
Therapeutic Vaccines against Hepatocellular Carcinoma in the Immune Checkpoint Inhibitor Era: Time for Neoantigens?

 ,
,
Abstract
1. Introduction
2. Immunotherapy of HCC
3. Tumor Microenvironment and Immune Response in Liver Cancer
3.1. Antigens Associated with Liver Cancer
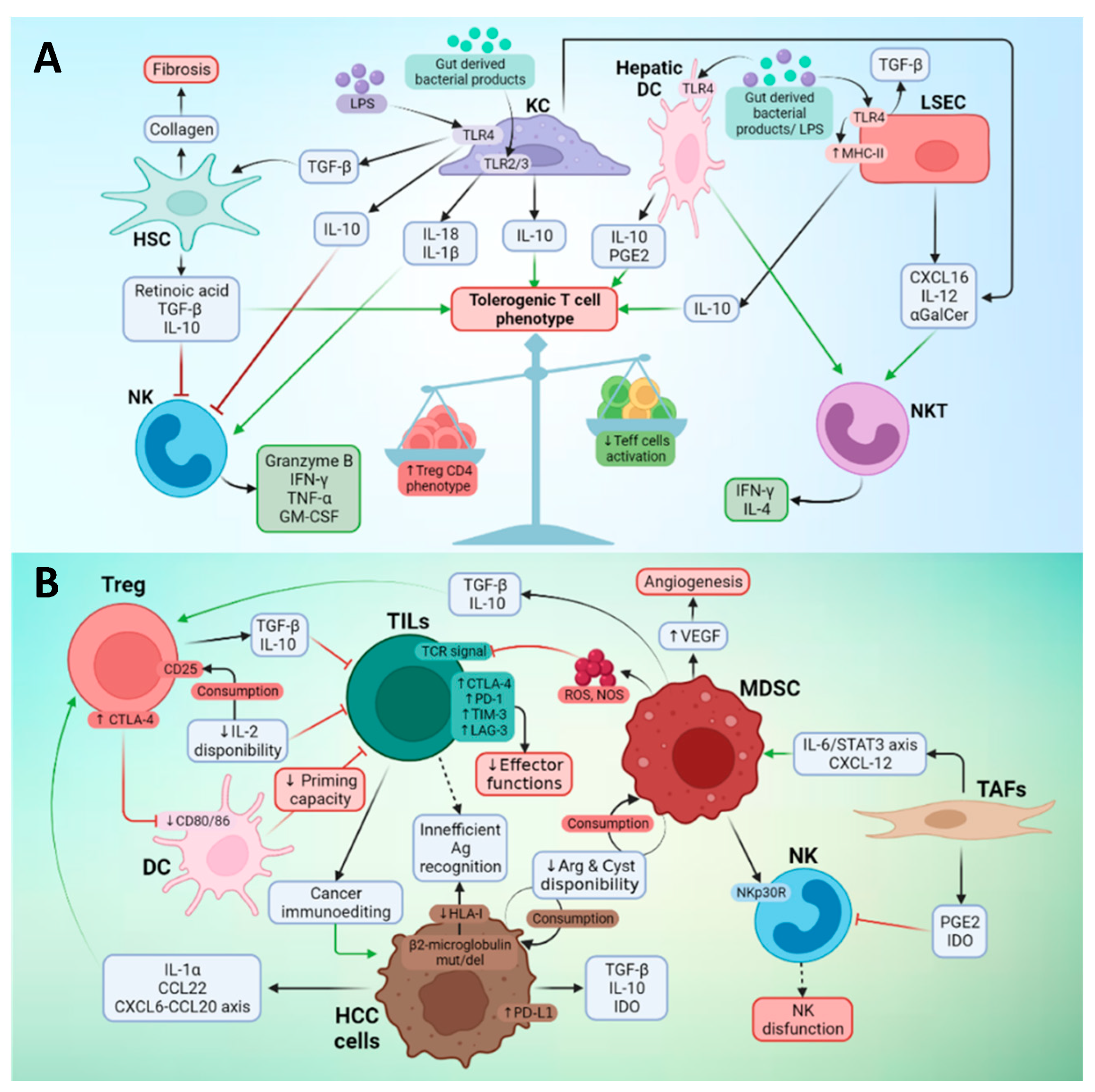
3.2. Effector Immune Cells in the TME of Liver Cancer
3.3. Suppressor Cells in the TME of Liver Cancer
3.4. Heterogeneity of TME in HCC
4. Vaccines against HCC
5. Neoantigens as New Targets for Vaccination
5.1. Mutations Involved in neoAg Generation
5.2. Factors Determining neoAg Immunogenicity
5.3. neoAg-Based Vaccines
6. Mutations in HCC as Elements for neoAg-Based Vaccines
7. neoAg-Based Vaccines as Combinatorial Partners with Immune Checkpoint Inhibitors
8. Concluding Remarks and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Suresh, D.; Srinivas, A.N.; Kumar, D.P. Etiology of Hepatocellular Carcinoma: Special Focus on Fatty Liver Disease. Front. Oncol. 2020, 10, 601710. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.H.; You, S.L.; Chen, C.J.; Liu, C.J.; Lee, C.M.; Lin, S.M.; Chu, H.C.; Wu, T.C.; Yang, S.S.; Kuo, H.S.; et al. Decreased incidence of hepatocellular carcinoma in hepatitis B vaccinees: A 20-year follow-up study. J. Natl. Cancer Inst. 2009, 101, 1348–1355. [Google Scholar] [CrossRef]
- Kilany, S.; Ata, L.; Gomaa, A.; Sabry, A.; Nada, A.; Tharwa, E.-S.; Badra, G.; Abogabal, A.; Elwaraky, M.; Moaz, E.; et al. Decreased Incidence of Hepatocellular Carcinoma after Directly Acting Antiviral Therapy in Patients with Hepatitis C–Related Advanced Fibrosis and Cirrhosis. J. Hepatocell. Carcinoma 2021, 8, 925–935. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Henry, L. Epidemiology of non-alcoholic fatty liver disease and hepatocellular carcinoma. JHEP Rep. 2021, 3, 100305. [Google Scholar] [CrossRef]
- Lee, J.-S.; Chu, I.-S.; Heo, J.; Calvisi, D.F.; Sun, Z.; Roskams, T.; Durnez, A.; Demetris, A.J.; Thorgeirsson, S.S. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology 2004, 40, 667–676. [Google Scholar] [CrossRef]
- Hoshida, Y.; Nijman, S.M.B.; Kobayashi, M.; Chan, J.A.; Brunet, J.-P.; Chiang, D.Y.; Villanueva, A.; Newell, P.; Ikeda, K.; Hashimoto, M.; et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009, 69, 7385–7392. [Google Scholar] [CrossRef]
- Boyault, S.; Rickman, D.S.; de Reyniès, A.; Balabaud, C.; Rebouissou, S.; Jeannot, E.; Hérault, A.; Saric, J.; Belghiti, J.; Franco, D.; et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 2007, 45, 42–52. [Google Scholar] [CrossRef]
- Rebouissou, S.; Nault, J.-C. Advances in molecular classification and precision oncology in hepatocellular carcinoma. J. Hepatol. 2020, 72, 215–229. [Google Scholar] [CrossRef]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; Castro de Moura, M.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Brú, C.; Bruix, J. Prognosis of hepatocellular carcinoma: The BCLC staging classification. Semin. Liver Dis. 1999, 19, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Real, M.I.; Montaña, X.; Planas, R.; Coll, S.; Aponte, J.; Ayuso, C.; Sala, M.; Muchart, J.; Solà, R.; et al. Arterial embolisation or chemoembolisation versus symptomatic treatment in patients with unresectable hepatocellular carcinoma: A randomised controlled trial. Lancet 2002, 359, 1734–1739. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Parks, A.L.; McWhirter, R.M.; Evason, K.; Kelley, R.K. Cases of spontaneous tumor regression in hepatobiliary cancers: Implications for immunotherapy? J. Gastrointest. Cancer 2015, 46, 161–165. [Google Scholar] [CrossRef]
- Kumar, A.; Le, D.T. Hepatocellular Carcinoma Regression after Cessation of Immunosuppressive Therapy. J. Clin. Oncol. 2016, 34, e90–e92. [Google Scholar] [CrossRef]
- Worns, M.A.; Weinmann, A.; Schuchmann, M.; Galle, P.R. Systemic therapies in hepatocellular carcinoma. Dig. Dis. 2009, 27, 175–188. [Google Scholar]
- Sangro, B.; Sarobe, P.; Hervás-Stubbs, S.; Melero, I. Advances in immunotherapy for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 525–543. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): A randomised, multicentre, open-label, phase 3 trial. Lancet. Oncol. 2022, 23, 77–90. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients with Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Ren, Z.; Meng, Z.; Chen, Z.; Chai, X.; Xiong, J.; Bai, Y.; Yang, L.; Zhu, H.; Fang, W.; et al. Camrelizumab in patients with previously treated advanced hepatocellular carcinoma: A multicentre, open-label, parallel-group, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 571–580. [Google Scholar] [CrossRef]
