 2014, 387, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, P.; Christ, T.; Hussain, R.I.; Engel, A.; Berk, E.; Gillette, K.T.; Chen, L.; Galindo-Tovar, A.; Krobert, K.A.; Ravens, U.; et al. PDE3, but not PDE4, reduces β1- and β2-adrenoceptor-mediated inotropic and lusitropic effects in failing ventricle from metoprolol-treated patients. Br. J. Pharmacol. 2013, 169, 528–538. [Google Scholar] [CrossRef] [Green Version]
- Molenaar, P.; Christ, T.; Berk, E.; Engel, A.; Gillette, K.T.; Galindo-Tovar, A.; Ravens, U.; Kaumann, A.J. Carvedilol induces greater control of β2- than β1-adrenoceptor-mediated inotropic and lusitropic effects by PDE3, while PDE4 has no effect in human failing myocardium. Naunyn Schmiedebergs Arch. Pharmacol. 2014, 387, 629–640. [Google Scholar] [CrossRef]
- Verde, I.; Vandecasteele, G.; Lezoualc’H, F.; Fischmeister, R. Characterization of the cyclic nucleotide phosphodiesterase subtypes involved in the regulation of the L-type Ca2+current in rat ventricular myocytes. J. Cereb. Blood Flow Metab. 1999, 127, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Mongillo, M.; McSorley, T.; Evellin, S.; Sood, A.; Lissandron, V.; Terrin, A.; Huston, E.; Hannawacker, A.; Lohse, M.J.; Pozzan, T.; et al. Fluorescence Resonance Energy Transfer–Based Analysis of cAMP Dynamics in Live Neonatal Rat Cardiac Myocytes Reveals Distinct Functions of Compartmentalized Phosphodiesterases. Circ. Res. 2004, 95, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Mika, D.; Bobin, P.; Lindner, M.; Boet, A.; Hodzic, A.; Lefebvre, F.; Lechène, P.; Sadoune, M.; Samuel, J.-L.; Algalarrondo, V.; et al. Synergic PDE3 and PDE4 control intracellular cAMP and cardiac excitation-contraction coupling in a porcine model. J. Mol. Cell. Cardiol. 2019, 133, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Cosson, M.-V.; Hiis, H.G.; Moltzau, L.R.; Levy, F.O.; Krobert, K.A. Knockout of adenylyl cyclase isoform 5 or 6 differentially modifies the β1-adrenoceptor-mediated inotropic response. J. Mol. Cell. Cardiol. 2019, 131, 132–145. [Google Scholar] [CrossRef]
- Vargas, M.L.; Hernandez, J.; Kaumann, A.J. Phosphodiesterase PDE3 blunts the positive inotropic and cyclic AMP enhancing effects of CGP12177 but not of noradrenaline in rat ventricle. Br. J. Pharmacol. 2006, 147, 158–163. [Google Scholar] [CrossRef] [Green Version]
- Galindo-Tovar, A.; Kaumann, A.J. Phosphodiesterase-4 blunts inotropism and arrhythmias but not sinoatrial tachycardia of (−)-adrenaline mediated through mouse cardiac β1-adrenoceptors. J. Cereb. Blood Flow Metab. 2008, 153, 710–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, P.T.; Bhogal, N.K.; Diakonov, I.; Pannell, L.M.K.; Perera, R.K.; Bork, N.I.; Schobesberger, S.; Lucarelli, C.; Faggian, G.; Alvarez-Laviada, A.; et al. Cardiomyocyte membrane structure and cAMP compartmentation produce anatomical variation in β2AR-cAMP responsiveness in murine hearts. Cell Rep. 2018, 23, 459–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprenger, J.U.; Perera, R.K.; Steinbrecher, J.H.; Lehnart, S.E.; Maier, L.S.; Hasenfuss, G.; Nikolaev, V. In vivo model with targeted cAMP biosensor reveals changes in receptor–microdomain communication in cardiac disease. Nat. Commun. 2015, 6, 6965. [Google Scholar] [CrossRef] [Green Version]
- Rudokas, M.W.; Post, J.P.; Sataray-Rodriguez, A.; Sherpa, R.T.; Moshal, K.S.; Agarwal, S.R.; Harvey, R.D. Compartmentation of β2-adrenoceptor stimulated cAMP responses by phosphodiesterase types 2 and 3 in cardiac ventricular myocytes. Br. J. Pharmacol. 2021, 178, 1574–1587. [Google Scholar] [CrossRef] [PubMed]
- Lehnart, S.E.; Wehrens, X.H.T.; Reiken, S.; Warrier, S.; Belevych, A.E.; Harvey, R.D.; Richter, W.; Jin, S.-L.C.; Conti, M.; Marks, A.R. Phosphodiesterase 4D Deficiency in the Ryanodine-Receptor Complex Promotes Heart Failure and Arrhythmias. Cell 2005, 123, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Beca, S.; Helli, P.B.; Simpson, J.A.; Zhao, D.; Farman, G.P.; Jones, P.; Tian, X.; Wilson, L.S.; Ahmad, F.; Chen, S.R.W.; et al. Phosphodiesterase 4D regulates baseline sarcoplasmic reticulum Ca2+ release and cardiac contractility, independently of L-type Ca2+ current. Circ. Res. 2011, 109, 1024–1030. [Google Scholar] [CrossRef] [Green Version]
- Bünemann, M.; Gerhardstein, B.L.; Gao, T.; Hosey, M.M. Functional Regulation of L-type Calcium Channels via Protein Kinase A-mediated Phosphorylation of the β2 Subunit. J. Biol. Chem. 1999, 274, 33851–33854. [Google Scholar] [CrossRef] [Green Version]
- Leroy, J.; Richter, W.; Mika, D.; Castro, L.R.; Abi-Gerges, A.; Xie, M.; Scheitrum, C.; Lefebvre, F.; Schittl, J.; Mateo, P.; et al. Phosphodiesterase 4B in the cardiac L-type Ca2+ channel complex regulates Ca2+ current and protects against ventricular arrhythmias in mice. J. Clin. Investig. 2011, 121, 2651–2661. [Google Scholar] [CrossRef] [Green Version]
- Berisha, F.; Götz, K.R.; Wegener, J.W.; Brandenburg, S.; Subramanian, H.; Molina, C.E.; Ruffer, A.; Petersen, J.; Bernhardt, A.; Girdauskas, E.; et al. cAMP imaging at ryanodine receptors reveals β2-adrenoceptor driven arrhythmias. Circ. Res. 2021, 129, 81–94. [Google Scholar] [CrossRef]
- Weninger, S.; De Maeyer, J.H.; Lefebvre, R.A. Study of the regulation of the inotropic response to 5-HT4 receptor activation via phosphodiesterases and its cross-talk with C-type natriuretic peptide in porcine left atrium. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2012, 385, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Melsom, C.B.; Hussain, R.I.; Ørstavik, Ø.; Aronsen, J.M.; Sjaastad, I.; Skomedal, T.; Osnes, J.-B.; Levy, F.O.; Krobert, K.A. Non-classical regulation of β1- and β2-adrenoceptor-mediated inotropic responses in rat heart ventricle by the G protein Gi. Naunyn Schmiedebergs Arch. Exp. Pathol. Pharmakol. 2014, 387, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Melsom, C.B.; Ørstavik, Ø.; Osnes, J.B.; Skomedal, T.; Levy, F.O.; Krobert, K.A. Gi proteins regulate adenylyl cyclase activity independent of receptor activation. PLoS ONE 2014, 9, e106608. [Google Scholar] [CrossRef] [Green Version]
