
Analysis of the Neutralizing Activity of Antibodies Targeting Open or Closed SARS-CoV-2 Spike Protein Conformations


Abstract
:1. Introduction
2. Methods
2.1. Dataset Construction
2.2. Identification of Spike Protein Epitopes and ACE2 Binding Residues
2.3. Estimation of Steric Clashes of nAbs with the Open/Closed Spike Protein Conformations
2.4. Prediction of nAb-spike Protein Binding Affinity Changes and Escape Fractions
2.5. Measured Escape Map of Human Polyclonal Plasma
3. Results
3.1. Epitopes in the Dataset
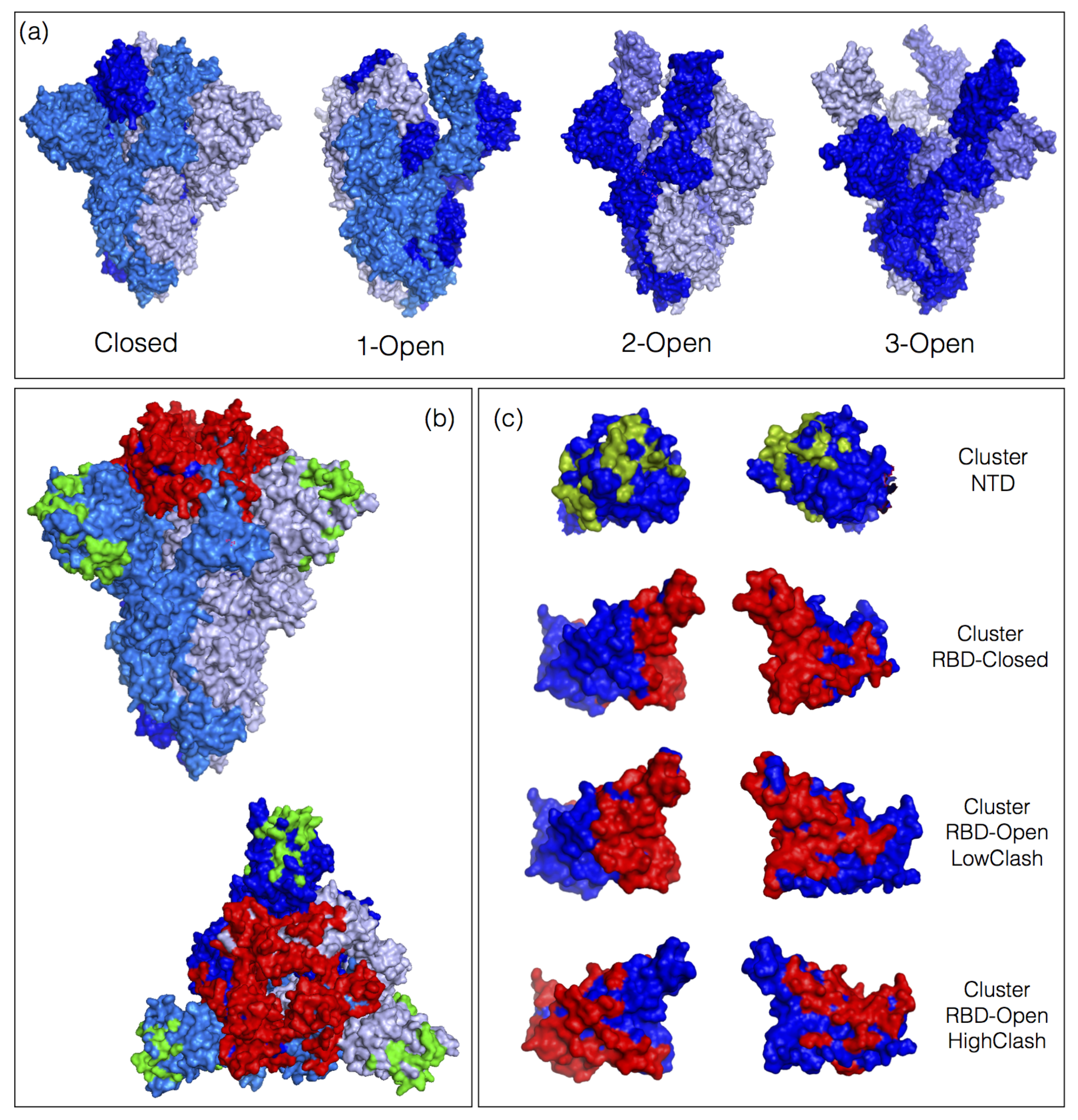
3.2. Classes of nAbs Based on their Ability to Bind Open or Closed Spike Protein Conformations
- The NTD class contains nAbs that bind to the NTD. The clash score is in this case zero as this domain is always fully accessible, both in the closed, 1-open and 3-open conformations. Some nAb-NTD complexes of this group are not affected by the considered NTD variants, while other complexes are strongly destabilized, for example by the variants G142D, E156G, Y145H or D253G.