- Kelley, R.K.; Sangro, B.; Harris, W.P.; Ikeda, M.; Okusaka, T.; Kang, Y.K.; Qin, S.; Taj, W.M.D.; Lim, H.Y.; Yau, T.; et al. Efficacy, tolerability, and biologic activity of a novel regimen of tremelimumab (T) in combination with durvalumab (D) for patients (pts) with advanced hepatocellular carcinoma (aHCC). J. Clin. Oncol 2020, 38, 4508. [Google Scholar] [CrossRef]
- Lee, M.; Ryoo, B.-Y.; Hsu, C.-H.; Numata, K.; Stein, S.; Verret, W.; Hack, S.; Spahn, J.; Liu, B.; Abdullah, H.; et al. Randomised efficacy and safety results for atezolizumab (Atezo) + bevacizumab (Bev) in patients (pts) with previously untreated, unresectable hepatocellular carcinoma (HCC). Ann. Oncol. 2019, 30, v875. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.K.; Kim, T.Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef]
- Finn, R.S.; Ikeda, M.; Zhu, A.X.; Sung, M.W.; Baron, A.D.; Kudo, M.; Okusaka, T.; Kobayashi, M.; Kumada, H.; Kaneko, S.; et al. Phase ib study of lenvatinib plus pembrolizumab in patients with unresectable hepatocellular carcinoma. J. Clin. Oncol. 2020, 38, 2960–2970. [Google Scholar] [CrossRef]
- Yau, T.; Zagonel, V.; Santoro, A.; Acosta-Rivera, M.; Choo, S.P.; Matilla, A.; He, A.R.; Gracián, A.C.; El-Khoueiry, A.B.; Sangro, B.; et al. Nivolumab (NIVO) + ipilimumab (IPI) + cabozantinib (CABO) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Results from CheckMate 040. J. Clin. Oncol. 2020, 38, 478. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef]
- Flecken, T.; Schmidt, N.; Hild, S.; Gostick, E.; Drognitz, O.; Zeiser, R.; Schemmer, P.; Bruns, H.; Eiermann, T.; Price, D.A.; et al. Immunodominance and functional alterations of tumor-associated antigen-specific CD8(+) T-cell responses in hepatocellular carcinoma. Hepatology 2014, 59, 1415–1426. [Google Scholar] [CrossRef] [PubMed]
- Bricard, G.; Bouzourene, H.; Martinet, O.; Rimoldi, D.; Halkic, N.; Gillet, M.; Chaubert, P.; MacDonald, H.R.; Romero, P.; Cerottini, J.-C.; et al. Naturally acquired MAGE-A10- and SSX-2-specific CD8+ T cell responses in patients with hepatocellular carcinoma. J. Immunol. 2005, 174, 1709–1716. [Google Scholar] [CrossRef] [PubMed]
- Zerbini, A.; Pilli, M.; Soliani, P.; Ziegler, S.; Pelosi, G.; Orlandini, A.; Cavallo, C.; Uggeri, J.; Scandroglio, R.; Crafa, P.; et al. Ex vivo characterization of tumor-derived melanoma antigen encoding gene-specific CD8+ cells in patients with hepatocellular carcinoma. J. Hepatol. 2004, 40, 102–109. [Google Scholar] [CrossRef]
- Komori, H.; Nakatsura, T.; Senju, S.; Yoshitake, Y.; Motomura, Y.; Ikuta, Y.; Fukuma, D.; Yokomine, K.; Harao, M.; Beppu, T.; et al. Identification of HLA-A2- or HLA-A24-restricted CTL epitopes possibly useful for glypican-3-specific immunotherapy of hepatocellular carcinoma. Clin. Cancer Res. 2006, 12, 2689–2697. [Google Scholar] [CrossRef]
- Korangy, F.; Ormandy, L.A.; Bleck, J.S.; Klempnauer, J.; Wilkens, L.; Manns, M.P.; Greten, T.F. Spontaneous tumor-specific humoral and cellular immune responses to NY-ESO-1 in hepatocellular carcinoma. Clin. Cancer Res. 2004, 10, 4332–4341. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Fu, J.; Zhang, Z.; Zhou, L.; Qi, Z.; Xing, S.; Lv, J.; Shi, J.; Fu, B.; Liu, Z.; Zhang, J.Y.; et al. Impairment of CD4+ cytotoxic T cells predicts poor survival and high recurrence rates in patients with hepatocellular carcinoma. Hepatology 2013, 58, 139–149. [Google Scholar] [CrossRef]
- Cariani, E.; Pilli, M.; Zerbini, A.; Rota, C.; Olivani, A.; Zanelli, P.; Zanetti, A.; Trenti, T.; Ferrari, C.; Missale, G. HLA and killer immunoglobulin-like receptor genes as outcome predictors of hepatitis C virus-related hepatocellular carcinoma. Clin. Cancer Res. 2013, 19, 5465–5473. [Google Scholar] [CrossRef]
- López-Vázquez, A.; Rodrigo, L.; Martínez-Borra, J.; Pérez, R.; Rodríguez, M.; Fdez-Morera, J.L.; Fuentes, D.; Rodríguez-Rodero, S.; González, S.; López-Larrea, C. Protective effect of the HLA-Bw4I80 epitope and the killer cell immunoglobulin-like receptor 3DS1 gene against the development of hepatocellular carcinoma in patients with hepatitis C virus infection. J. Infect. Dis. 2005, 192, 162–165. [Google Scholar] [CrossRef]
- Cariani, E.; Pilli, M.; Barili, V.; Porro, E.; Biasini, E.; Olivani, A.; Dalla Valle, R.; Trenti, T.; Ferrari, C.; Missale, G. Natural killer cells phenotypic characterization as an outcome predictor of HCV-linked HCC after curative treatments. Oncoimmunology 2016, 5, e1154249. [Google Scholar] [CrossRef] [PubMed]