- Abi-Gerges, A.; Castro, L.; Leroy, J.; Domergue, V.; Fischmeister, R.; Vandecasteele, G. Selective changes in cytosolic β-adrenergic cAMP signals and L-type Calcium Channel regulation by Phosphodiesterases during cardiac hypertrophy. J. Mol. Cell. Cardiol. 2020, 150, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Weninger, S.; De Maeyer, J.H.; Lefebvre, R.A. Influence of phosphodiesterases and cGMP on cAMP generation and on phosphorylation of phospholamban and troponin I by 5-HT4 receptor activation in porcine left atrium. Naunyn-Schmiedebergs Arch. Exp. Pathol. Pharmakol. 2013, 386, 671–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolce, B.; Christ, T.; Pavlidou, N.G.; Yildirim, Y.; Reichenspurner, H.; Eschenhagen, T.; Nikolaev, V.O.; Kaumann, A.J.; Molina, C.E. Impact of phosphodiesterases PDE3 and PDE4 on 5-hydroxytryptamine receptor4-mediated increase of cAMP in human atrial fibrillation. Naunyn Schmiedebergs Arch. Exp. Pathol. Pharmakol. 2020, 394, 291–298. [Google Scholar] [CrossRef]
- Zhao, C.Y.; Greenstein, J.L.; Winslow, R.L. Interaction between phosphodiesterases in the regulation of the cardiac β-adrenergic pathway. J. Mol. Cell. Cardiol. 2015, 88, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Soderling, S.H.; Bayuga, S.J.; Beavo, J.A. Cloning and characterization of a cAMP-specific cyclic nucleotide phosphodiesterase. Proc. Natl. Acad. Sci. USA 1998, 95, 8991–8996. [Google Scholar] [CrossRef] [Green Version]
- Galperin, M.Y.; Nikolskaya, A.N.; Koonin, E.V. Novel domains of the prokaryotic two-component signal transduction systems. FEMS Microbiol. Lett. 2001, 203, 11–21. [Google Scholar] [CrossRef]
- Gilles-Gonzalez, M.-A.; Gonzalez, G.; Sieck, G.C. Signal transduction by heme-containing PAS-domain proteins. J. Appl. Physiol. 2004, 96, 774–783. [Google Scholar] [CrossRef]
- Patrucco, E.; Albergine, M.S.; Santana, L.; Beavo, J.A. Phosphodiesterase 8A (PDE8A) regulates excitation–contraction coupling in ventricular myocytes. J. Mol. Cell. Cardiol. 2010, 49, 330–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mery, P.F.; Pavoine, C.; Belhassen, L.; Pecker, F.; Fischmeister, R. Nitric oxide regulates cardiac Ca2+ current. Involvement of cGMP-inhibited and cGMP-stimulated phosphodiesterases through guanylyl cyclase activation. J. Biol. Chem. 1993, 268, 26286–26295. [Google Scholar] [CrossRef]
- Weyrich, A.; Ma, X.L.; Buerke, M.; Murohara, T.; Armstead, V.E.; Lefer, A.M.; Nicolas, J.M.; Thomas, A.; Lefer, D.J.; Vinten-Johansen, J. Physiological concentrations of nitric oxide do not elicit an acute negative inotropic effect in unstimulated cardiac muscle. Circ. Res. 1994, 75, 692–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawrath, H.; Bäumner, D.; Rupp, J.; Oelert, H. The ineffectiveness of the NO-cyclic GMP signaling pathway in the atrial myocardium. J. Cereb. Blood Flow Metab. 1995, 116, 3061–3067. [Google Scholar] [CrossRef] [Green Version]
- Kojda, G.; Kottenberg, K.; Nix, P.; Schlüter, K.D.; Piper, H.M.; Noack, E. Low Increase in cGMP Induced by Organic Nitrates and Nitrovasodilators Improves Contractile Response of Rat Ventricular Myocytes. Circ. Res. 1996, 78, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Flesch, M.; Kilter, H.; Cremers, B.; Lenz, O.; Südkamp, M.; Kuhn-Regnier, F.; Böhm, M. Acute effects of nitric oxide and cyclic GMP on human myocardial contractility. J. Pharmacol. Exp. Ther. 1997, 281, 1340–1349. [Google Scholar]
- Takimoto, E.; Belardi, D.; Tocchetti, C.G.; Vahebi, S.; Cormaci, G.; Ketner, E.A.; Moens, A.L.; Champion, H.C.; Kass, D.A. Compartmentalization of Cardiac β-Adrenergic Inotropy Modulation by Phosphodiesterase Type 5. Circulation 2007, 115, 2159–2167. [Google Scholar] [CrossRef] [Green Version]
- Meier, S.; Andressen, K.W.; Aronsen, J.M.; Sjaastad, I.; Hougen, K.; Skomedal, T.; Osnes, J.B.; Qvigstad, E.; Levy, F.O.; Moltzau, L.R. PDE3 inhibition by C-type natriuretic peptide-induced cGMP enhances cAMP-mediated signaling in both non-failing and failing hearts. Eur. J. Pharmacol. 2017, 812, 174–183. [Google Scholar] [CrossRef]
- Tajima, M.; Bartunek, J.; Weinberg, E.; Ito, N.; Lorell, B.H. Atrial Natriuretic Peptide Has Different Effects on Contractility and Intracellular pH in Normal and Hypertrophied Myocytes from Pressure-Overloaded Hearts. Circulation 1998, 98, 2760–2764. [Google Scholar] [CrossRef] [Green Version]
- Frantz, S.; Klaiber, M.; Baba, H.A.; Oberwinkler, H.; Völker, K.; Gaβner, B.; Bayer, B.; Abeβer, M.; Schuh, K.; Feil, R.; et al. Stress-dependent dilated cardiomyopathy in mice with cardiomyocyte-restricted inactivation of cyclic GMP-dependent protein kinase I. Eur. Heart, J. 2011, 34, 1233–1244. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.S.; Lau, A.; Tu, R.; Lue, T.F. Expression of three isoforms of cGMP-binding cGMP-specific phosphodiesterase (PDE5) in human penile cavernosum. Biochem. Biophys. Res. Commun. 2000, 268, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Castro, L.R.; Schittl, J.; Fischmeister, R. Feedback control through cGMP-dependent protein kinase contributes to differential regulation and compartmentation of cGMP in rat cardiac myocytes. Circ. Res. 2010, 107, 1232–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takimoto, E.; Champion, H.C.; Belardi, D.; Moslehi, J.; Mongillo, M.; Mergia, E.; Montrose, D.C.; Isoda, T.; Aufiero, K.; Zaccolo, M.; et al. cGMP Catabolism by Phosphodiesterase 5A Regulates Cardiac Adrenergic Stimulation by NOS3-Dependent Mechanism. Circ. Res. 2005, 96, 100–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, L.R.V.; Verde, I.; Cooper, D.M.; Fischmeister, R. Cyclic Guanosine Monophosphate Compartmentation in Rat Cardiac Myocytes. Circulation 2006, 113, 2221–2228. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.I.; Zhu, G.; Sasaki, T.; Cho, G.-S.; Hamdani, N.; Holewinski, R.J.; Jo, S.-H.; Danner, T.; Zhang, M.; Rainer, P.P.; et al. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 2015, 519, 472–476. [Google Scholar] [CrossRef]