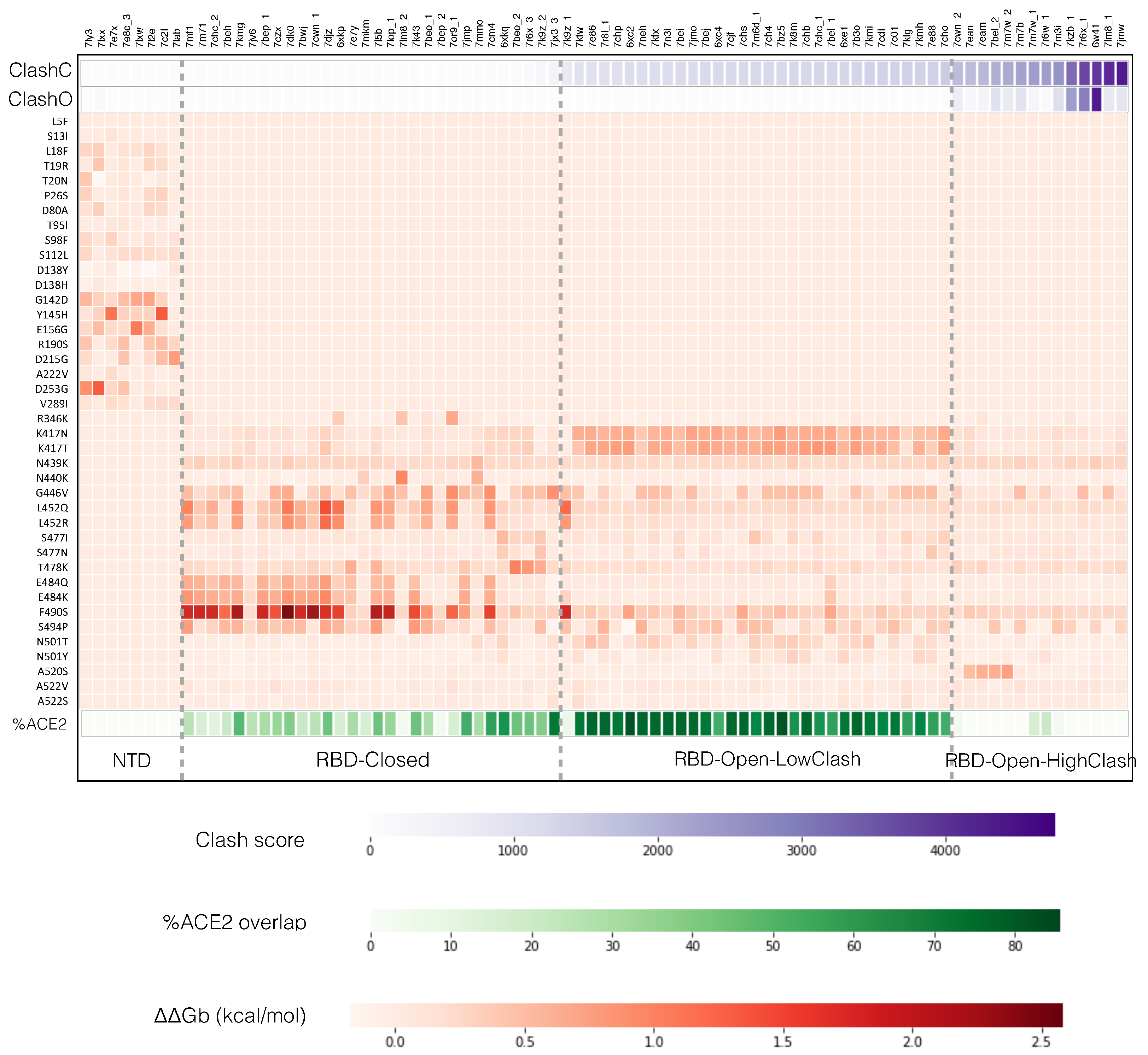
- The RBD-Closed class corresponds to nAbs that can bind the RBD in its closed conformation, given their low clash score with the full trimeric spike protein in closed conformation. Note that all these nAbs can also bind the open conformation. RBD variants that abolish the neutralizing activity of this class are mainly E484K, E484Q, F490S, L452R, L452Q and G446V.