- Hoechst, B.; Voigtlaender, T.; Ormandy, L.; Gamrekelashvili, J.; Zhao, F.; Wedemeyer, H.; Lehner, F.; Manns, M.P.; Greten, T.F.; Korangy, F. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 2009, 50, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.F.; Yin, W.W.; Xia, Y.; Yi, Y.Y.; He, Q.F.; Wang, X.; Ren, H.; Zhang, D.Z. Liver-infiltrating CD11b—CD27—NK subsets account for NK-cell dysfunction in patients with hepatocellular carcinoma and are associated with tumor progression. Cell. Mol. Immunol. 2017, 14, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Wu, K.; Kryczek, I.; Chen, L.; Zou, W.; Welling, T.H. Kupffer cell suppression of CD8+ T cells in human hepatocellular carcinoma is mediated by B7-H1/programmed death-1 interactions. Cancer Res. 2009, 69, 8067–8075. [Google Scholar] [CrossRef]
- Ormandy, L.A.; Hillemann, T.; Wedemeyer, H.; Manns, M.P.; Greten, T.F.; Korangy, F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. 2005, 65, 2457–2464. [Google Scholar] [CrossRef]
- Shetty, S.; Lalor, P.F.; Adams, D.H. Liver sinusoidal endothelial cells—Gatekeepers of hepatic immunity. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 555–567. [Google Scholar] [CrossRef]
- Schildberg, F.A.; Hegenbarth, S.I.; Schumak, B.; Limmer, A.; Knolle, P.A. Liver sinusoidal endothelial cells veto CD8 T cell activation by antigen-presenting dendritic cells. Eur. J. Immunol. 2008, 38, 957–967. [Google Scholar] [CrossRef]
- Dou, L.; Ono, Y.; Chen, Y.F.; Thomson, A.W.; Chen, X.P. Hepatic Dendritic Cells, the Tolerogenic Liver Environment, and Liver Disease. Semin. Liver Dis. 2018, 38, 170–180. [Google Scholar] [CrossRef]
- Höchst, B.; Schildberg, F.A.; Sauerborn, P.; Gäbel, Y.A.; Gevensleben, H.; Goltz, D.; Heukamp, L.C.; Türler, A.; Ballmaier, M.; Gieseke, F.; et al. Activated human hepatic stellate cells induce myeloid derived suppressor cells from peripheral blood monocytes in a CD44-dependent fashion. J. Hepatol. 2013, 59, 528–535. [Google Scholar] [CrossRef]
- Dunham, R.M.; Thapa, M.; Velazquez, V.M.; Elrod, E.J.; Denning, T.L.; Pulendran, B.; Grakoui, A. Hepatic stellate cells preferentially induce Foxp3+ regulatory T cells by production of retinoic acid. J. Immunol. 2013, 190, 2009–2016. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.C.; Chen, C.H.; Liang, X.; Wang, L.; Gandhi, C.R.; Fung, J.J.; Lu, L.; Qian, S. Inhibition of T-cell responses by hepatic stellate cells via B7-H1-mediated T-cell apoptosis in mice. Hepatology 2004, 40, 1312–1321. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Hoechst, B.; Ormandy, L.A.; Ballmaier, M.; Lehner, F.; Kruger, C.; Manns, M.P.; Greten, T.F.; Korangy, F. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology 2008, 135, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Arihara, F.; Mizukoshi, E.; Kitahara, M.; Takata, Y.; Arai, K.; Yamashita, T.; Nakamoto, Y.; Kaneko, S. Increase in CD14+HLA-DR-/low myeloid-derived suppressor cells in hepatocellular carcinoma patients and its impact on prognosis. Cancer Immunol. Immunother. 2013, 62, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.J.; Lin, S.Z.; Zhou, L.; Xie, H.Y.; Zhou, W.H.; Taki-Eldin, A.; Zheng, S. Sen Selective recruitment of regulatory T cell through CCR6-CCL20 in hepatocellular carcinoma fosters tumor progression and predicts poor prognosis. PLoS ONE 2011, 6, e24671. [Google Scholar] [CrossRef]
- Wiedemann, G.M.; Knott, M.M.L.; Vetter, V.K.; Rapp, M.; Haubner, S.; Fesseler, J.; Kühnemuth, B.; Layritz, P.; Thaler, R.; Kruger, S.; et al. Cancer cell-derived IL-1α induces CCL22 and the recruitment of regulatory T cells. Oncoimmunology 2016, 5, e1175794. [Google Scholar] [CrossRef]
- Fu, J.; Xu, D.; Liu, Z.; Shi, M.; Zhao, P.; Fu, B.; Zhang, Z.; Yang, H.; Zhang, H.; Zhou, C.; et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology 2007, 132, 2328–2339. [Google Scholar] [CrossRef]
- Zhou, J.; Ding, T.; Pan, W.; Zhu, L.Y.; Li, L.; Zheng, L. Increased intratumoral regulatory T cells are related to intratumoral macrophages and poor prognosis in hepatocellular carcinoma patients. Int. J. Cancer 2009, 125, 1640–1648. [Google Scholar] [CrossRef] [PubMed]
- Bricard, G.; Cesson, V.; Devevre, E.; Bouzourene, H.; Barbey, C.; Rufer, N.; Im, J.S.; Alves, P.M.; Martinet, O.; Halkic, N.; et al. Enrichment of human CD4+ V(alpha)24/Vbeta11 invariant NKT cells in intrahepatic malignant tumors. J. Immunol. 2009, 182, 5140–5151. [Google Scholar] [CrossRef] [PubMed]