- Götz, K.R.; Sprenger, J.U.; Perera, R.K.; Steinbrecher, J.H.; Lehnart, S.E.; Kuhn, M.; Gorelik, J.; Balligand, J.-L.; Nikolaev, V.O. Transgenic Mice for Real-Time Visualization of cGMP in Intact Adult Cardiomyocytes. Circ. Res. 2014, 114, 1235–1245. [Google Scholar] [CrossRef] [Green Version]
- Borlaug, B.A.; Melenovsky, V.; Marhin, T.; Fitzgerald, P.; Kass, D.A. Sildenafil Inhibits β-Adrenergic–Stimulated Cardiac Contractility in Humans. Circulation 2005, 112, 2642–2649. [Google Scholar] [CrossRef]
- Isidori, A.M.; Cornacchione, M.; Barbagallo, F.; Di Grazia, A.; Barrios, F.; Fassina, L.; Monaco, L.; Giannetta, E.; Gianfrilli, D.; Garofalo, S.; et al. Inhibition of type 5 phosphodiesterase counteracts β2-adrenergic signalling in beating cardiomyocytes. Cardiovasc. Res. 2015, 106, 408–420. [Google Scholar] [CrossRef] [Green Version]
- Schobesberger, S.; Wright, P.T.; Poulet, C.; Sanchez Alonso Mardones, J.L.; Mansfield, C.; Friebe, A.; Harding, S.E.; Balligand, J.L.; Nikolaev, V.O.; Gorelik, J. β3-Adrenoceptor redistribution impairs NO/cGMP/PDE2 signalling in failing cardiomyocytes. Elife 2020, 9, e52221. [Google Scholar] [CrossRef]
- Mongillo, M.; Tocchetti, C.G.; Terrin, A.; Lissandron, V.; Cheung, Y.F.; Dostmann, W.R.; Pozzan, T.; Kass, D.A.; Paolocci, N.; Houslay, M.D.; et al. Compartmentalized phosphodiesterase-2 activity blunts β-adrenergic cardiac inotropy via an NO/cGMP-dependent pathway. Circ. Res. 2006, 98, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.; Sadagopan, N.; Dunkerly-Eyring, B.; Rodriguez, S.; Sarver, D.C.; Ceddia, R.P.; Murphy, S.A.; Knutsdottir, H.; Jani, V.P.; Ashok, D.; et al. Inhibition of phosphodiesterase type 9 reduces obesity and cardiometabolic syndrome in mice. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Snyder, P.B.; Florio, V.A.; Ferguson, K.; Loughney, K. Isolation, expression and analysis of splice variants of a human Ca2+/calmodulin-stimulated phosphodiesterase (PDE1A). Cell. Signal. 1999, 11, 535–544. [Google Scholar] [CrossRef]
- Sonnenburg, W.K.; Seger, D.; Kwak, K.S.; Huang, J.; Charbonneau, H.; Beavo, J.A. Identification of inhibitory and calmodulin-binding domains of the PDE1A1 and PDE1A2 calmodulin-stimulated cyclic nucleotide phosphodiesterases. J. Biol. Chem. 1995, 270, 30989–31000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.K. Diversity of calcium action in regulation of mammalian calmodulin-dependent cyclic nucleotide phosphodiesterase. Indian J. Biochem. Biophys. 2003, 40, 77–91. [Google Scholar] [PubMed]
- Geremia, R.; Rossi, P.; Mocini, D.; Pezzotti, R.; Conti, M. Characterization of a calmodulin-dependent high-affinity cyclic AMP and cyclic GMP phosphodiesterase from male mouse germ cells. Biochem. J. 1984, 217, 693–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florio, V.A.; Sonnenburg, W.K.; Johnson, R.; Kwak, K.S.; Jensen, G.S.; Walsh, K.A.; Beavo, J.A. Phosphorylation of the 61-kDa Calmodulin-Stimulated Cyclic Nucleotide Phosphodiesterase at Serine 120 Reduces Its Affinity for Calmodulin. Biochemistry 1994, 33, 8948–8954. [Google Scholar] [CrossRef]
- Vandeput, F.; Wolda, S.L.; Krall, J.; Hambleton, R.; Uher, L.; McCaw, K.N.; Radwanski, P.B.; Florio, V.; Movsesian, M.A. Cyclic Nucleotide Phosphodiesterase PDE1C1 in Human Cardiac Myocytes. J. Biol. Chem. 2007, 282, 32749–32757. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.L.; Oikawa, M.; Cai, Y.; Wojtovich, A.P.; Nagel, D.J.; Xu, X.; Xu, H.; Florio, V.; Rybalkin, S.D.; Beavo, J.A.; et al. Role of Ca2+/calmodulin-stimulated cyclic nucleotide phosphodiesterase 1 in mediating cardiomyocyte hypertrophy. Circ. Res. 2009, 105, 956–964. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, T.; Kim, G.E.; Tunin, R.S.; Adesiyun, T.; Hsu, S.; Nakagawa, R.; Zhu, G.; O’Brien, J.J.; Hendrick, J.P.; Davis, R.E.; et al. Acute enhancement of cardiac function by phosphodiesterase type 1 inhibition. Circulation 2018, 138, 1974–1987. [Google Scholar] [CrossRef]
- Knight, W.E.; Chen, S.; Zhang, Y.; Oikawa, M.; Wu, M.; Zhou, Q.; Miller, C.L.; Cai, Y.; Mickelsen, D.M.; Moravec, C.; et al. PDE1C deficiency antagonizes pathological cardiac remodeling and dysfunction. Proc. Natl. Acad. Sci. USA 2016, 113, E7116–E7125. [Google Scholar] [CrossRef] [Green Version]
- Muller, G.K.; Song, J.; Jani, V.; Wu, Y.; Liu, T.; Jeffreys, W.P.; O’Rourke, B.; Anderson, M.E.; Kass, D.A. PDE1 Inhibition Modulates Cav1.2 Channel to Stimulate Cardiomyocyte Contraction. Circ. Res. 2021, 129, 872–886. [Google Scholar] [CrossRef] [PubMed]
- Martins, T.J.; Mumby, M.C.; Beavo, J.A. Purification and characterization of a cyclic GMP-stimulated cyclic nucleotide phosphodiesterase from bovine tissues. J. Biol. Chem. 1982, 257, 1973–1979. [Google Scholar] [CrossRef]
- Rosman, G.J.; Martins, T.J.; Sonnenburg, W.K.; Beavo, J.A.; Ferguson, K.; Loughney, K. Isolation and characterization of human cDNAs encoding a cGMP-stimulated 3′,5′-cyclic nucleotide phosphodiesterase. Gene 1997, 191, 89–95. [Google Scholar] [CrossRef]
- Weishaar, R.E.; Burrows, S.D.; Kobylarz, D.C.; Quade, M.M.; Evans, D.B. Multiple molecular forms of cyclic nucleotide phosphodiesterase in cardiac and smooth muscle and in platelets: Isolation, characterization, and effects of various reference phosphodiesterase inhibitors and cardiotonic agents. Biochem. Pharmacol. 1986, 35, 787–800. [Google Scholar] [CrossRef]
- Pyne, N.; Cooper, M.E.; Houslay, M. Identification and characterization of both the cytosolic and particulate forms of cyclic GMP-stimulated cyclic AMP phosphodiesterase from rat liver. Biochem. J. 1986, 234, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Russwurm, C.; Zoidl, G.; Koesling, D.; Russwurm, M. Dual Acylation of PDE2A Splice Variant 3: Targeting to Synaptic Membranes. J. Biol. Chem. 2009, 284, 25782–25790. [Google Scholar] [CrossRef] [Green Version]
- Sadhu, K.; Hensley, K.; Florio, V.A.; Wolda, S.L. Differential expression of the cyclic GMP-stimulated phosphodiesterase PDE2A in human venous and capillary endothelial cells. J. Histochem. Cytochem. 1999, 47, 895–905. [Google Scholar] [CrossRef] [Green Version]