- The RBD-Open-LowClash corresponds to nAbs that cannot bind the closed spike protein conformation but that do bind the open conformation, as shown by their intermediary clash score (500–2000) with the closed conformation and low clash score with the 1-open conformation. The escape pattern of this class strongly differs from that of the previous class: the neutralizing activity is reduced by RBD mutations such as K417N, K417T and G446V.
- The RBD-Open-HighClash contains nAbs that seem able to bind neither the closed nor the 1-open spike protein conformation, as demonstrated by their high clash score with both conformations. They can bind only the 2-open or 3-open conformations. Again, this class has a very different escape pattern, with little changes in neutralizing activity for the considered variants.
3.3. Escaping Mechanisms and Key Residue Interactions
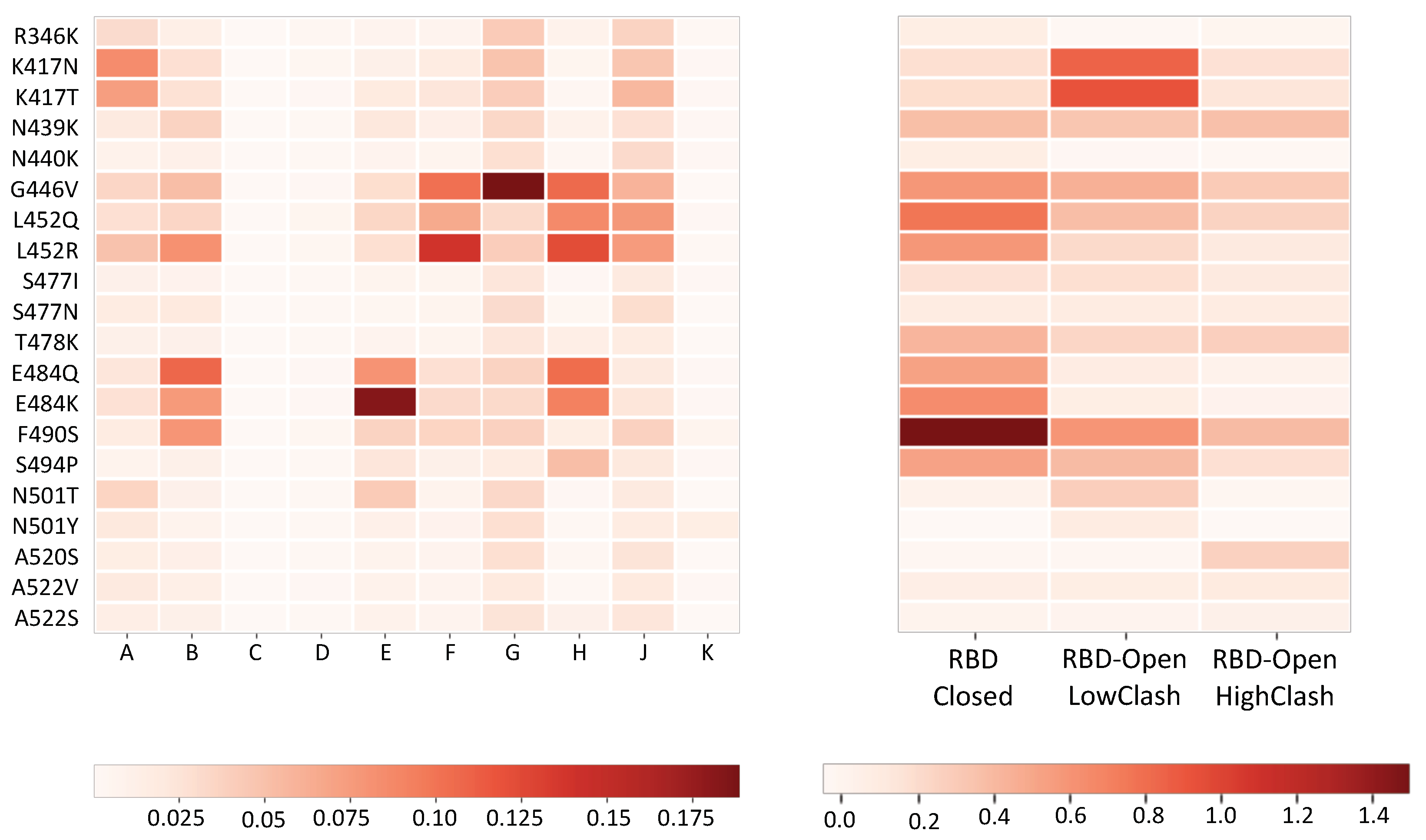
- Aromatic-involving interactions play a major role in nAb-spike protein binding, which is a general trend in antigen-antibody interfaces [19].