- Cariani, E.; Pilli, M.; Zerbini, A.; Rota, C.; Olivani, A.; Pelosi, G.; Schianchi, C.; Soliani, P.; Campanini, N.; Silini, E.M.; et al. Immunological and molecular correlates of disease recurrence after liver resection for hepatocellular carcinoma. PLoS ONE 2012, 7, e32493. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Lao, X.M.; Chen, M.M.; Liu, R.X.; Wei, Y.; Ouyang, F.Z.; Chen, D.P.; Zhao, X.Y.; Zhao, Q.; Li, X.F.; et al. PD-1hi Identifies a Novel Regulatory B-cell Population in Human Hepatoma That Promotes Disease Progression. Cancer Discov. 2016, 6, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Hoechst, B.; Gamrekelashvili, J.; Ormandy, L.A.; Voigtländer, T.; Wedemeyer, H.; Ylaya, K.; Wang, X.W.; Hewitt, S.M.; Manns, M.P.; et al. Human CCR4+ CCR6+ Th17 cells suppress autologous CD8+ T cell responses. J. Immunol. 2012, 188, 6055–6062. [Google Scholar] [CrossRef]
- Matsubara, T.; Kanto, T.; Kuroda, S.; Yoshio, S.; Higashitani, K.; Kakita, N.; Miyazaki, M.; Sakakibara, M.; Hiramatsu, N.; Kasahara, A.; et al. TIE2-expressing monocytes as a diagnostic marker for hepatocellular carcinoma correlates with angiogenesis. Hepatology 2013, 57, 1416–1425. [Google Scholar] [CrossRef]
- Han, Y.; Chen, Z.; Yang, Y.; Jiang, Z.; Gu, Y.; Liu, Y.; Lin, C.; Pan, Z.; Yu, Y.; Jiang, M.; et al. Human CD14+ CTLA-4+ regulatory dendritic cells suppress T-cell response by cytotoxic T-lymphocyte antigen-4-dependent IL-10 and indoleamine-2,3-dioxygenase production in hepatocellular carcinoma. Hepatology 2014, 59, 567–579. [Google Scholar] [CrossRef]
- Zhou, S.L.; Zhou, Z.J.; Hu, Z.Q.; Huang, X.W.; Wang, Z.; Chen, E.B.; Fan, J.; Cao, Y.; Dai, Z.; Zhou, J. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology 2016, 150, 1646–1658.e17. [Google Scholar] [CrossRef]
- Affo, S.; Yu, L.X.; Schwabe, R.F. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu. Rev. Pathol. 2017, 12, 153–186. [Google Scholar] [CrossRef]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef]
- Ally, A.; Balasundaram, M.; Carlsen, R.; Chuah, E.; Clarke, A.; Dhalla, N.; Holt, R.A.; Jones, S.J.M.; Lee, D.; Ma, Y.; et al. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Kurebayashi, Y.; Ojima, H.; Tsujikawa, H.; Kubota, N.; Maehara, J.; Abe, Y.; Kitago, M.; Shinoda, M.; Kitagawa, Y.; Sakamoto, M. Landscape of immune microenvironment in hepatocellular carcinoma and its additional impact on histological and molecular classification. Hepatology 2018, 68, 1025–1041. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lou, Y.; Yang, J.; Wang, J.; Feng, J.; Zhao, Y.; Wang, L.; Huang, X.; Fu, Q.; Ye, M.; et al. Integrated multiomic analysis reveals comprehensive tumour heterogeneity and novel immunophenotypic classification in hepatocellular carcinomas. Gut 2019, 68, 2019–2031. [Google Scholar] [CrossRef] [PubMed]
- Buonaguro, L. Developments in cancer vaccines for hepatocellular carcinoma. Cancer Immunol. Immunother. 2016, 65, 93–99. [Google Scholar] [CrossRef]
- Buonaguro, L. New vaccination strategies in liver cancer. Cytokine Growth Factor Rev. 2017, 36, 125–129. [Google Scholar] [CrossRef]
- Butterfield, L.H.; Ribas, A.; Meng, W.S.; Dissette, V.B.; Amarnani, S.; Vu, H.T.; Seja, E.; Todd, K.; Glaspy, J.A.; McBride, W.H.; et al. T-cell responses to HLA-A*0201 immunodominant peptides derived from alpha-fetoprotein in patients with hepatocellular cancer. Clin. Cancer Res. 2003, 9, 5902–5908. [Google Scholar]
- Butterfield, L.H.; Ribas, A.; Dissette, V.B.; Lee, Y.; Yang, J.Q.; De la Rocha, P.; Duran, S.D.; Hernandez, J.; Seja, E.; Potter, D.M.; et al. A phase I/II trial testing immunization of hepatocellular carcinoma patients with dendritic cells pulsed with four alpha-fetoprotein peptides. Clin. Cancer Res. 2006, 12, 2817–2825. [Google Scholar] [CrossRef]
- Sawada, Y.; Yoshikawa, T.; Ofuji, K.; Yoshimura, M.; Tsuchiya, N.; Takahashi, M.; Nobuoka, D.; Gotohda, N.; Takahashi, S.; Kato, Y.; et al. Phase II study of the GPC3-derived peptide vaccine as an adjuvant therapy for hepatocellular carcinoma patients. Oncoimmunology 2016, 5, e1129483. [Google Scholar] [CrossRef]