- Aravind, L.; Ponting, C.P. The GAF domain: An evolutionary link between diverse phototransducing proteins. Trends Biochem. Sci. 1997, 22, 458–459. [Google Scholar] [CrossRef]
- Martinez, S.E.; Wu, A.Y.; Glavas, N.A.; Tang, X.B.; Turley, S.; Hol, W.G.; Beavo, J.A. The two GAF domains in phosphodiesterase 2A have distinct roles in dimerization and in cGMP binding. Proc. Natl. Acad. Sci. USA 2002, 99, 13260–13265. [Google Scholar] [CrossRef] [Green Version]
- Zaccolo, M.; Movsesian, M.A. cAMP and cGMP signaling cross-talk: Role of phosphodiesterases and implications for cardiac pathophysiology. Circ. Res. 2007, 100, 1569–1578. [Google Scholar] [CrossRef] [Green Version]
- Mika, D.; Bobin, P.; Pomérance, M.; Lechêne, P.; Westenbroek, R.E.; Catterall, W.A.; Vandecasteele, G.; Leroy, J.; Fischmeister, R. Differential regulation of cardiac excitation–contraction coupling by cAMP phosphodiesterase subtypes. Cardiovasc. Res. 2013, 100, 336–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soler, F.; Fernández-Belda, F.; Pérez-Schindler, J.; Handschin, C.; Fuente, T.; Hernandez-Cascales, J. PDE2 activity differs in right and left rat ventricular myocardium and differentially regulates β2 adrenoceptor-mediated effects. Exp. Biol. Med. 2014, 240, 1205–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méry, P.-F.; Pavoine, C.; Pecker, F.; Fischmeister, R. Erythro-9-(2-hydroxy-3-nonyl)adenine inhibits cyclic GMP-stimulated phosphodiesterase in isolated cardiac myocytes. Mol. Pharmacol. 1995, 48, 121–130. [Google Scholar]
- Mehel, H.; Emons, J.; Vettel, C.; Wittköpper, K.; Seppelt, D.; Dewenter, M.; Lutz, S.; Sossalla, S.; Maier, L.S.; Lechêne, P.; et al. Phosphodiesterase-2 Is Up-Regulated in Human Failing Hearts and Blunts β-Adrenergic Responses in Cardiomyocytes. J. Am. Coll. Cardiol. 2013, 62, 1596–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, H.; Froese, A.; Jönsson, P.; Schmidt, H.; Gorelik, J.; Nikolaev, V.O. Distinct submembrane localisation compartmentalises cardiac NPR1 and NPR2 signalling to cGMP. Nat. Commun. 2018, 9, 2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivet-Bastide, M.; Vandecasteele, G.; Hatem, S.; Verde, I.; Benardeau, A.; Mercadier, J.J.; Fischmeister, R. cGMP-stimulated cyclic nucleotide phosphodiesterase regulates the basal calcium current in human atrial myocytes. J. Clin. Investig. 1997, 99, 2710–2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittrich, M.; Jurevičius, J.; Georget, M.; Rochais, F.; Fleischmann, B.K.; Hescheler, J.; Fischmeister, R. Local response of L-type Ca2+current to nitric oxide in frog ventricular myocytes. J. Physiol. 2001, 534, 109–121. [Google Scholar] [CrossRef]
- Vandecasteele, G.; Verde, I.; Rücker-Martin, C.; Donzeau-Gouge, P.; Fischmeister, R. Cyclic GMP regulation of the L-type Ca2+channel current in human atrial myocytes. J. Physiol. 2001, 533, 329–340. [Google Scholar] [CrossRef]
- Stangherlin, A.; Gesellchen, F.; Zoccarato, A.; Terrin, A.; Fields, L.A.; Berrera, M.; Surdo, N.C.; Craig, M.A.; Smith, G.; Ham-ilton, G.; et al. cGMP signals modulate cAMP levels in a compartment-specific manner to regulate catecholamine-dependent signaling in cardiac myocytes. Circ. Res. 2011, 108, 929–939. [Google Scholar] [CrossRef] [Green Version]
- Fischmeister, R.; Hartzell, H.C. Cyclic guanosine 3′,5′-monophosphate regulates the calcium current in single cells from frog ventricle. J. Physiol. 1987, 387, 453–472. [Google Scholar] [CrossRef]
- Perera, R.K.; Sprenger, J.U.; Steinbrecher, J.H.; Hubscher, D.; Lehnart, S.E.; Abesser, M.; Schuh, K.; El-Armouche, A.; Nikolaev, V.O. Microdomain switch of cGMP-regulated phosphodiesterases leads to ANP-induced augmentation of β-adrenoceptor-stimulated contractility in early cardiac hypertrophy. Circ. Res. 2015, 116, 1304–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinhardt, R.R.; Chin, E.; Zhou, J.; Taira, M.; Murata, T.; Manganiello, V.C.; Bondy, C.A. Distinctive anatomical patterns of gene expression for cGMP-inhibited cyclic nucleotide phosphodiesterases. J. Clin. Investig. 1995, 95, 1528–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wechsler, J.; Choi, Y.H.; Krall, J.; Ahmad, F.; Manganiello, V.C.; Movsesian, M.A. Isoforms of cyclic nucleotide phosphodiesterase PDE3A in cardiac myocytes. J. Biol. Chem. 2002, 277, 38072–38078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hambleton, R.; Krall, J.; Tikishvili, E.; Honeggar, M.; Ahmad, F.; Manganiello, V.C.; Movsesian, M.A. Isoforms of Cyclic Nucleotide Phosphodiesterase PDE3 and Their Contribution to cAMP Hydrolytic Activity in Subcellular Fractions of Human Myocardium. J. Biol. Chem. 2005, 280, 39168–39174. [Google Scholar] [CrossRef] [Green Version]
- Rascon, A.; Lindgren, S.; Stavenow, L.; Belfrage, P.; Andersson, K.E.; Manganiello, V.C.; Degerman, E. Purification and properties of the cGMP-inhibited cAMP phosphodiesterase from bovine aortic smooth muscle. Biochim. Biophys. Acta 1992, 1134, 149–156. [Google Scholar] [CrossRef]
- Chung, Y.W.; Lagranha, C.; Chen, Y.; Sun, J.; Tong, G.; Hockman, S.C.; Ahmad, F.; Esfahani, S.G.; Bae, D.H.; Polidovitch, N.; et al. Targeted disruption of PDE3B, but not PDE3A, protects murine heart from ischemia/reperfusion injury. Proc. Natl. Acad. Sci. USA 2015, 112, E2253–E2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakur, Y.; Holst, L.S.; Landstrom, T.R.; Movsesian, M.; Degerman, E.; Manganiello, V. Regulation and function of the cyclic nucleotide phosphodiesterase (PDE3) gene family. Prog. Nucleic Acid Res. Mol. Biol. 2000, 66, 241–277. [Google Scholar] [CrossRef]
- Palmer, D.; Jimmo, S.L.; Raymond, D.R.; Wilson, L.S.; Carter, R.L.; Maurice, D.H. Protein Kinase A Phosphorylation of Human Phosphodiesterase 3B Promotes 14-3-3 Protein Binding and Inhibits Phosphatase-catalyzed Inactivation. J. Biol. Chem. 2007, 282, 9411–9419. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, F.; Shen, W.; Vandeput, F.; Szabo-Fresnais, N.; Krall, J.; Degerman, E.; Goetz, F.; Klussmann, E.; Movsesian, M.; Manganiello, V. Regulation of sarcoplasmic reticulum Ca2+ ATPase 2 (SERCA2) activity by phosphodiesterase 3A (PDE3A) in human myocardium: Phosphorylation-dependent interaction of PDE3A1 with SERCA2. J. Biol. Chem. 2015, 290, 6763–6776. [Google Scholar] [CrossRef] [Green Version]