- Each class of nAbs targets different patterns of spike protein residues. However, there is a partial overlap between residues targeted by the RBD-Closed and RBD-Open-LowClash nAb classes, which results from the overlap between the two epitope regions, visible in Figure 1.
- Residues that are important for the binding of nAbs from the RBD-Closed class, such as E484, F490 and L452, are often mutated in the circulating strains (Figure 2). This indicates that the RBD-Closed epitope region is under strong selective pressure. The selective pressure on the RBD-Open-LowClash epitope region seems less strong as only K417 is found mutated in circulating strains.
- Residues that are important for spike protein binding to ACE2 (Table S2 and Figure S2) are bound by nAbs of both the RBD-Closed and RBD-Open-LowClash classes despite the fact that the epitope region from the latter overlaps more with the ACE2 binding region (see next section).
3.4. Contributions of the nAb Classes to the Immune Response

{kind=link}
{kind=link}
{kind=link}
| NTD | RBD-Closed | RBD-LowClash | RBD-HighClash | |
|---|---|---|---|---|
| in closed state (N) | 9 | 58 | 1412 | 3080 |
| in 1-open state (N) | 23 | 1 | 0 | 947 |
| in 3-open state (N) | 35 | 30 | 45 | 319 |
| (%) | 0.0 | 36.0 | 72.8 | 3.7 |
| -escape fraction COVID-19) | - | 0.64 | 0.24 | 0.15 |
| p-value | 0.003 | 0.32 | 0.52 | |
| -escape fraction mRNA-1273) | - | 0.20 | 0.59 | 0.18 |
| p-value | 0.39 | 0.005 | 0.45 |
3.5. Circulating Strains, nAb Classes and Immune Evasion
4. Discussion
- The spike protein evolution must be analyzed in terms of classes of nAbs that recognize different epitopes [14], but also different conformations of the spike protein.
- It must be noted that nAb-RBD crystal structures and their in vitro binding affinity only give partial indications of the neutralizing activity of nAbs. Indeed, they do not account for the different conformations of the spike protein trimer [37].
- Our results show that nAbs recognizing the RBD in the closed state of the spike protein trimer play a pivotal role in shaping the immune response and are thus important for protection (see also [14,30]). This is in agreement with experimental results suggesting that, in the state preceding ACE2 binding and entrance of the virus into the cells, the spike protein trimer spends the majority of the time in closed conformation [38,39], at least when attached to the virus. This suggests to design highly stable closed spike protein trimers as improved vaccine immunogens [30,39,40].
- The contribution of the different classes of nAbs to the neutralizing activity of polyclonal plasma is challenging to analyze. Indeed, it is highly variable among COVID-19 patients, as shown in Figure 3 and Figure S1 of Supplementary Material, and moreover, it evolves over time [26]. For example, even though the immune response of the majority of infected patients appears to be driven by the RBD-Closed nAb class, for a few of them, it is driven by the RBD-Open-LowClash class.