- Greten, T.F.; Forner, A.; Korangy, F.; N’Kontchou, G.; Barget, N.; Ayuso, C.; Ormandy, L.A.; Manns, M.P.; Beaugrand, M.; Bruix, J. A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer 2010, 10, 209. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, Y.; Lee, M.; Heo, M.K.; Song, J.S.; Kim, K.H.; Lee, H.; Yi, N.J.; Lee, K.W.; Suh, K.S.; et al. A phase I/IIa study of adjuvant immunotherapy with tumour antigen-pulsed dendritic cells in patients with hepatocellular carcinoma. Br. J. Cancer 2015, 113, 1666–1676. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.C.; Wang, H.C.; Hung, C.F.; Huang, P.F.; Lia, C.R.; Chen, M.F. Vaccination of advanced hepatocellular carcinoma patients with tumor lysate-pulsed dendritic cells: A clinical trial. J. Immunother. 2005, 28, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.H.; Midgley, R.S.; Mirza, N.; Torr, E.E.; Ahmed, F.; Steele, J.C.; Steven, N.M.; Kerr, D.J.; Young, L.S.; Adams, D.H. A phase II study of adoptive immunotherapy using dendritic cells pulsed with tumor lysate in patients with hepatocellular carcinoma. Hepatology 2009, 49, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Mizukoshi, E.; Kobayashi, E.; Tamai, T.; Hamana, H.; Ozawa, T.; Kishi, H.; Kitahara, M.; Yamashita, T.; Arai, K.; et al. Association between High-Avidity T-Cell Receptors, Induced by α-Fetoprotein-Derived Peptides, and Anti-Tumor Effects in Patients with Hepatocellular Carcinoma. Gastroenterology 2017, 152, 1395–1406.e10. [Google Scholar] [CrossRef]
- Sawada, Y.; Yoshikawa, T.; Nobuoka, D.; Shirakawa, H.; Kuronuma, T.; Motomura, Y.; Mizuno, S.; Ishii, H.; Nakachi, K.; Konishi, M.; et al. Phase I trial of a glypican-3-derived peptide vaccine for advanced hepatocellular carcinoma: Immunologic evidence and potential for improving overall survival. Clin. Cancer Res. 2012, 18, 3686–3696. [Google Scholar] [CrossRef]
- Iwashita, Y.; Tahara, K.; Goto, S.; Sasaki, A.; Kai, S.; Seike, M.; Chen, C.L.; Kawano, K.; Kitano, S. A phase I study of autologous dendritic cell-based immunotherapy for patients with unresectable primary liver cancer. Cancer Immunol. Immunother. 2003, 52, 155–161. [Google Scholar] [CrossRef]
- Campillo-Davo, D.; Flumens, D.; Lion, E. The Quest for the Best: How TCR Affinity, Avidity, and Functional Avidity Affect TCR-Engineered T-Cell Antitumor Responses. Cells 2020, 9, 1720. [Google Scholar] [CrossRef]
- Kastenmuller, W.; Kastenmuller, K.; Kurts, C.; Seder, R.A. Dendritic cell-targeted vaccines—Hope or hype? Nat. Rev. Immunol. 2014, 14, 705–711. [Google Scholar] [CrossRef]
- Shang, N.; Figini, M.; Shangguan, J.; Wang, B.; Sun, C.; Pan, L.; Ma, Q.; Zhang, Z. Dendritic cells based immunotherapy. Am. J. Cancer Res. 2017, 7, 2091–2102. [Google Scholar]
- Silva, L.; Egea, J.; Villanueva, L.; Ruiz, M.; Llopiz, D.; Repáraz, D.; Aparicio, B.; Lasarte-cia, A.; Lasarte, J.J.; de Galarreta, M.R.; et al. Cold-Inducible RNA Binding Protein as a Vaccination Platform to Enhance Immunotherapeutic Responses against Hepatocellular Carcinoma. Cancers 2020, 12, 3397. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, A.; Gao, M.; Yan, Y.; Zhang, W. Potential therapeutic value of dendritic cells loaded with NY-ESO-1 protein for the immunotherapy of advanced hepatocellular carcinoma. Int. J. Mol. Med. 2013, 32, 1366–1372. [Google Scholar] [CrossRef][Green Version]
- Charneau, J.; Suzuki, T.; Shimomura, M.; Fujinami, N.; Nakatsura, T. Peptide-Based Vaccines for Hepatocellular Carcinoma: A Review of Recent Advances. J. Hepatocell. Carcinoma 2021, 8, 1035–1054. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef]
- Van Buuren, M.M.; Calis, J.J.A.; Schumacher, T.N.M. High sensitivity of cancer exome-based CD8 T cell neo-antigen identification. Oncoimmunology 2014, 3, e28836. [Google Scholar] [CrossRef]
- Coulie, P.G.; Lehmann, F.; Lethé, B.; Herman, J.; Lurquin, C.; Andrawiss, M.; Boon, T. A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc. Natl. Acad. Sci. USA 1995, 92, 7976–7980. [Google Scholar] [CrossRef]
- Linnemann, C.; Van Buuren, M.M.; Bies, L.; Verdegaal, E.M.E.; Schotte, R.; Calis, J.J.A.; Behjati, S.; Velds, A.; Hilkmann, H.; Atmioui, D.E.; et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat. Med. 2015, 21, 81–85. [Google Scholar] [CrossRef]
- Wang, R.F.; Wang, X.; Atwood, A.C.; Topalian, S.L.; Rosenberg, S.A. Cloning genes encoding MHC class II-restricted antigens: Mutated CDC27 as a tumor antigen. Science 1999, 284, 1351–1354. [Google Scholar] [CrossRef]
- Lennerz, V.; Fatho, M.; Gentilini, C.; Frye, R.A.; Lifke, A.; Ferel, D.; Wolfel, C.; Huber, C.; Wolfel, T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc. Natl. Acad. Sci. USA 2005, 102, 16013–16018. [Google Scholar] [CrossRef]
- Hunger, R.E.; Brand, C.U.; Streit, M.; Eriksen, J.A.; Gjertsen, M.K.; Saeterdal, I.; Braathen, L.R.; Gaudernack, G. Successful induction of immune responses against mutant ras in melanoma patients using intradermal injection of peptides and GM-CSF as adjuvant. Exp. Dermatol. 2001, 10, 161–167. [Google Scholar] [CrossRef]