- Weishaar, R.E.; Kobylarz-Singer, D.C.; Steffen, R.P.; Kaplan, H.R. Subclasses of cyclic AMP-specific phosphodiesterase in left ventricular muscle and their involvement in regulating myocardial contractility. Circ. Res. 1987, 61, 539–547. [Google Scholar] [CrossRef] [Green Version]
- Beca, S.; Ahmad, F.; Shen, W.; Liu, J.; Makary, S.; Polidovitch, N.; Sun, J.; Hockman, S.; Chung, Y.W.; Movsesian, M.; et al. Phosphodiesterase Type 3A Regulates Basal Myocardial Contractility Through Interacting with Sarcoplasmic Reticulum Calcium ATPase Type 2a Signaling Complexes in Mouse Heart. Circ. Res. 2013, 112, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Movsesian, M.A.; Smith, C.J.; Krall, J.; Bristow, M.R.; Manganiello, V.C. Sarcoplasmic reticulum-associated cyclic adenosine 5′-monophosphate phosphodiesterase activity in normal and failing human hearts. J. Clin. Investig. 1991, 88, 15–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christ, T.; Engel, A.; Ravens, U.; Kaumann, A.J. Cilostamide potentiates more the positive inotropic effects of (−)-adrenaline through β2-adrenoceptors than the effects of (−)-noradrenaline through β1-adrenoceptors in human atrial myocardium. Naunyn Schmiedebergs Arch. Exp. Pathol. Pharmakol. 2006, 374, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Eschenhagen, T. PDE4 in the human heart—Major player or little helper? Br. J. Pharmacol. 2013, 169, 524–527. [Google Scholar] [CrossRef]
- Jurevicius, J.; Skeberdis, V.A.; Fischmeister, R. Role of cyclic nucleotide phosphodiesterase isoforms in cAMP compartmentation following β2-adrenergic stimulation of ICa,L in frog ventricular myocytes. J. Physiol. 2003, 551, 239–252. [Google Scholar] [CrossRef]
- Levy, F.O. Cardiac PDEs and crosstalk between cAMP and cGMP signalling pathways in the regulation of contractility. Naunyn Schmiedebergs Arch. Exp. Pathol. Pharmakol. 2013, 386, 665–670. [Google Scholar] [CrossRef] [Green Version]
- Fujishige, K.; Kotera, J.; Michibata, H.; Yuasa, K.; Takebayashi, S.-I.; Okumura, K.; Omori, K. Cloning and Characterization of a Novel Human Phosphodiesterase That Hydrolyzes Both cAMP and cGMP (PDE10A). J. Biol. Chem. 1999, 274, 18438–18445. [Google Scholar] [CrossRef] [Green Version]
- Soderling, S.H.; Bayuga, S.J.; Beavo, J.A. Isolation and characterization of a dual-substrate phosphodiesterase gene family: PDE10A. Proc. Natl. Acad. Sci. USA 1999, 96, 7071–7076. [Google Scholar] [CrossRef] [Green Version]
- Loughney, K.; Snyder, P.; Uher, L.; Rosman, G.; Ferguson, K.; Florio, V. Isolation and characterization of PDE10A, a novel human 3′, 5′-cyclic nucleotide phosphodiesterase. Gene 1999, 234, 109–117. [Google Scholar] [CrossRef]
- McMurray, J.J.; Pfeffer, M.A. Heart failure. Lancet 2005, 365, 1877–1889. [Google Scholar] [CrossRef]
- Mudd, J.O.; Kass, D.A. Tackling heart failure in the twenty-first century. Nature 2008, 451, 919–928. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Celutkiene, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.J.; Engelhardt, S.; Eschenhagen, T. What is the role of β-adrenergic signaling in heart failure? Circ. Res. 2003, 93, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Holmes, S.J.; Espiner, E.A.; Richards, A.M.; Yandle, T.; Frampton, C. Renal, endocrine, and hemodynamic effects of human brain natriuretic peptide in normal man. J. Clin. Endocrinol. Metab. 1993, 76, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Wegener, J.W.; Nawrath, H.; Wolfsgruber, W.; Kühbandner, S.; Werner, C.; Hofmann, F.; Feil, R. cGMP-Dependent Protein Kinase I Mediates the Negative Inotropic Effect of cGMP in the Murine Myocardium. Circ. Res. 2002, 90, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, E.; Champion, H.C.; Li, M.; Belardi, D.; Ren, S.; Rodriguez, E.R.; Bedja, D.; Gabrielson, K.L.; Wang, Y.; Kass, D.A. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat. Med. 2005, 11, 214–222. [Google Scholar] [CrossRef]
- Kuhn, M. Function and Dysfunction of Mammalian Membrane Guanylyl Cyclase Receptors: Lessons from Genetic Mouse Models and Implications for Human Diseases. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelber, Germany, 2009; pp. 47–69. [Google Scholar] [CrossRef]
- Patrucco, E.; Domes, K.; Sbroggio, M.; Blaich, A.; Schlossmann, J.; Desch, M.; Rybalkin, S.D.; Beavo, J.A.; Lukowski, R.; Hofmann, F. Roles of cGMP-dependent protein kinase I (cGKI) and PDE5 in the regulation of Ang II-induced cardiac hypertrophy and fibrosis. Proc. Natl. Acad. Sci. USA 2014, 111, 12925–12929. [Google Scholar] [CrossRef] [Green Version]
- Michel, K.; Herwig, M.; Werner, F.; Spes, K.; Abeßer, M.; Schuh, K.; Dabral, S.; Mügge, A.; Baba, H.A.; Skryabin, B.V.; et al. C-type natriuretic peptide moderates titin-based cardiomyocyte stiffness. JCI Insight 2020, 5, e139910. [Google Scholar] [CrossRef]
- Baliga, R.S.; Preedy, M.E.J.; Dukinfield, M.S.; Chu, S.M.; Aubdool, A.A.; Bubb, K.J.; Moyes, A.J.; Tones, M.A.; Hobbs, A.J. Phosphodiesterase 2 inhibition preferentially promotes NO/guanylyl cyclase/cGMP signaling to reverse the development of heart failure. Proc. Natl. Acad. Sci. USA 2018, 115, E7428–E7437. [Google Scholar] [CrossRef] [Green Version]
- Ghofrani, H.A.; D’Armini, A.M.; Grimminger, F.; Hoeper, M.M.; Jansa, P.; Kim, N.H.; Mayer, E.; Simonneau, G.; Wilkins, M.R.; Fritsch, A.; et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N. Engl. J. Med. 2013, 369, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, P.W.; Pieske, B.; Anstrom, K.J.; Ezekowitz, J.; Hernandez, A.F.; Butler, J.; Lam, C.S.; Ponikowski, P.; Voors, A.A.; Jia, G.; et al. Vericiguat in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2020, 382, 1883–1893. [Google Scholar] [CrossRef] [PubMed]