- We found differences in the dominant nAb classes in SARS-CoV-2 infected patients and vaccinated healthy individuals (Table 2 and Figure S1): the RBD-Closed class for the former and the RBD-Open-LowClash class for the latter. This result could be due to the fact that these two classes are elicited by different spike protein conformations, and that the most frequent conformation adopted by the spike protein could differ according to whether it is administered as a vaccine immunogen or attached to the viral membrane. Indeed, although the spike protein trimer in the viral envelope is known to spend most of the time in closed conformation [38,39], the free spike protein could possibly prefer open conformations.
- The analysis of how the contribution of the different classes of nAbs to the immune response has changed as a result of recently circulating viral strains [1], such as the B.1.617.2 (Delta) or B.1.1.529 (Omicron) strains, is very instructive. Indeed, mutations affecting the affinity of immunodominant nAbs can lead to an enhancement of subdominant nAb contributions and to a complex interplay between different nAbs classes.
- The fact that natural infection and mRNA vaccine elicit a different class of nAb could explain why "hydrid" immunization obtained via the combined action of vaccine injection and natural infection elicits a stronger and more robust immune response [41].
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Acronyms
| PDB | Protein Data Bank |
| nAbs | neutralizing antibodies |
| RBD | receptor binding domain |
| NTD | N-terminal domain |
| ACE2 | angiotensin-converting enzyme 2 |
| RSA | relative solvent accessibility |
| ΔΔGb | change in binding affinity upon mutation |
| RMSD | root mean square deviation |
References
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Wintjens, R.; Bifani, A.M.; Bifani, P. Impact of glycan cloud on the B-cell epitope prediction of SARS-CoV-2 Spike protein. NPJ Vaccines 2020, 5, 81. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Gao, G.F. Viral targets for vaccines against COVID-19. Nat. Rev. Immunol. 2021, 21, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Gaebler, C.; Wang, Z.; Lorenzi, J.C.; Muecksch, F.; Finkin, S.; Tokuyama, M.; Cho, A.; Jankovic, M.; Schaefer-Babajew, D.; Oliveira, T.Y.; et al. Evolution of antibody immunity to SARS-CoV-2. Nature 2021, 591, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Post, N.; Eddy, D.; Huntley, C.; van Schalkwyk, M.C.; Shrotri, M.; Leeman, D.; Rigby, S.; Williams, S.V.; Bermingham, W.H.; Kellam, P.; et al. Antibody response to SARS-CoV-2 infection in humans: A systematic review. PLoS ONE 2020, 15, e0244126. [Google Scholar] [CrossRef] [PubMed]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.M.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Robbiani, D.F.; Gaebler, C.; Muecksch, F.; Lorenzi, J.C.; Wang, Z.; Cho, A.; Agudelo, M.; Barnes, C.O.; Gazumyan, A.; Finkin, S.; et al. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature 2020, 584, 437–442. [Google Scholar] [CrossRef]
- McCallum, M.; De Marco, A.; Lempp, F.A.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.; Zatta, F.; et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell 2021, 184, 2332–2347. [Google Scholar] [CrossRef]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. [Google Scholar] [CrossRef]
- Henderson, R.; Edwards, R.J.; Mansouri, K.; Janowska, K.; Stalls, V.; Gobeil, S.M.; Kopp, M.; Li, D.; Parks, R.; Hsu, A.L.; et al. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 2020, 27, 925–933. [Google Scholar] [CrossRef]