- Andersen, M.H.; Fensterle, J.; Ugurel, S.; Reker, S.; Houben, R.; Guldberg, P.; Berger, T.G.; Schadendorf, D.; Trefzer, U.; Bröcker, E.B.; et al. Immunogenicity of constitutively active V599EBRaf. Cancer Res. 2004, 64, 5456–5460. [Google Scholar] [CrossRef]
- Holmström, M.O.; Hjortsø, M.D.; Ahmad, S.M.M.; Martinenaite, E.; Riley, C.; Straten, P.; Svane, I.M.; Hasselbalch, H.C.; Andersen, M.H. The JAK2V617F mutation is a target for specific T cells in the JAK2V617F-positive myeloproliferative neoplasms. Leukemia 2017, 31, 495–498. [Google Scholar] [CrossRef]
- Inderberg, E.M.; Wälchli, S.; Myhre, M.R.; Trachsel, S.; Almåsbak, H.; Kvalheim, G.; Gaudernack, G. T cell therapy targeting a public neoantigen in microsatellite instable colon cancer reduces in vivo tumor growth. Oncoimmunology 2017, 6, e1302631. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef]
- Fritsch, E.F.; Rajasagi, M.; Ott, P.A.; Brusic, V.; Hacohen, N.; Wu, C.J. HLA-binding properties of tumor neoepitopes in humans. Cancer Immunol. Res. 2014, 2, 522–529. [Google Scholar] [CrossRef]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al Bakir, M.; et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol. 2017, 18, 1009–1021. [Google Scholar] [CrossRef]
- Laumont, C.M.; Daouda, T.; Laverdure, J.P.; Bonneil, É.; Caron-Lizotte, O.; Hardy, M.P.; Granados, D.P.; Durette, C.; Lemieux, S.; Thibault, P.; et al. Global proteogenomic analysis of human MHC class I-associated peptides derived from non-canonical reading frames. Nat. Commun. 2016, 7, 10238. [Google Scholar] [CrossRef]
- Yotnda, P.; Garcia, F.; Peuchmaur, M.; Grandchamp, B.; Duval, M.; Lemonnier, F.; Vilmer, E.; Langlade-Demoyen, P. Cytotoxic T cell response against the chimeric ETV6-AML1 protein in childhood acute lymphoblastic leukemia. J. Clin. Investig. 1998, 102, 455–462. [Google Scholar] [CrossRef]
- Popović, J.; Li, A.P.; Kloetzel, P.M.; Leisegang, M.; Uckert, W.; Blankenstein, T. The only proposed T-cell epitope derived from the TEL-AML1 translocation is not naturally processed. Blood 2011, 118, 946–954. [Google Scholar] [CrossRef]
- Dalet, A.; Robbins, P.F.; Stroobant, V.; Vigneron, N.; Li, Y.F.; El-Gamil, M.; Hanada, K.I.; Yang, J.C.; Rosenberg, S.A.; Van Den Eyndea, B.J. An antigenic peptide produced by reverse splicing and double asparagine deamidation. Proc. Natl. Acad. Sci. USA 2011, 108, E323–E331. [Google Scholar] [CrossRef]
- Cobbold, M.; De La Peña, H.; Norris, A.; Polefrone, J.M.; Qian, J.; English, A.M.; Cummings, K.L.; Penny, S.; Turner, J.E.; Cottine, J.; et al. MHC class I-associated phosphopeptides are the targets of memory-like immunity in leukemia. Sci. Transl. Med. 2013, 5, 203ra125. [Google Scholar] [CrossRef]
- Liepe, J.; Marino, F.; Sidney, J.; Jeko, A.; Bunting, D.E.; Sette, A.; Kloetzel, P.M.; Stumpf, M.P.H.; Heck, A.J.R.; Mishto, M. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science 2016, 354, 354–358. [Google Scholar] [CrossRef]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef]
- Verdegaal, E.M.E.; De Miranda, N.F.C.C.; Visser, M.; Harryvan, T.; Van Buuren, M.M.; Andersen, R.S.; Hadrup, S.R.; Van Der Minne, C.E.; Schotte, R.; Spits, H.; et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature 2016, 536, 91–95. [Google Scholar] [CrossRef]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Lower, M.; van de Roemer, N.; de Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012, 72, 1081–1091. [Google Scholar] [CrossRef]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Schumacher, T.; Bunse, L.; Pusch, S.; Sahm, F.; Wiestler, B.; Quandt, J.; Menn, O.; Osswald, M.; Oezen, I.; Ott, M.; et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014, 512, 324–327. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Fujimoto, A.; Furuta, M.; Totoki, Y.; Tsunoda, T.; Kato, M.; Shiraishi, Y.; Tanaka, H.; Taniguchi, H.; Kawakami, Y.; Ueno, M.; et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 2016, 48, 500–509. [Google Scholar] [CrossRef]
- Cai, Z.; Su, X.; Qiu, L.; Li, Z.; Li, X.; Dong, X.; Wei, F.; Zhou, Y.; Luo, L.; Chen, G.; et al. Personalized neoantigen vaccine prevents postoperative recurrence in hepatocellular carcinoma patients with vascular invasion. Mol. Cancer 2021, 20, 164. [Google Scholar] [CrossRef]
- Löffler, M.W.; Mohr, C.; Bichmann, L.; Freudenmann, L.K.; Walzer, M.; Schroeder, C.M.; Trautwein, N.; Hilke, F.J.; Zinser, R.S.; Mühlenbruch, L.; et al. Multi-omics discovery of exome-derived neoantigens in hepatocellular carcinoma. Genome Med. 2019, 11, 28. [Google Scholar] [CrossRef]