- Mcmurray, J.J.V.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin–Neprilysin Inhibition versus Enalapril in Heart Failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurray, J.J.V.; Jackson, A.M.; Lam, C.S.P.; Redfield, M.M.; Anand, I.S.; Ge, J.; Lefkowitz, M.P.; Maggioni, A.P.; Martinez, F.; Packer, M.; et al. Effects of sacubitril-valsartan versus valsartan in women compared with men with heart failure and preserved ejection fraction: Insights from PARAGON-HF. Circulation 2020, 141, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Krüger, M.; Kötter, S.; Grützner, A.; Lang, P.; Andresen, C.; Redfield, M.M.; Butt, E.; Dos Remedios, C.G.; Linke, W.A. Protein Kinase G Modulates Human Myocardial Passive Stiffness by Phosphorylation of the Titin Springs. Circ. Res. 2009, 104, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Sollie, S.J.; Moltzau, L.R.; Hernandez-Valladares, M.; Berven, F.; Levy, F.O.; Andressen, K.W. C-type natriuretic peptide increases titin phosphorylation and decreases passive stiffness in rat cardiomyocytes; Abstracts from the 8th International Conference on cGMP Generators, Effectors and Therapeutic Implications. BMC Pharmacol. Toxicol. 2017, 18 (Suppl. S1), 64. [Google Scholar]
- Krüger, M.; Linke, W.A. Protein kinase-A phosphorylates titin in human heart muscle and reduces myofibrillar passive tension. J. Muscle Res. Cell Motil. 2006, 27, 435–444. [Google Scholar] [CrossRef]
- van Heerebeek, L.; Borbély, A.; Niessen, H.; Bronzwaer, J.G.; van der Velden, J.; Stienen, G.; Linke, W.; Laarman, G.J.; Paulus, W.J. Myocardial Structure and Function Differ in Systolic and Diastolic Heart Failure. Circulation 2006, 113, 1966–1973. [Google Scholar] [CrossRef] [Green Version]
- Borbély, A.; Falcão-Pires, I.; van Heerebeek, L.; Hamdani, N.; Édes, I.; Gavina, C.; Leite-Moreira, A.; Bronzwaer, J.G.; Papp, Z.; van der Velden, J.; et al. Hypophosphorylation of the Stiff N2B Titin Isoform Raises Cardiomyocyte Resting Tension in Failing Human Myocardium. Circ. Res. 2009, 104, 780–786. [Google Scholar] [CrossRef] [Green Version]
- Yanaka, N.; Kurosawa, Y.; Minami, K.; Kawai, E.; Omori, K. cGMP-phosphodiesterase activity is up-regulated in response to pressure overload of rat ventricles. Biosci. Biotechnol. Biochem. 2003, 67, 973–979. [Google Scholar] [CrossRef] [Green Version]
- Silver, P.J.; Allen, P.; Etzler, J.H.; Hamel, L.T.; Bentley, R.G.; Pagani, E.D. Cellular distribution and pharmacological sensitivity of low Km cyclic nucleotide phosphodiesterase isozymes in human cardiac muscle from normal and cardiomyopathic subjects. Second. Messengers Phosphoprot. 1990, 13, 13–25. [Google Scholar]
- Smith, C.J.; Huang, R.; Sun, D.; Ricketts, S.; Hoegler, C.; Ding, J.Z.; Moggio, R.A.; Hintze, T.H. Development of decompensated dilated cardiomyopathy is associated with decreased gene expression and activity of the milrinone-sensitive cAMP phosphodiesterase PDE3A. Circulation 1997, 96, 3116–3123. [Google Scholar] [CrossRef]
- Ding, B.; Abe, J.I.; Wei, H.; Huang, Q.; Walsh, R.A.; Molina, C.A.; Zhao, A.; Sadoshima, J.; Blaxall, B.C.; Berk, B.C.; et al. Functional role of phosphodiesterase 3 in cardiomyocyte apoptosis: Implication in heart failure. Circulation 2005, 111, 2469–2476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abi-Gerges, A.; Richter, W.; Lefebvre, F.; Mateo, P.; Varin, A.; Heymes, C.; Samuel, J.-L.; Lugnier, C.; Conti, M.; Fischmeister, R.; et al. Decreased Expression and Activity of cAMP Phosphodiesterases in Cardiac Hypertrophy and Its Impact on β-Adrenergic cAMP Signals. Circ. Res. 2009, 105, 784–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Osanai, T.; Nakano, T.; Wakui, M.; Okumura, K. Enhanced activities and gene expression of phosphodiesterase types 3 and 4 in pressure-induced congestive heart failure. Heart Vessel. 2002, 16, 249–256. [Google Scholar] [CrossRef]
- Holbrook, M.; Coker, S.J. Effects of zaprinast and rolipram on platelet aggregation and arrhythmias following myocardial ischaemia and reperfusion in anaesthetized rabbits. J. Cereb. Blood Flow Metab. 1991, 103, 1973–1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandenwijngaert, S.; Pokreisz, P.; Hermans, H.; Gillijns, H.; Pellens, M.; Bax, N.; Coppiello, G.; Oosterlinck, W.; Balogh, A.; Papp, Z.; et al. Increased Cardiac Myocyte PDE5 Levels in Human and Murine Pressure Overload Hypertrophy Contribute to Adverse LV Remodeling. PLoS ONE 2013, 8, e58841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senzaki, H.; Smith, C.J.; Juang, G.J.; Isoda, T.; Mayer, S.P.; Ohler, A.; Paolocci, N.; Tomaselli, G.F.; Hare, J.M.; Kass, D.A. Cardiac phosphodiesterase 5 (cGMP-specific) modulates β-adrenergic signaling in vivo and is down-regulated in heart failure. FASEB J. 2001, 15, 1718–1726. [Google Scholar] [CrossRef]
- Pokreisz, P.; Vandenwijngaert, S.; Bito, V.; Van Den Bergh, A.; Lenaerts, I.; Busch, C.; Marsboom, G.; Gheysens, O.; Vermeersch, P.; Biesmans, L.; et al. Ventricular phosphodiesterase-5 expression is increased in patients with advanced heart failure and contributes to adverse ventricular remodeling after myocardial infarction in mice. Circulation 2009, 119, 408–416. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Xu, X.; Hu, X.; Lee, S.; Traverse, J.H.; Zhu, G.; Fassett, J.; Tao, Y.; Zhang, P.; dos Remedios, C.; et al. Oxidative Stress Regulates Left Ventricular PDE5 Expression in the Failing Heart. Circulation 2010, 121, 1474–1483. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Takimoto, E.; Lee, D.-I.; Santos, C.X.; Nakamura, T.; Hsu, S.; Jiang, A.; Nagayama, T.; Bedja, D.; Yuan, Y.; et al. Pathological Cardiac Hypertrophy Alters Intracellular Targeting of Phosphodiesterase Type 5 From Nitric Oxide Synthase-3 to Natriuretic Peptide Signaling. Circulation 2012, 126, 942–951. [Google Scholar] [CrossRef]
- Vettel, C.; Lindner, M.; Dewenter, M.; Lorenz, K.; Schanbacher, C.; Riedel, M.; Lammle, S.; Meinecke, S.; Mason, F.E.; Sossalla, S.; et al. Phosphodiesterase 2 protects against catecholamine-induced arrhythmia and preserves contractile function after myocardial infarction. Circ. Res. 2017, 120, 120–132. [Google Scholar] [CrossRef]