- Focosi, D.; Maggi, F.; Franchini, M.; McConnell, S.; Casadevall, A. Analysis of Immune Escape Variants from Antibody-Based Therapeutics against COVID-19: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 29. [Google Scholar] [CrossRef] [PubMed]
- Pucci, F.; Rooman, M. Prediction and Evolution of the Molecular Fitness of SARS-CoV-2 Variants: Introducing SpikePro. Viruses 2021, 13, 935. [Google Scholar] [CrossRef] [PubMed]
- Hastie, K.M.; Li, H.; Bedinger, D.; Schendel, S.L.; Dennison, S.M.; Li, K.; Rayaprolu, V.; Yu, X.; Mann, C.; Zandonatti, M.; et al. Defining variant-resistant epitopes targeted by SARS-CoV-2 antibodies: A global consortium study. Science 2021, 374, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Starr, T.N.; Barnes, C.O.; Weisblum, Y.; Schmidt, F.; Caskey, M.; Gaebler, C.; Cho, A.; Agudelo, M.; Finkin, S.; et al. Mapping mutations to the SARS-CoV-2 RBD that escape binding by different classes of antibodies. Nat. Commun. 2021, 12, 4196. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Uchil, P.D.; Li, W.; Terry, D.S.; Gorman, J.; Zhang, B.; Zhou, T.; Ding, S.; Liu, L.; Ho, D.D.; et al. Real-Time Conformational Dynamics of SARS-CoV-2 Spikes on Virus Particles. Biophys. J. 2021, 120, 276a. [Google Scholar] [CrossRef]
- Raybould, M.I.; Kovaltsuk, A.; Marks, C.; Deane, C.M. CoV-AbDab: The coronavirus antibody database. Bioinformatics 2021, 37, 734–735. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Godzik, A. CD-HIT: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Dalkas, G.A.; Teheux, F.; Kwasigroch, J.M.; Rooman, M. Cation–π, amino–π, π–π, and H-bond interactions stabilize antigen–antibody interfaces. Proteins Struct. Funct. Bioinform. 2014, 82, 1734–1746. [Google Scholar] [CrossRef] [Green Version]
- Lv, Z.; Deng, Y.Q.; Ye, Q.; Cao, L.; Sun, C.Y.; Fan, C.; Huang, W.; Sun, S.; Sun, Y.; Zhu, L.; et al. Structural basis for neutralization of SARS-CoV-2 and SARS-CoV by a potent therapeutic antibody. Science 2020, 369, 1505–1509. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 183, 1735. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, Y.; Kwasigroch, J.M.; Rooman, M.; Gilis, D. BeAtMuSiC: Prediction of changes in protein–protein binding affinity on mutations. Nucleic Acids Res. 2013, 41, W333–W339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data–from vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 2021, 29, 463–476. [Google Scholar] [CrossRef]
- Greaney, A.J.; Loes, A.N.; Gentles, L.E.; Crawford, K.H.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Antibodies elicited by mRNA-1273 vaccination bind more broadly to the receptor binding domain than do those from SARS-CoV-2 infection. Sci. Transl. Med. 2021, 13, eabi9915. [Google Scholar] [CrossRef]
- Tina, K.; Bhadra, R.; Srinivasan, N. PIC: Protein interactions calculator. Nucleic Acids Res. 2007, 35, W473–W476. [Google Scholar] [CrossRef] [Green Version]
- Ge, J.; Wang, R.; Ju, B.; Zhang, Q.; Sun, J.; Chen, P.; Zhang, S.; Tian, Y.; Shan, S.; Cheng, L.; et al. Antibody neutralization of SARS-CoV-2 through ACE2 receptor mimicry. Nat. Commun. 2021, 12, 250. [Google Scholar] [CrossRef]
- Carnell, G.W.; Ciazynska, K.A.; Wells, D.A.; Xiong, X.; Aguinam, E.T.; McLaughlin, S.H.; Mallery, D.; Ebrahimi, S.; Ceron-Gutierrez, L.; Asbach, B.; et al. SARS-CoV-2 spike protein stabilized in the closed state induces potent neutralizing responses. J. Virol. 2021, 95, e00203-21. [Google Scholar] [CrossRef]