- Vrecko, S.; Guenat, D.; Mercier-Letondal, P.; Faucheu, H.; Dosset, M.; Royer, B.; Galaine, J.; Boidot, R.; Kim, S.; Jary, M.; et al. Personalized identification of tumor-associated immunogenic neoepitopes in hepatocellular carcinoma in complete remission after sorafenib treatment. Oncotarget 2018, 9, 35394–35407. [Google Scholar] [CrossRef]
- Chen, P.; Fang, Q.X.; Chen, D.B.; Chen, H.S. Neoantigen vaccine: An emerging immunotherapy for hepatocellular carcinoma. World J. Gastrointest. Oncol. 2021, 13, 673–683. [Google Scholar] [CrossRef]
- Liu, X.; Li, Z.; Cai, Z.; Chen, G.; Liu, J. Neoantigen profile of hepatocellular carcinoma reveals its correlation with tumour progression and clonal evolution. Ann. Oncol. 2019, 30, ix110–ix111. [Google Scholar] [CrossRef]
- Petrizzo, A.; Tagliamonte, M.; Mauriello, A.; Costa, V.; Aprile, M.; Esposito, R.; Caporale, A.; Luciano, A.; Arra, C.; Tornesello, M.L.; et al. Unique true predicted neoantigens (TPNAs) correlates with anti-tumor immune control in HCC patients. J. Transl. Med. 2018, 16, 286. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Jiang, J.; Zhan, M.; Zhang, H.; Wang, Q.T.; Sun, S.N.; Guo, X.K.; Yin, H.; Wei, Y.; Liu, J.O.; et al. Targeting Neoantigens in Hepatocellular Carcinoma for Immunotherapy: A Futile Strategy? Hepatology 2021, 73, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Vercher, E.; Tamayo, I.; Mancheño, U.; Elizalde, E.; Conde, E.; Reparaz, D.; Lasarte, J.J.; Villar, V.; Iñarrairaegui, M.; Sangro, B.; et al. Identification of neoantigen-reactive T cells in hepatocellular carcinoma: Implication in adoptive T cell therapy. J. Hepatol. 2020, 73, S39–S40. [Google Scholar] [CrossRef]
- Liu, T.; Tan, J.; Wu, M.; Fan, W.; Wei, J.; Zhu, B.; Guo, J.; Wang, S.; Zhou, P.; Zhang, H.; et al. High-affinity neoantigens correlate with better prognosis and trigger potent antihepatocellular carcinoma (HCC) activity by activating CD39+CD8+T cells. Gut 2021, 70, 1965–1977. [Google Scholar] [CrossRef]
- Riviere, P.; Goodman, A.M.; Okamura, R.; Barkauskas, D.A.; Whitchurch, T.J.; Lee, S.; Khalid, N.; Collier, R.; Mareboina, M.; Frampton, G.M.; et al. High Tumor Mutational Burden Correlates with Longer Survival in Immunotherapy-Naïve Patients with Diverse Cancers. Mol. Cancer Ther. 2020, 19, 2139–2145. [Google Scholar] [CrossRef]
- Mauriello, A.; Zeuli, R.; Cavalluzzo, B.; Petrizzo, A.; Tornesello, M.L.; Buonaguro, F.M.; Ceccarelli, M.; Tagliamonte, M.; Buonaguro, L. High Somatic Mutation and Neoantigen Burden Do Not Correlate with Decreased Progression-Free Survival in HCC Patients not Undergoing Immunotherapy. Cancers 2019, 11, 1824. [Google Scholar] [CrossRef]
- Yang, H.; Sun, L.; Guan, A.; Yin, H.; Liu, M.; Mao, X.; Xu, H.; Zhao, H.; Lu, X.; Sang, X.; et al. Unique TP53 neoantigen and the immune microenvironment in long-term survivors of Hepatocellular carcinoma. Cancer Immunol. Immunother. 2021, 70, 667–677. [Google Scholar] [CrossRef]
- Yarchoan, M.; Gane, E.; Marron, T.; Rochestie, S.; Cooch, N.; Peters, J.; Csiki, I.; Perales-Puchalt, A.; Sardesai, N. Personalized DNA neoantigen vaccine (GNOS-PV02) in combination with plasmid IL-12 and pembrolizumab for the treatment of patients with advanced hepatocellular carcinoma. Proc. J. ImmunoTherapy Cancer 2021, 9, A481. [Google Scholar] [CrossRef]
- Cheever, M.A.; Higano, C.S. PROVENGE (Sipuleucel-T) in prostate cancer: The first FDA-approved therapeutic cancer vaccine. Clin. Cancer Res. 2011, 17, 3520–3526. [Google Scholar] [CrossRef]
- Massarelli, E.; William, W.; Johnson, F.; Kies, M.; Ferrarotto, R.; Guo, M.; Feng, L.; Lee, J.J.; Tran, H.; Kim, Y.U.; et al. Combining Immune Checkpoint Blockade and Tumor-Specific Vaccine for Patients with Incurable Human Papillomavirus 16-Related Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 67–73. [Google Scholar] [CrossRef]
- Youn, J.W.; Hur, S.Y.; Woo, J.W.; Kim, Y.M.; Lim, M.C.; Park, S.Y.; Seo, S.S.; No, J.H.; Kim, B.G.; Lee, J.K.; et al. Pembrolizumab plus GX-188E therapeutic DNA vaccine in patients with HPV-16-positive or HPV-18-positive advanced cervical cancer: Interim results of a single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 1653–1660. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanović, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu-Lieskovan, S.; Chmielowski, B.; Govindan, R.; Naing, A.; Bhardwaj, N.; Margolin, K.; Awad, M.M.; Hellmann, M.D.; Lin, J.J.; et al. A Phase Ib Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients with Advanced Melanoma, Non-small Cell Lung Cancer, or Bladder Cancer. Cell 2020, 183, 347–362.e24. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Treatment | Patients (n) | Setting | ORR% (CRR%) | mOS (Months) |