- Vettel, C.; Lämmle, S.; Ewens, S.; Cervirgen, C.; Emons, J.; Ongherth, A.; Dewenter, M.; Lindner, D.; Westermann, D.; Nikolaev, V.O.; et al. PDE2-mediated cAMP hydrolysis accelerates cardiac fibroblast to myofibroblast conversion and is antagonized by exogenous activation of cGMP signaling pathways. Am. J. Physiol. Circ. Physiol. 2014, 306, H1246–H1252. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Aroonsakool, N.; Yokoyama, U.; Patel, H.H.; Insel, P.A. Increase in cellular cyclic AMP concentrations reverses the profibrogenic phenotype of cardiac myofibroblasts: A novel therapeutic approach for cardiac fibrosis. Mol. Pharmacol. 2013, 84, 787–793. [Google Scholar] [CrossRef] [Green Version]
- Sassi, Y.; Ahles, A.; Truong, D.-J.J.; Baqi, Y.; Lee, S.-Y.; Husse, B.; Hulot, J.-S.; Foinquinos, A.; Thum, T.; Müller, C.E.; et al. Cardiac myocyte–secreted cAMP exerts paracrine action via adenosine receptor activation. J. Clin. Investig. 2014, 124, 5385–5397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koser, F.; Loescher, C.; Linke, W.A. Posttranslational modifications of titin from cardiac muscle: How, where, and what for? FEBS J. 2019, 286, 2240–2260. [Google Scholar] [CrossRef]
- Bubb, K.J.; Trinder, S.L.; Baliga, R.S.; Patel, J.; Clapp, L.H.; MacAllister, R.J.; Hobbs, A.J. Inhibition of Phosphodiesterase 2 Augments cGMP and cAMP Signaling to Ameliorate Pulmonary Hypertension. Circulation 2014, 130, 496–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoccarato, A.; Surdo, N.C.; Aronsen, J.M.; Fields, L.A.; Mancuso, L.; Dodoni, G.; Stangherlin, A.; Livie, C.; Jiang, H.; Sin, Y.Y.; et al. Cardiac Hypertrophy Is Inhibited by a Local Pool of cAMP Regulated by Phosphodiesterase Circ. Res. 2015, 117, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Dibianco, R.; Shabetai, R.; Kostuk, W.; Moran, J.; Schlant, R.C.; Wright, R. A Comparison of Oral Milrinone, Digoxin, and Their Combination in the Treatment of Patients with Chronic Heart Failure. N. Engl. J. Med. 1989, 320, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Cuffe, M.S.; Califf, R.M.; Adams Jr, K.F.; Benza, R.; Bourge, R.; Colucci, W.S.; Massie, B.M.; O’Connor, C.M.; Pina, I.; Quigg, R. Short-term intravenous milrinone for acute exacerbation of chronic heart failure: A randomized controlled trial. JAMA 2002, 287, 1541–1547. [Google Scholar] [CrossRef] [Green Version]
- Bristow, M.R.; Ginsburg, R.; Minobe, W.; Cubicciotti, R.S.; Sageman, W.S.; Lurie, K.; Billingham, M.E.; Harrison, D.C.; Stinson, E.B. Decreased Catecholamine Sensitivity and β-Adrenergic-Receptor Density in Failing Human Hearts. N. Engl. J. Med. 1982, 307, 205–211. [Google Scholar] [CrossRef]
- Amsallem, E.; Kasparian, C.; Haddour, G.; Boissel, J.-P.; Nony, P. Phosphodiesterase III inhibitors for heart failure. Cochrane Database Syst. Rev. 2005. [Google Scholar] [CrossRef] [PubMed]
- Nanayakkara, S.; Byrne, M.; Mak, V.; Carter, K.; Dean, E.; Kaye, D.M. Extended-Release Oral Milrinone for the Treatment of Heart Failure with Preserved Ejection Fraction. J. Am. Heart Assoc. 2020, 9, e015026. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Li, H.; Shakur, Y.; Hensley, J.; Hockman, S.; Kambayashi, J.; Manganiello, V.C.; Liu, Y. Role of phosphodiesterase type 3A and 3B in regulating platelet and cardiac function using subtype-selective knockout mice. Cell. Signal. 2007, 19, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.; Katugampola, S.; Able, S.; Napier, C.; Harding, S. Profiling of cAMP and cGMP phosphodiesterases in isolated ventricular cardiomyocytes from human hearts: Comparison with rat and guinea pig. Life Sci. 2012, 90, 328–336. [Google Scholar] [CrossRef]
- Karam, S.; Margaria, J.P.; Bourcier, A.; Mika, D.; Varin, A.; Bedioune, I.; Lindner, M.; Bouadjel, K.; Dessillons, M.; Gaudin, F.; et al. Cardiac Overexpression of PDE4B Blunts β-Adrenergic Response and Maladaptive Remodeling in Heart Failure. Circulation 2020, 142, 161–174. [Google Scholar] [CrossRef]
- Nagendran, J.; Archer, S.L.; Soliman, D.; Gurtu, V.; Moudgil, R.; Haromy, A.; Aubin, C.S.; Webster, L.; Rebeyka, I.M.; Ross, D.B.; et al. Phosphodiesterase Type 5 Is Highly Expressed in the Hypertrophied Human Right Ventricle, and Acute Inhibition of Phosphodiesterase Type 5 Improves Contractility. Circulation 2007, 116, 238–248. [Google Scholar] [CrossRef]
- Shan, X.; Quaile, M.P.; Monk, J.K.; French, B.; Cappola, T.P.; Margulies, K.B. Differential Expression of PDE5 in Failing and Nonfailing Human Myocardium. Circ. Heart Fail. 2012, 5, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Garcia, A.M.; Nakano, S.J.; Karimpour-Fard, A.; Nunley, K.; Blain-Nelson, P.; Stafford, N.M.; Stauffer, B.L.; Sucharov, C.C.; Miyamoto, S.D. Phosphodiesterase-5 is elevated in failing single ventricle myocardium and affects cardiomyocyte remodeling in vitro. Circ. Heart Fail. 2018, 11, e004571. [Google Scholar] [CrossRef]
- Zhang, M.; Koitabashi, N.; Nagayama, T.; Rambaran, R.; Feng, N.; Takimoto, E.; Koenke, T.; O’Rourke, B.; Champion, H.C.; Crow, M.T.; et al. Expression, activity, and pro-hypertrophic effects of PDE5A in cardiac myocytes. Cell. Signal. 2008, 20, 2231–2236. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-K.; Chen, B.-Y.; Guo, A.; Chen, R.; Zhu, Y.-Q.; Kutschke, W.; Hong, J.; Song, L.-S. Sildenafil ameliorates left ventricular T-tubule remodeling in a pressure overload-induced murine heart failure model. Acta Pharmacol. Sin. 2016, 37, 473–482. [Google Scholar] [CrossRef] [Green Version]
- Lawless, M.; Caldwell, J.L.; Radcliffe, E.J.; Smith, C.E.R.; Madders, G.W.P.; Hutchings, D.C.; Woods, L.S.; Church, S.J.; Unwin, R.D.; Kirkwood, G.J.; et al. Phosphodiesterase 5 inhibition improves contractile function and restores transverse tubule loss and catecholamine responsiveness in heart failure. Sci. Rep. 2019, 9, 6801. [Google Scholar] [CrossRef] [Green Version]
- Lewis, G.D.; Shah, R.; Shahzad, K.; Camuso, J.M.; Pappagianopoulos, P.P.; Hung, J.; Tawakol, A.; Gerszten, R.E.; Systrom, D.; Bloch, K.D.; et al. Sildenafil Improves Exercise Capacity and Quality of Life in Patients With Systolic Heart Failure and Secondary Pulmonary Hypertension. Circulation 2007, 116, 1555–1562. [Google Scholar] [CrossRef] [Green Version]