- Wang, Z.; Schmidt, F.; Weisblum, Y.; Muecksch, F.; Barnes, C.O.; Finkin, S.; Schaefer-Babajew, D.; Cipolla, M.; Gaebler, C.; Lieberman, J.A.; et al. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Nature 2021, 592, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 2021, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Iketani, S.; Guo, Y.; Chan, J.F.W.; Wang, M.; Liu, L.; Luo, Y.; Chu, H.; Huang, Y.; Nair, M.S.; et al. Striking Antibody Evasion Manifested by the Omicron Variant of SARS-CoV-2. Nature 2021, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Planas, D.; Saunders, N.; Maes, P.; Benhassine, F.G.; Planchais, C.; Porrot, F.; Staropoli, I.; Lemoine, F.; Pere, H.; Veyer, D.; et al. Considerable escape of SARS-CoV-2 variant Omicron to antibody neutralization. Nature 2021, 1–7. [Google Scholar] [CrossRef]
- Ledford, H. How severe are Omicron infections? Nature 2021, 600, 577–578. [Google Scholar] [CrossRef]
- Barnes, C.O.; West, A.P., Jr.; Huey-Tubman, K.E.; Hoffmann, M.A.; Sharaf, N.G.; Hoffman, P.R.; Koranda, N.; Gristick, H.B.; Gaebler, C.; Muecksch, F.; et al. Structures of human antibodies bound to SARS-CoV-2 spike reveal common epitopes and recurrent features of antibodies. Cell 2020, 182, 828–842. [Google Scholar] [CrossRef]
- Ke, Z.; Oton, J.; Qu, K.; Cortese, M.; Zila, V.; McKeane, L.; Nakane, T.; Zivanov, J.; Neufeldt, C.J.; Cerikan, B.; et al. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature 2020, 588, 498–502. [Google Scholar] [CrossRef]
- Juraszek, J.; Rutten, L.; Blokland, S.; Bouchier, P.; Voorzaat, R.; Ritschel, T.; Bakkers, M.J.; Renault, L.L.; Langedijk, J.P. Stabilizing the closed SARS-CoV-2 spike trimer. Nat. Commun. 2021, 12, 244. [Google Scholar] [CrossRef]
- Xiong, X.; Qu, K.; Ciazynska, K.A.; Hosmillo, M.; Carter, A.P.; Ebrahimi, S.; Ke, Z.; Scheres, S.H.; Bergamaschi, L.; Grice, G.L.; et al. A thermostable, closed SARS-CoV-2 spike protein trimer. Nat. Struct. Mol. Biol. 2020, 27, 934–941. [Google Scholar] [CrossRef]
- Bates, T.A.; McBride, S.K.; Leier, H.C.; Guzman, G.; Lyski, Z.L.; Schoen, D.; Winders, B.; Lee, J.Y.; Lee, D.X.; Messer, W.B.; et al. Vaccination before or after SARS-CoV-2 infection leads to robust humoral response and antibodies that effectively neutralize variants. Sci. Immunol. 2022, eabn8014. [Google Scholar] [CrossRef] [PubMed]
| Escape | Escape | |||||
|---|---|---|---|---|---|---|
| NTD | RBD-Closed | RBD-Open- | RBD-Open- | COVID-19 | Vaccine | |
| nAbs | nAbs | LowClash nAbs | HighClash nAbs | Plasma | mRNA | |
| B.1.1.7 (Alpha) | 0.0 | 0.0 | 0.1 | 0.0 | 0% | 0% |
| B.1.351 (Beta) | 0.8 | 0.8 | 1.0 | 0.2 | 5% | 2% |
| B.1.617.2 (Delta) | 1.2 | 1.0 | 0.5 | 0.4 | 6% | 2% |
| B.1.1.529 (Omicron) | 1.7 | 4.1 | 5.1 | 1.6 | 42% | 10% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cia, G.; Pucci, F.; Rooman, M. Analysis of the Neutralizing Activity of Antibodies Targeting Open or Closed SARS-CoV-2 Spike Protein Conformations. Int. J. Mol. Sci. 2022, 23, 2078. https://doi.org/10.3390/ijms23042078
Cia G, Pucci F, Rooman M. Analysis of the Neutralizing Activity of Antibodies Targeting Open or Closed SARS-CoV-2 Spike Protein Conformations. International Journal of Molecular Sciences. 2022; 23(4):2078. https://doi.org/10.3390/ijms23042078
Chicago/Turabian StyleCia, Gabriel, Fabrizio Pucci, and Marianne Rooman. 2022. "Analysis of the Neutralizing Activity of Antibodies Targeting Open or Closed SARS-CoV-2 Spike Protein Conformations" International Journal of Molecular Sciences 23, no. 4: 2078. https://doi.org/10.3390/ijms23042078
APA StyleCia, G., Pucci, F., & Rooman, M. (2022). Analysis of the Neutralizing Activity of Antibodies Targeting Open or Closed SARS-CoV-2 Spike Protein Conformations. International Journal of Molecular Sciences, 23(4), 2078. https://doi.org/10.3390/ijms23042078