|---|---|---|---|---|
| Nivolumab [21] | 371 | 1 L | 15(4) | 16.4 |
| Pembrolizumab [22] | 278 | 2 L | 18 (2) | 13.9 |
| Camrelizumab [23] | 217 | 2 L | 15 (0) | 13.8 |
| Durvalumab [24] | 104 | 1 L/2 L | 11 (0) | 13.6 |
| Tremelimumab [24] | 69 | 1 L/2 L | 7 (0) | 15.1 |
| Atezolizumab [25] | 59 | 1 L | 17 (5) | NA |
| Durvalumab and Tremelimumab (different doses) [24] | 159 | 1 L/2 L | 9.5–24 (1–2) NA | 11.3–18.7 |
| Nivolumab and Ipilimumab (different doses) [26] | 148 | 2 L | 31–32 (0–8) | 12.5–22.8 |
| Pembrolizumab and Levantinib [27] | 100 | 1 L | 36 (1) | 22 |
| Nivolumab and Cabozantinib [28] | 36 | 1 L/2 L | 14 (3) | 21.5 |
| Nivolumab, Ipilimumab and Cabozantinib [28] | 35 | 1 L/2 L | 31 (6) | NE |
| Atezolizumab and Bevacizumab [29] | 336 | 1 L | 27 (6) | NE |
| Vaccine | Patient Inclusion Criteria | Patients (n) | Immune Response (%) | Clinical Response CR/PR/SD/PD | Observations |
|---|---|---|---|---|---|
| AFP HLA-A*02 restricted peptides+IFA | AFP+ tumors from (stage IV patients) [77] | 6 | 66 | 0/0/0/6 | Increased CTL response |
| AFP HLA-A*24:02 restricted peptides+IFA | Stage B/C tumors [84] | 15 | 33 | 1/0/8/6 | Increased CTL response |
| GPC3 HLA-A*24:02 and HLA-A*02- restricted peptides+IFA | Advanced or metastatic HCC [85] | 33 | 91 | 0/1/19/13 | Antitumor efficacy |
| GPC3 HLA-A*24:02 and HLA-A*02- restricted peptides+IFA | Patients undergone curative resection Vaccines as Adjuvant therapy [79] | 41 | 85 | Not applicable | Improved recurrence rate |
| Gv1001 peptide + GM-CSF + cyclophosphamide | Advanced-stage HCC with no previous antitumor treatment [80] | 37 | 0 | 0/0/17/20 | None clinical nor detected immunological response |
| DCs pulsed with AFP HLA-A*02 restricted peptides | Stage IV patients pretreated with surgery and/or chemotherapy [78] | 10 | 60 | 0/1/0/9 | No objective clinical responses |
| DCs pulsed with fused recombinant proteins (AFP, MAGE-1 and GPC-3) | After surgical resection and locoregional therapy [81] | 12 | 92 | Not applicable | Trend to improved survival |
| DCs pulsed with autologous tumor lysate | Advanced HCC [82] | 31 | 0 * | 0/4/17/10 | Improved survival |
| DCs pulsed with autologous tumor lysate | Unresectable HCC [86] | 8 | 62 | 0/0/4/3 | Immune response generation |
| DCs pulsed with hepatoma cell-line (HEP-G2) lysate | No other therapeutic option [83] | 35 | 11.4 | 0/1/6/18 | Evidence of antitumor efficacy |
| Vaccine Type | Advantages | Disadvantages | |
|---|---|---|---|
| Peptides | Easy preparation Known target Ag | Adjuvants required HLA restricted Limited Ag repertoire | |
| DCs | Does not require adjuvants | Labor-intensive in CMCF Individualized manufacture | |
| Peptide pulsed | Known target Ag | HLA restricted Limited Ag repertoire | |
| Protein pulsed | Not HLA restricted Known target Ag | Protein synthesis is more challenging Limited Ag repertoire | |
| Tumor lysate pulsed | Not HLA restricted Full Ag repertoire available | Tumor samples not always available Predominance of self-antigens that may eclipse tumor antigens | |
| Cell line pulsed | Not HLA restricted Unlimited Ag source | Ag repertoire may not coincide Responses against cell line-specific Ags | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Repáraz, D.; Aparicio, B.; Llopiz, D.; Hervás-Stubbs, S.; Sarobe, P. Therapeutic Vaccines against Hepatocellular Carcinoma in the Immune Checkpoint Inhibitor Era: Time for Neoantigens? Int. J. Mol. Sci. 2022, 23, 2022. https://doi.org/10.3390/ijms23042022
Repáraz D, Aparicio B, Llopiz D, Hervás-Stubbs S, Sarobe P. Therapeutic Vaccines against Hepatocellular Carcinoma in the Immune Checkpoint Inhibitor Era: Time for Neoantigens? International Journal of Molecular Sciences. 2022; 23(4):2022. https://doi.org/10.3390/ijms23042022
Chicago/Turabian StyleRepáraz, David, Belén Aparicio, Diana Llopiz, Sandra Hervás-Stubbs, and Pablo Sarobe. 2022. "Therapeutic Vaccines against Hepatocellular Carcinoma in the Immune Checkpoint Inhibitor Era: Time for Neoantigens?" International Journal of Molecular Sciences 23, no. 4: 2022. https://doi.org/10.3390/ijms23042022
APA StyleRepáraz, D., Aparicio, B., Llopiz, D., Hervás-Stubbs, S., & Sarobe, P. (2022). Therapeutic Vaccines against Hepatocellular Carcinoma in the Immune Checkpoint Inhibitor Era: Time for Neoantigens? International Journal of Molecular Sciences, 23(4), 2022. https://doi.org/10.3390/ijms23042022