- Guazzi, M.; Vicenzi, M.; Arena, R.; Guazzi, M.D. Pulmonary hypertension in heart failure with preserved ejection fraction: A target of phosphodiesterase-5 inhibition in a 1-year study. Circulation 2011, 124, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Redfield, M.M.; Chen, H.H.; Borlaug, B.A.; Semigran, M.J.; Lee, K.L.; Lewis, G.; Lewinter, M.M.; Rouleau, J.L.; Bull, D.A.; Mann, D.L.; et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: A randomized clinical trial. JAMA 2013, 309, 1268–1277. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.-D.; Long, M.; Li, F.; Hu, X.; Liao, X.-X.; Du, Z.-M. PDE5 inhibitor sildenafil in the treatment of heart failure: A meta-analysis of randomized controlled trials. Int. J. Cardiol. 2014, 172, 581–587. [Google Scholar] [CrossRef] [PubMed]
- De Vecchis, R.; Cesaro, A.; Ariano, C.; Giasi, A.; Cioppa, C. Phosphodiesterase-5 Inhibitors Improve Clinical Outcomes, Exercise Capacity and Pulmonary Hemodynamics in Patients with Heart Failure with Reduced Left Ventricular Ejection Fraction: A Meta-Analysis. J. Clin. Med. Res. 2017, 9, 488–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunkerly-Eyring, B.; Kass, D.A. Myocardial Phosphodiesterases and Their Role in cGMP Regulation. J. Cardiovasc. Pharmacol. 2020, 75, 483–493. [Google Scholar] [CrossRef]
- Shah, S.J.; Kitzman, D.W.; Borlaug, B.A.; Van Heerebeek, L.; Zile, M.R.; Kass, D.A.; Paulus, W.J. Phenotype-specific treatment of heart failure with preserved ejection fraction: A multiorgan roadmap. Circulation 2016, 134, 73–90. [Google Scholar] [CrossRef] [Green Version]
- Richards, D.A.; Aronovitz, M.J.; Liu, P.; Martin, G.L.; Tam, K.; Pande, S.; Karas, R.H.; Bloomfield, D.M.; Mendelsohn, M.E.; Blanton, R.M. CRD-733, a novel PDE9 (phosphodiesterase 9) inhibitor, reverses pressure overload-induced heart failure. Circ. Heart Fail. 2021, 14, e007300. [Google Scholar] [CrossRef]
- Zaccolo, M.; Zerio, A.; Lobo, M.J. Subcellular Organization of the cAMP Signaling Pathway. Pharmacol. Rev. 2020, 73, 278–309. [Google Scholar] [CrossRef]
- Koschinski, A.; Zaccolo, M. Activation of PKA in cell requires higher concentration of cAMP than in vitro: Implications for compartmentalization of cAMP signalling. Sci. Rep. 2017, 7, 14090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Øgreid, D.; Døskeland, S.O. Activation of protein kinase isoenzymes under near physiological conditions. Evidence that both types (A and B) of cAMP binding sites are involved in the activation of protein kinase by cAMP and 8-N3-cAMP. FEBS Lett. 1982, 150, 161–166. [Google Scholar] [CrossRef] [Green Version]
- Adams, S.R.; Harootunian, A.T.; Buechler, Y.J.; Taylor, S.S.; Tsien, R.Y. Fluorescence ratio imaging of cyclic AMP in single cells. Nature 1991, 349, 694–697. [Google Scholar] [CrossRef] [PubMed]
- Iancu, R.V.; Ramamurthy, G.; Warrier, S.; Nikolaev, V.O.; Lohse, M.J.; Jones, S.W.; Harvey, R.D. Cytoplasmic cAMP concentrations in intact cardiac myocytes. Am. J. Physiol. Physiol. 2008, 295, C414–C422. [Google Scholar] [CrossRef]
- Nikolaev, V.O.; Bünemann, M.; Hein, L.; Hannawacker, A.; Lohse, M.J. Novel single chain cAMP sensors for receptor-induced signal propagation. J. Biol. Chem. 2004, 279, 37215–37218. [Google Scholar] [CrossRef] [Green Version]
- Richards, M.; Lomas, O.; Jalink, K.; Ford, K.L.; Vaughan-Jones, R.D.; Lefkimmiatis, K.; Swietach, P. Intracellular tortuosity underlies slow cAMP diffusion in adult ventricular myocytes. Cardiovasc. Res. 2016, 110, 395–407. [Google Scholar] [CrossRef] [Green Version]
- McCabe, K.J.; Rangamani, P. Computational modeling approaches to cAMP/PKA signaling in cardiomyocytes. J. Mol. Cell. Cardiol. 2021, 154, 32–40. [Google Scholar] [CrossRef]
- Bock, A.; Annibale, P.; Konrad, C.; Hannawacker, A.; Anton, S.E.; Maiellaro, I.; Zabel, U.; Sivaramakrishnan, S.; Falcke, M.; Lohse, M.J. Optical Mapping of cAMP Signaling at the Nanometer Scale. Cell 2020, 182, 1519–1530.e17. [Google Scholar] [CrossRef]
- Zhang, J.Z.; Lu, T.-W.; Stolerman, L.M.; Tenner, B.; Yang, J.R.; Zhang, J.-F.; Falcke, M.; Rangamani, P.; Taylor, S.S.; Mehta, S.; et al. Phase Separation of a PKA Regulatory Subunit Controls cAMP Compartmentation and Oncogenic Signaling. Cell 2020, 182, 1531–1544.e15. [Google Scholar] [CrossRef]
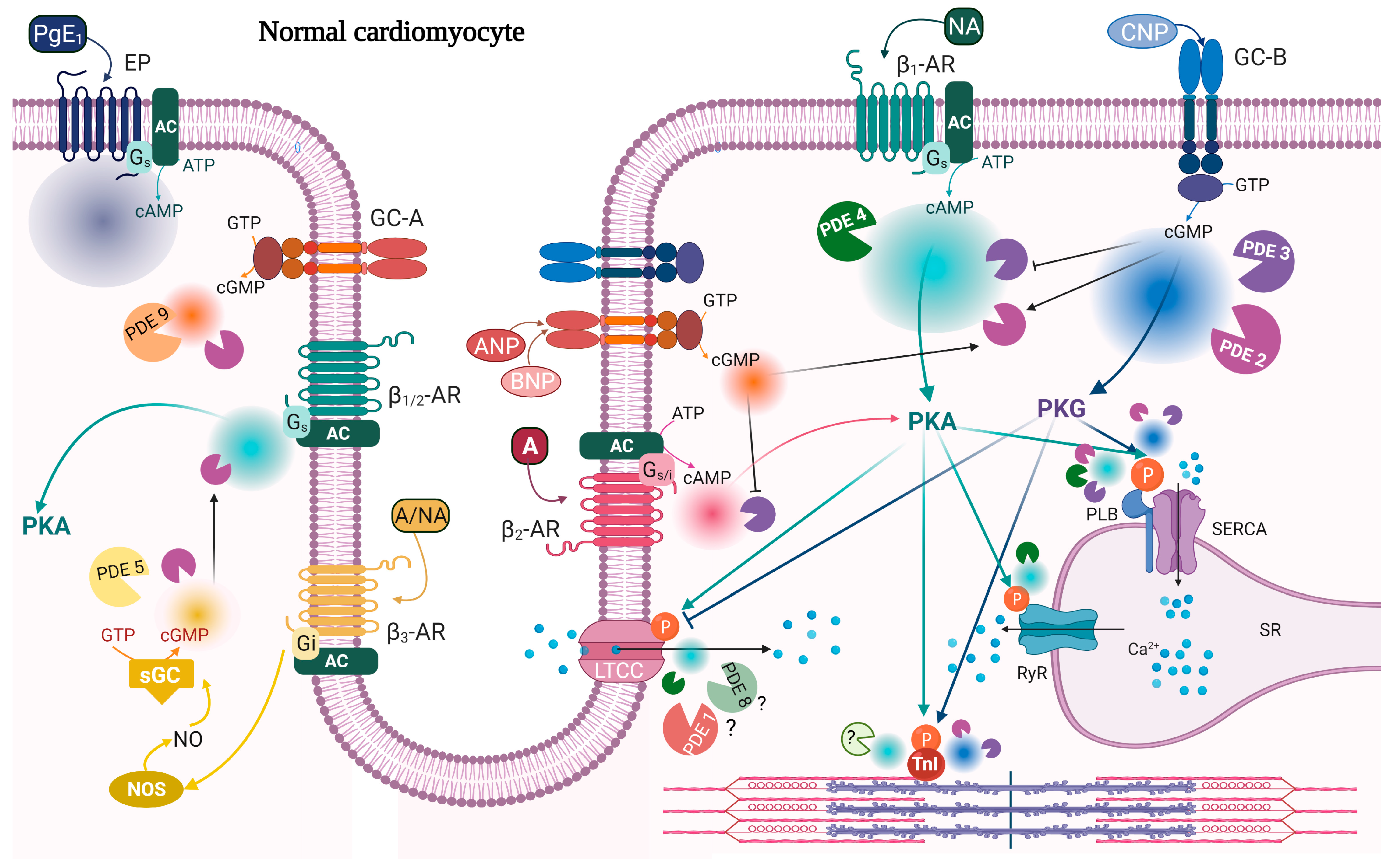
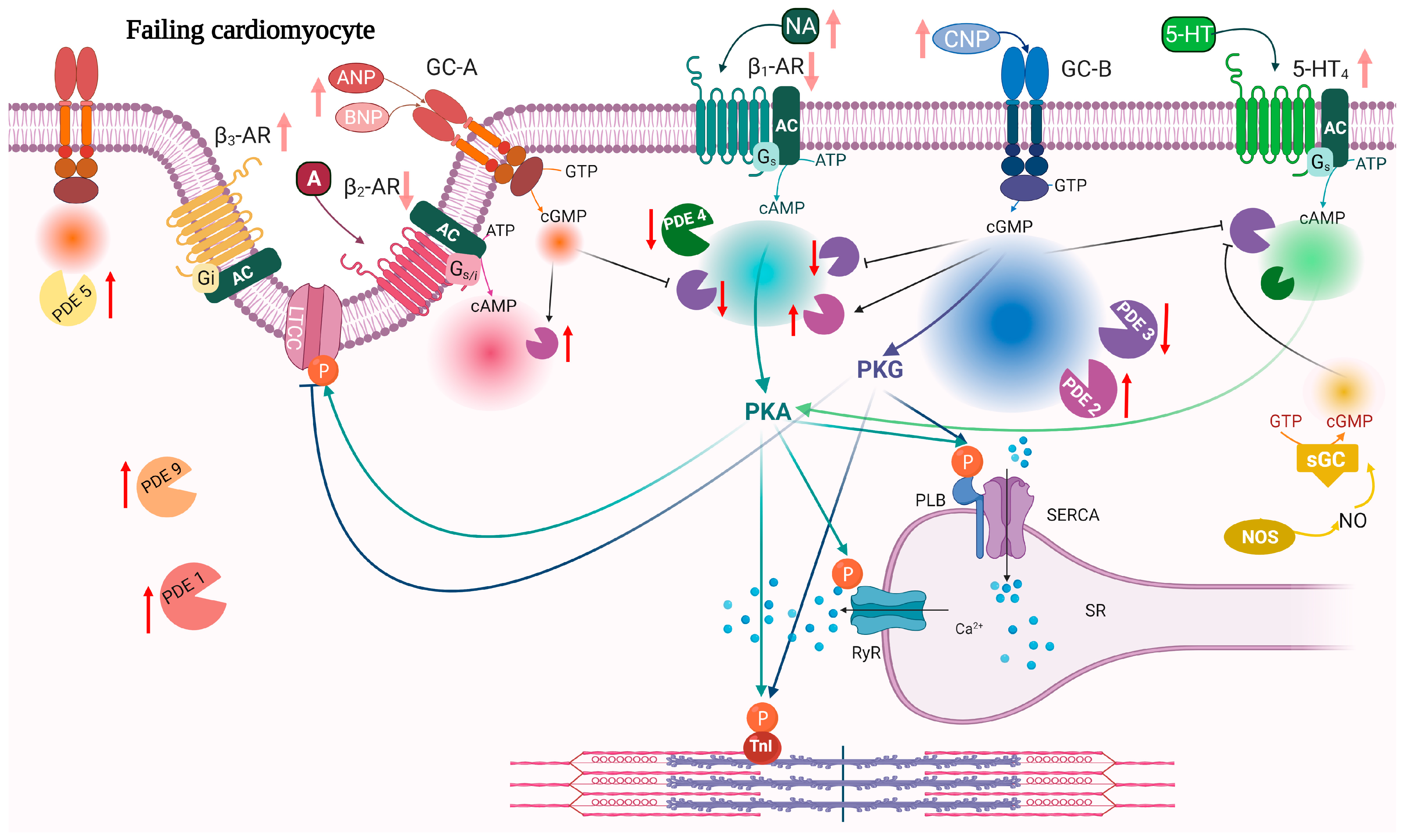

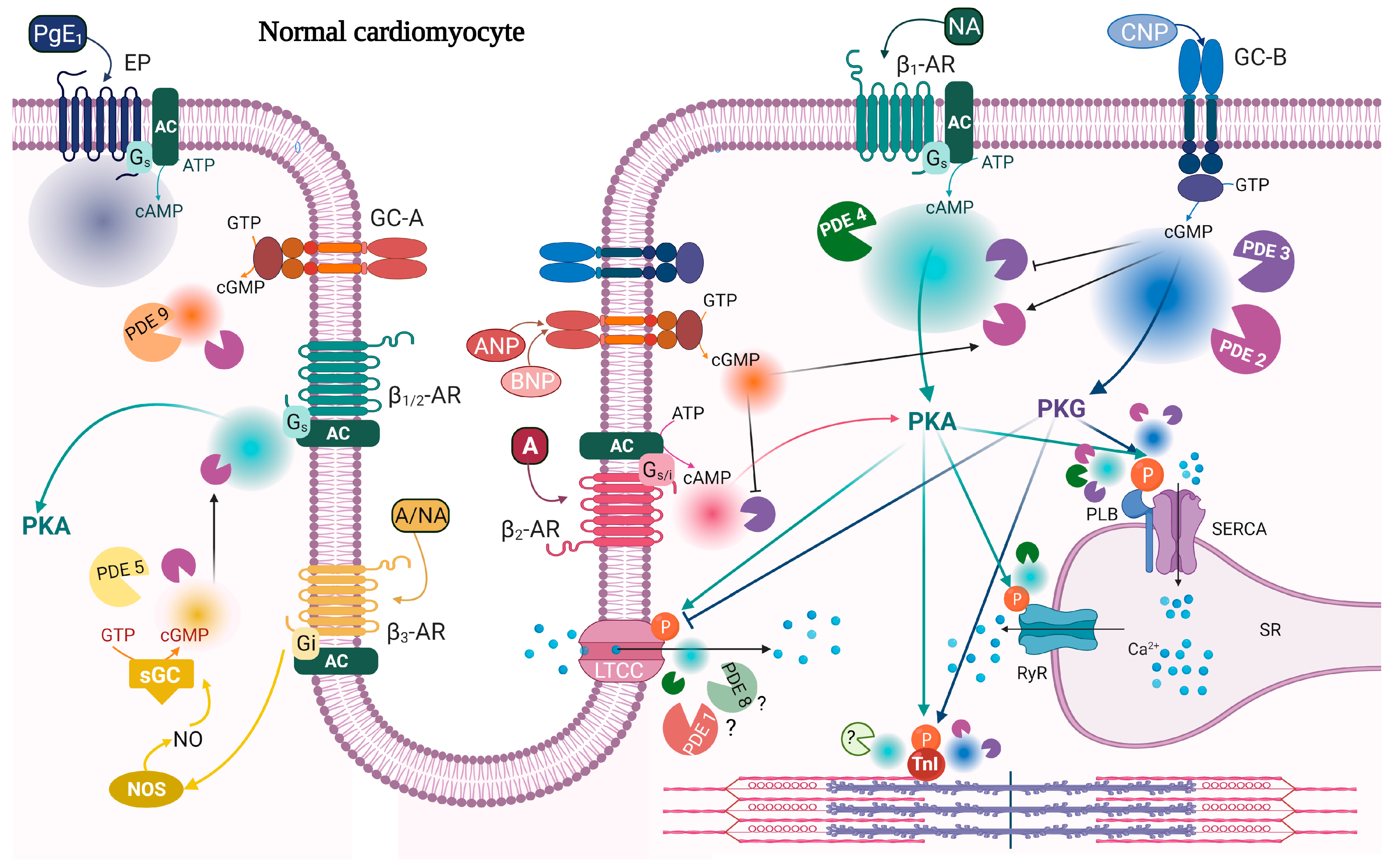{kind=link}
{kind=link}
| PDE | Expression | Activity | Localization | References |
|---|---|---|---|---|
| PDE2 | ↑ | ↑ | ↑ activity around β2-AR and PLB↓activity around β1-AR | [134,141,180] |
| PDE3 | ↓ ↑ | ↓ ↑ | ↑ activity around β1-AR | [141,181,182,183,184,185] |
| PDE4 | ↓ | ↓ | ↓PDE4D activity around RyR | [40,63,76,79,183,186] |
| PDE5 | ↑ | ↑ | From Z-line localization to diffuse localization | [187,188,189,190,191] |
| PDE9 | ↑ | ↑ | [105] | |
| PDE10 | ↑ | ↑ | [3] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calamera, G.; Moltzau, L.R.; Levy, F.O.; Andressen, K.W. Phosphodiesterases and Compartmentation of cAMP and cGMP Signaling in Regulation of Cardiac Contractility in Normal and Failing Hearts. Int. J. Mol. Sci. 2022, 23, 2145. https://doi.org/10.3390/ijms23042145
Calamera G, Moltzau LR, Levy FO, Andressen KW. Phosphodiesterases and Compartmentation of cAMP and cGMP Signaling in Regulation of Cardiac Contractility in Normal and Failing Hearts. International Journal of Molecular Sciences. 2022; 23(4):2145. https://doi.org/10.3390/ijms23042145
Chicago/Turabian StyleCalamera, Gaia, Lise Román Moltzau, Finn Olav Levy, and Kjetil Wessel Andressen. 2022. "Phosphodiesterases and Compartmentation of cAMP and cGMP Signaling in Regulation of Cardiac Contractility in Normal and Failing Hearts" International Journal of Molecular Sciences 23, no. 4: 2145. https://doi.org/10.3390/ijms23042145
APA StyleCalamera, G., Moltzau, L. R., Levy, F. O., & Andressen, K. W. (2022). Phosphodiesterases and Compartmentation of cAMP and cGMP Signaling in Regulation of Cardiac Contractility in Normal and Failing Hearts. International Journal of Molecular Sciences, 23(4), 2145. https://doi.org/10.3390/ijms23042145